

Abstract
Alzheimer’s disease (AD) is one of the most common causes of dementia in elderly populations. A multifactorial and complex etiology has hindered the establishment of successful disease-modifying and retarding treatments. An important molecular target that has a close link with the disease’s pathophysiology is glycogen synthase kinase 3β (GSK-3β). GSK-3β is thought to be an important bridge between amyloid and tau pathologies, the two principle pathogenic hallmarks of the disease. In particular, its kinase activity is thought to be a contributing factor for initiating aberrant tau hyperphosphorylation toward neurodegenerative progression. To identify potential inhibitors for GSK-3β, a pharmacophore-based virtual screening was used on the VITAS-M Lab database, a large database of small molecules. A co-crystal ligand employed as the template allowed the screening of roughly 200,000 compounds. Compounds successfully screened were selected on the basis of the Phase screen score combining vector alignments, volume scores, and site matching parameters. Using a cutoff score of 1.7, 174 compounds were docked using the Glide tool for molecular docking to further identify potential high-affinity binding ligands. Finally, four chemicals with the best binding scores (cutoff Glide GScore values of − 8 kcal/mol) were identified. Among these, 3-(2-oxo-2H-chromen-3-yl)-N-(4-sulfamoylphenyl) benzamide (VL-1) and trimethylsilyl trifluoromethanesulfonate (VL-2) showed strong and stable binding interactions, as evidenced by molecular dynamics simulation (MDS). The results suggest that VL-1 and VL-2 may serve as promising lead compounds for GSK-3β-based anti-AD therapeutics. However, further in vivo mechanistic validation is warrantied to confirm their therapeutic applicability.
Supplementary Information
The online version contains supplementary material available at 10.1007/s10571-025-01568-8.
Keywords: Alzheimer’s disease, Dementia, Glycogen synthase kinase 3β, Virtual screening, ADMET, Molecular simulation
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that leads to memory impairment, cognitive decline, and overall functional deterioration (Monteiro et al. 2023). As the global population ages and life expectancy increases, AD has become a major health concern worldwide. It is now the sixth leading cause of death, responsible for almost a third of fatalities among older adults in the United States (Alzheimer’s Association Report 2024). The disease typically manifests in its later stages in life and is marked by reduced levels of cerebrospinal fluid amyloid β (CSF Aβ) and an enhanced accumulation of Aβ in the central nervous system (CNS). In addition, levels of oligomerized hyperphosphorylated tau are significantly upregulated. Hence, pathogenic amyloid and tau species cumulatively result in synaptic, bioenergetic, redox and neuroinflammatory deficits, resulting in progressive neuronal degeneration and loss (Gallardo and Holtzman 2019). In spite of appreciable advancements in our understanding of the pathophysiology of AD, current therapeutic regimens are largely limited to providing symptomatic relief, with no effective disease-modifying or retarding therapies currently available. A major cause of this may be the multifactorial origin and heterogeneous nature of the disease etiology (Ferrari and Sorbi 2021; Jellinger 2022). This highlights the need for combinatorial therapeutic regimens, based upon pharmacological, behavioral, and environmental interventions, aimed at beneficially altering the course of AD cases in a patient-specific manner.
Glycogen synthase kinase 3β, abbreviated as GSK-3β, is a serine/threonine kinase crucial to various physiological processes of carbohydrate metabolism, cell growth and survival, inflammation, microtubule dynamics, gene expression, and protein homeostasis (Mandlik et al. 2024). Aberrant changes in the functions of GSK-3β are associated with all pathological hallmarks of AD, including amyloid and tau pathologies, synaptic signaling and plasticity deficits, and memory and cognition imapriments (Lauretti et al. 2020). GSK-3β possesses a well-conserved ATP-binding pocket within its catalytic domain, the primary site of ligand binding. This pocket is defined by significant residues such as Val135, Asp181, Lys183, and Arg141, which are engaged in substrate binding. The ATP-binding site is flanked by a deep hydrophobic pocket and a phosphate-binding loop, making it an appealing target for small-molecule inhibitors. Involvement of GSK-3β in tau hyperphosphorylation has made selective inhibition of this enzymatic pocket a prime interest in the development of potential inhibitors. Indeed, because of the multimodal contributions of GSK-3β to the disease pathogenic mechanisms, it has increasingly been envisioned as a notable therapeutic target for pharmacological interventions against AD (Lauretti et al. 2020; Mandlik et al. 2024).
Direct involvement of GSK-3β in the hyperphosphorylation of tau fragments which increases the latter’s propensity for oligomerization (Hernandez et al. 2012) indicates that GSK-3β-tau interaction may also contribute to tauopathies other than AD, and α-synucleinopathies which are closely associated with the pathology of CNS disorders such as Parkinson’s disease (PD) and stroke (Mehta et al. 2023). While studies in the past have evaluated the efficacy of multiple pharmacological agents as GSK-3β inhibitors, there has been a critical lack of clinically applicable drugs. This study is hence directed at the initial identification of high-affinity binding ligands for the active site of GSK-3β using extensive virtual screening of 200,000 chemicals based on the pharmacophore model, followed by confirmation by ADMET and molecular dynamics simulation (MDS) analyses (Fig. 1). Our final identification of two novel ligands for GSK-3β may pave the way for the development of pharmacological interventions against AD, however further experimental studies will be required to confirm the applicability of our findings under clinical settings.
Fig. 1.
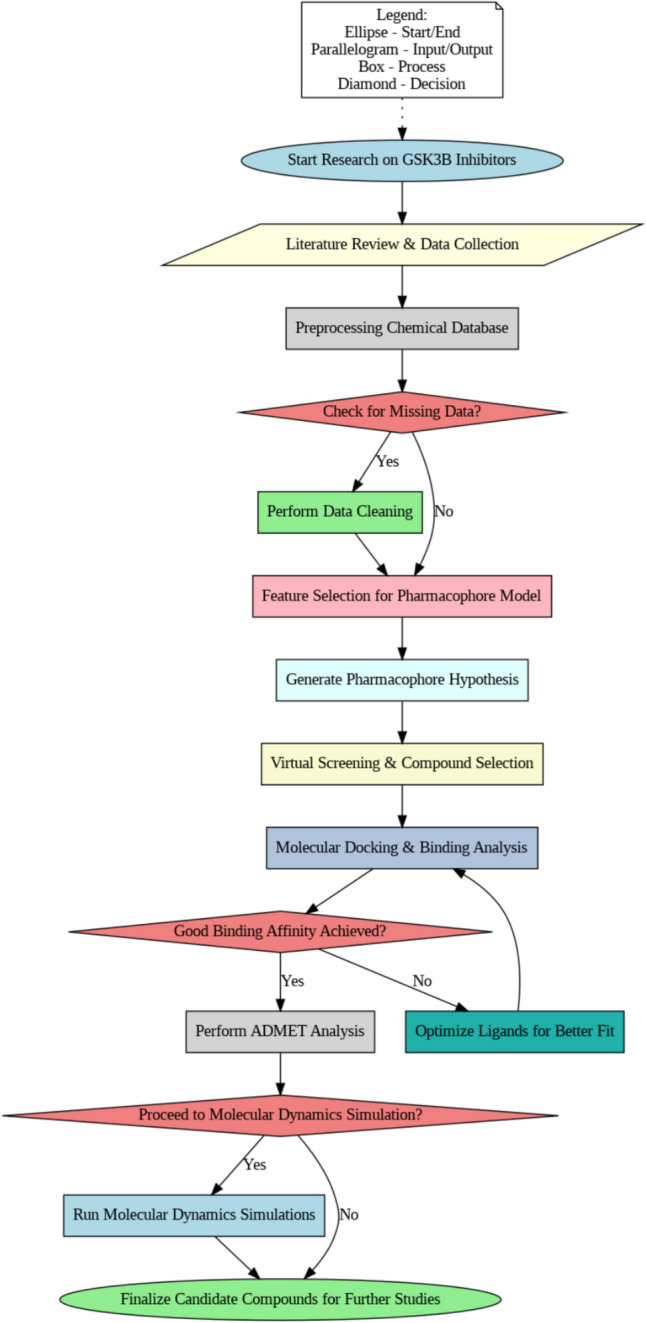
Schematic flowchart of the study methodology
Methodology
Pharmacophore Hypothesis Development and Validation
Co-crystal structures of GSK-3β for the development of the pharmacophore model query were obtained from the Protein Data Bank (PDB; https://www.rcsb.org/). For this, one co-crystal ligand, specifically PDB entry 6LQ, was selected as the reference because of its strong IC50 value of 6.9 nM as a measure of GSK-3β inhibition with an ATP-competitive inhibitor. The structure possessed key pharmacophoric features essential for interaction, demonstrated structural conformability with similar inhibitors, and followed a well-defined set of interaction rules. The structure’s preorganization for fixed binding and its experimentally verified configuration establish it as the best template available for pharmacophore modeling and virtual screening. Further, the corresponding crystal structure of GSK-3β (for the corresponding PDB ID: 4ACG) is of high resolution (1.78 Å) with a well defined ATP-binding site, and good structure-ligand matching. This ligand-bound conformation of GSK-3β is very close to its physiological state, serving as a valid model for virtual screening, ensuring accurate docking and pharmacophore development. To examine how each co-crystal structure interacts with its binding site, the structures were overlaid using PyMOL (Yuan et al. 2017). The ligands from these co-crystals were aligned with their corresponding parent proteins to identify common features across them. The 6LQ ligand possessed all the features typically associated with the aligned compounds, thus positioning it as the optimal basis for developing the pharmacophore hypothesis using Phase (Dixon et al. 2006). Before proceeding to virtual screening, the query was validated based on area under the curve (AUC) values. This AUC-based validation for the pharmacophore model used a dataset that included co-crystal compounds from the active dataset and 477 compounds from a decoy dataset. The decoy set was built from the ChEMBL database (https://www.ebi.ac.uk/chembl/) in order to test the selectivity of our pharmacophore model. The physicochemical properties of the 6LQ ligand were used as a reference point to filter through a decoy dataset. Selection criterion removed compounds that had very high structural similarity to the known inhibitors, while retaining similar properties, such as molecular weight, logP, and rotatable bonds. Quantitative measures for statistical validation such as BEDROC160.9 (Boltzman-enhanced discrimination of receiver operating characteristics), AUAC (area under accumulation curve), and total actives against ranked actives, were applied to measure discriminative potency of the model.
Database Preparation and Virtual Screening
A subset of 200,000 compounds was selected from 1.4 million compounds in the VITAS-M Lab database, based on chemical diversity, drug-likeness criteria, and structural relevance to the pharmacophore model. Filtering steps included removing highly similar compounds, ensuring compliance with Lipinski’s Rule of Five, and optimizing molecular properties using Phase software (Dixon et al. 2006) for efficient and representative virtual screening. For every ligand, ten conformers were generated so as to allow for appropriate chemical space exploration. To explain the various possible states at pH 7, Epik software (Shelley et al. 2007) was used to produce different protonation forms, and high-energy tautomeric structures were removed. This filtered set was used for virtual screening, which was based on a hypothesis that had been previously established (Hu et al. 2024). The results of the screening were ranked based on the Phase screen score, a metric that combines vector alignments, volume scores, and root mean square deviation (RMSD) site matching. Only those compounds with a Phase score greater than 1.7 were selected for subsequent molecular docking studies. The selection of the Phase score cutoff of 1.7 was done by statistical validation using active and decoy sets. Benchmarking was done using a known dataset of GSK-3β inhibitors and the cutoff was optimized based upon the enrichment factor at 1% (EF1%). EF1% was calculated to ascertain the validity of the virtual screening method. This metric evaluates how effectively the pharmacophore-based screening ranks active compounds within the top 1% of the candidate list. Thus, out of a total of 200,000 compounds, 1% (2000 compounds) were selected based on their Phase screen scores.
Molecular Docking
The crystal structure of GSK-3β (PDB ID: 4ACG) was prepared in Maestro Protein Preparation Wizard (Galande and Rohane 2021). This involved a variety of steps including adding hydrogen atoms, stripping off solvent molecules, adjusting side-chain atoms of amino acid residues, deleting unwanted chains, and generating tautomeric states at pH 7.0 applying PROPKA (Rostkowski et al. 2011). The protein target was optimized and reduced using the OPLS_2000 force field, which is proven to perform high-quality protein–ligand simulations (Shivakumar et al. 2010). We utilized the Nosé-Hoover thermostat and Martyna-Tobias-Klein barostat under NPT ensemble conditions. Equilibration was performed for 2 ns before production runs. A particular ligand was selected to create a docking grid for site-specific docking. To this end, the van der Waals radii of the receptor atoms were set to 1.0, the potential of the non-polar regions was reduced, and a charge value of 0.25 was assigned, with X–Y–Z coordinates specified as 23.09, 19.46, and 10.55, respectively. LigPreps tool in Maestro was utilized for preparing the ligands for docking (Chen and Foloppe 2010). Ten conformers per ligand were generated during LigPrep, and 174 compounds were subjected to molecular docking. All three docking modes (high-throughput virtual screening; HTVS, standard precision SP, and extra precision XP) were used sequentially for compound selection. Program Epik analyzes potential ionization states at a pH of 7. All stereoisomers of major molecules with set chirality were provided using OPLS force field. The final ligands were docked into the improved receptor using the Glide docking tool and validated for their binding positions using the Glide G-Score (Halgren et al. 2004).
ADMET Analysis
The hits with higher binding affinity at the receptor level were analyzed based on their ADMET profiles, which incorporate features such as absorption, distribution, metabolism, excretion, and toxicity (Ferreira and Andricopulo 2019). This process involves the use of QikProp (Laoui and Polyakov 2011), which is implemented in Maestro. Based both upon the docking results and the ADMET profiles, which determine brain penetration, metabolic stability, and toxicity, selected compounds were further carried forward for performing MDS. Drug-likeness was evaluated as molecular weight (≤ 500 Da), hydrogen bond donors (≤ 5), and hydrogen bond acceptors (≤ 10). Other specific parameters included lipophilicity (QPlogPo/w scores), intestinal absorption (QPPCaco scores), blood–brain barrier permeability (QPlogBB scores), cardiotoxicity (QPlogHERG values), and plasma protein (human serum albumin) binding efficiency (QPlogKhsa scores).
Molecular Dynamics Simulation (MDS)
MDS of the selected protein–ligand complexes were carefully carried out for a period of 100 ns using Desmond (Chow et al. 2008) of the Schrodinger suite. Starting structures for these simulations were obtained from molecular docking results and were used to study dynamic interactions between the protein and the ligands in a detailed manner. Before carrying out the simulations, preprocessing phase consisting of structural optimization followed by energy minimization of complexes with the OPLS force field was carried out. An effective system builder tool was utilized for applying transferable intermolecular potential based on a support TIP3P water model (Onufriev and Izadi 2018) in an orthorhombic simulation box having the dimensions of 10 Å × 10 Å × 10 Å. Proper counterions were added to make the system neutrally charged. Physiological conditions were also mimicked by adding 0.15 M NaCl. The calculations were done using constant-pressure, constant-temperature (NPT) ensemble conditions of 1 atm and 300 K, respectively. After setting up, the entire complex was carefully relaxed for the simulation run. During analyses, snapshots were taken after every 50 ps for presentation of the temporal trajectory.
Binding Free Energies Calculations
Molecular mechanics/generalized born surface area (MM/GBSA) was used for the calculations of binding free energies (Genheden and Ryde 2015). This approach has been previously used in drug design research for calculating the binding affinities of ligands to a protein target with high accuracy and precision (Wang et al. 2019; Tuccinardi 2021). The following equation was used:
where ΔGcomp, ΔGpro, and ΔGlig depict binding energies of the complex, protein, and ligands, respectively, ΔEeleand ΔEvdW represent the components for electrostatic forces and van der Waals interactions, while ΔGgb is the polar solvation energy calculated by the GB model (Onufriev et al. 2004). Lastly, ΔGnonpol is the free energy change accompanying nonpolar interactions, and TΔS represents entropy change linked to ligand binding.
Principal Component Analysis (PCA)
PCA was employed for identifying coordinated motions for conformational structures obtained from MDS data, which allows evaluation of the effects of ligand binding-induced conformational changes in the target protein (Stein et al. 2006). For PCA analyses, atomic coordinates obtained from the simulation data were employed for construction of covariance matrices, and subjected to diagonalization in order to deliver a set of eigenvalues and eigenvectors. Eigenvectors indicate the movement direction in the protein domains’ conformational spaces, while the associated eigenvalues represent the square mean fluctuations. For our study, R program-based BIO3D was used for the prediction of dynamic movements (Grant et al. 2021).
Results
Data Retrieval
A total of 20 co-crystal structures of GSK-3β were downloaded from the PDB (Table 1). Taking into account the observed interactions between the individual ligand and its respective binding pocket, the structures were aligned to all co-crystal ligands, and features common to all such ligands were identified using PyMOL. Binding poses of each aligned ligand are shown in Fig. 2A. There were five common features among all the residues that interacted with the key residues as presented in Fig. 2B.
Table 1.
The co-crystal structures used in the study
| No | PDB ID | PDB ligands | No | PDB ID | PDB ligands |
|---|---|---|---|---|---|
| 1 | 3I4B | Z48 | 11 | 3SAY | 0FT |
| 2 | 7U36 | L7W | 12 | 4ACG | 6LQ |
| 3 | 4DIT | 0KD | 13 | 4IQ6 | IQ6 |
| 4 | 4JIR | I5R | 14 | 4PTC | 2WE |
| 5 | 4PTE | 2WF | 15 | 4PTG | 2WG |
| 6 | 5F94 | 3UO | 16 | 5F95 | 3UP |
| 7 | 5HLN | 65C | 17 | 6GJO | F1B |
| 8 | 6GN1 | F4N | 18 | 6TCU | NIQ |
| 9 | 6V6L | QQA | 19 | 6Y9S | OHK |
| 10 | 7B6F | SZW | 20 | 7U2Z | L7C |
Fig. 2.

A The alignment of co-crystal ligands to extract common pharmacophoric features. B The common pharmacophoric features represented on template ligand (6LQ). Orange circles show aromatic rings, cyan spheres show hydrogen bond donors, and red sphere shows hydrogen bond acceptor
Pharmacophore Query Generation and Validation
A pharmacophore hypothesis was created with a co-crystal ligand (PDB ID: 6LQ) as a template. The crystal template was brought into the Maestro workspace, and shared pharmacophoric features were determined based on ligand–protein interactions. The query that was developed was validated using the AUC value, which effectively discriminated active ligands from decoys with an AUC score of 0.82. A set of 200,000 compounds in the VITAS-M Lab database were screened against this confirmed pharmacophore hypothesis. The analysis conducted at EF1% showed that pharmacophore-based screening is suitable for enriching active inhibitors for GSK-3β. About 50 out of 174 identified actives were retrieved in the top 2000 compounds from the virtual screening results. This gave us our calculated value for EF1% of − 28.73, indicating that the top 1% of ranked compounds had 28.73 times the number of active ligands than would be expected from random selection. Hence, this validated that our virtual screening approach successfully prioritized high-affinity ligands, thereby ascertaining the strength of the pharmacophore model for the discovery of potential ATP-competitive GSK-3β inhibitors. Compounds were designated as potential hits if they passed at least four critical pharmacophoric features. Screening outcomes were ordered by the Phase screen score, which integrated vector alignment, volume score, and RMSD site match. The Phase screen score was between − 1.0 and 2.0, and higher values were indicative of improved alignment. Using a cutoff score of 1.7, 174 compounds were chosen for further molecular docking experiments. These highest-scoring compounds were structurally compatible with the pharmacophore model to ensure a precise selection for additional binding affinity measurements. The refined pharmacophore hypothesis and screening outcome are shown in Fig. 3.
Fig. 3.

A The area under curve of validated query differentiating active ligands from decoys with a value of 0.82. B The optimized pharmacophore hypothesis
Molecular Docking
Molecular docking of the selected ligands against GSK-3β were conducted with Glide GScore and RMSD serving as parameters for the strength of the binding poses. Based upon the GScores, four compounds (VL-1, 2, 3, and 4) were chosen, and their molecular interactions were analyzed (Table 2 and Fig. 4; see also Supplementary Figure for superimposition image). VL-1 formed four hydrogen bonds with Val135, Asp181, Lys183, and Asp200, along with five pi-alkyl interactions and one pi-sulfur bond. VL-2 created three hydrogen bonds with Lys85, Asp133, and Val135, along with four alkyl bonds and one pi-sulfur bond. VL-3 established one hydrogen bond with Val135, and VL-4 formed two hydrogen bonds with Lys85 and Asp200. Additional exploration of potential binding modes is depicted in Fig. 5.
Table 2.
The glide gscore and RMSD values of the selected docked compounds
| No | Hit ID | Compound ID | Structure | Phase screen score | Glide GScore |
|---|---|---|---|---|---|
| 1 | VL-1 | STK183871 |  |
1.85 | − 8.63 |
| 2 | VL-2 | STK036472 |  |
1.76 | − 8.49 |
| 3 | VL-3 | STK220387 |  |
1.90 | − 8.31 |
| 4 | VL-4 | STK364448 |  |
1.75 | − 8.20 |
Fig. 4.

The molecular interactions of hit compounds with the binding pocket of GSK-3β enzyme. Hydrogen bonds are shown with green color, Pi-Sulfur bonds with orange, hydrophobic interactions with pink color spheres
Fig. 5.

The potential binding configurations of the active compounds within the GSK-3β binding pocket. The residues of the binding site are illustrated in soft green sticks, while the hit ligands are represented with sticks in various colors
ADMET Properties
Subsequent to the docking analyses, the four compounds were subjected to assessment of their physicochemical and ADMET (absorption, distribution, metabolism, excretion, and toxicity) parameters. The physicochemical parameters were calculated using QikProp software (Ioakimidis et al. 2008). All compounds adhered to Lipinski’s Rule of Five (Pollastri 2010), indicating good drug-like potential. These included characteristics such as optimal molecular weights (≤ 500 Da), hydrogen bond donors (≤ 5), and acceptors (≤ 10). Further, the predicted octanol/water partition coefficient (QPlogPo/w) values were ≤ 5, indicating optimal lipophilicity. The permeability through Caco-2 cells (QPPCaco) was found to be moderate to high for the four drugs, while the human serum albumin binding (QPKhsa scores) was also in the acceptable range. The brain-to-blood partition coefficient (QPlogBB) values ranged from − 2.3 to − 0.53, while the hERG K+ channel blocking activity (QPloghERG values) was measured between -7.1 and -5.1.A full summary of the physicochemical and ADMET properties of the docked compounds is presented in Table 3.
Table 3.
The physiochemical and ADMET properties of the docked hits
| No | Hits | MW | HBD | HBA | QPlogPo/w | QPlogHERG | QPPCaco | QPlogBB | QPlogKhsa |
|---|---|---|---|---|---|---|---|---|---|
| 1 | VL-1 | 420.43 | 3 | 9 | 1.637 | − 7.123 | 51.667 | − 2.311 | − 0.132 |
| 2 | VL-2 | 351.31 | 1 | 5 | 2.008 | − 5.337 | 62.958 | − 1.915 | 0.121 |
| 3 | VL-3 | 355.39 | 1 | 5 | 4.186 | − 6.821 | 1004.955 | − 0.717 | 0.566 |
| 4 | VL-4 | 253.25 | 0 | 4 | 2.166 | − 5.079 | 800.499 | − 0.531 | − 0.139 |
MW Molecular Weight (in Da), HBD Hydrogen bond donor, HBA Hydrogen bond acceptor
MDS
After ADMET analysis, top two hits (VL-1 and 2) with best binding modes and ADMET properties, were selected for the evaluation of their stability with GSK-3β protein. The binding modes of selected hits were aligned on template co-crystal ligand and are depicted in Fig. 6.
Fig. 6.

The binding configurations of the active compounds as they align with the co-crystal ligand. In panel (A), VL-1 is depicted with blue sticks, perfectly aligned alongside the red co-crystal ligand. In panel (B), VL-2 is showcased using red sticks
The RMSD analysis was conducted by examining the trajectories, ensuring the stability of the complexes in comparison to solid protein–ligand complexes. The results illustrated that both complexes equilibrated by the 10 ns time point (Fig. 7A). After this equilibration, RMSD for the VL-1 complex fluctuated marginally from 20 to 40 ns, and stabilized at around 1.5–2 Å at 70 ns. After this equilibration period, only minor fluctuations could be seen, and the RMSD increased to around 2.25 Å at 80 ns, after which it leveled off and was stable for the rest of the simulation time. The RMSD of the VL-2 complex also leveled off after equilibration and remained between 1.75 and 2 Å until 80 ns. The root mean square fluctuation (RMSF) values were obtained for evaluating the flexibility of residues within the protein upon its interaction with the ligands. Throughout most of the simulation, the RMSF values for the individual residues stayed below 2 Å except for the loop regions, as shown in Fig. 7B. This indicates that protein residues were stable and showed minimal fluctuation during the simulation, confirming the stability of the ligand–protein complexes.
Fig. 7.

A The RMSD graphs illustrating the behavior of the protein and ligands throughout the 100 ns simulation period. B The variations in protein residues during the simulations, assessed through RMSF values. The blue graph represents the RMSF of the VL-1 complex, while the green graph depicts the VL-2 complex
Protein–Ligand Contacts
The following were identified as the most significant categories of interactions between the target protein and the ligands, as determined by MDS: hydrogen bonds, ionic bonds, and hydrophobic interactions. The hydrogen bonding side chains with the VL-1 complex included Ser66, Lys85, Val135, and Lys183 (Fig. 8A). In contrast, Lys85, Asp133, Val135, and Asp200 made hydrogen bonds with the VL-2 compound (Fig. 8B).
Fig. 8.

The interaction of protein–ligand during MDS. A The protein–ligand contacts of VL-1 complex. B The protein–ligand contacts of VL-2 complex. The interacting residues are shown are shown as stacked bars
PCA
Principal Component Analysis (PCA) was utilized to examine the dynamic behavior of proteins in both complexes, focusing on the collective motions observed in the MD trajectories. The variance proportion was plotted against the eigenvalues to illustrate the dynamic motions in hyperspace. Only the first three principal components (PCs) were considered, as they accounted for the majority of the fluctuations. For the VL-1 complex, PC1 exhibited the highest variation, accounting for 20.21%, while PC2 and PC3 showed variations of 12.84% and 7%, respectively (Fig. 9A). In the case of the VL-2 complex, PC1 displayed the largest variation at 19.14%, followed by PC2 and PC3 with variations of 10.75% and 7.47%, respectively (Fig. 9B). PCA analysis also revealed conformational changes across all clusters in the PC subspace, with the blue regions indicating the most significant movements, while the red regions corresponded to areas with less flexibility and movement.
Fig. 9.

Principal component analysis of VL-1 and VL-2 complexes. A The PCA plot of VL-1 complex with overall flexibility of 40.05%. B The PCA plot of VL-2 complex with overall flexibility of 37.36% in three hyper spaces
Cross Correlation
Protein residue interaction analysis was conducted using cross correlation matrix, with cyan and magenta colors representing positively correlated and anticorrelated residues. Most of the residues were positive correlated, as can be observed from the diagonal lines which show the positive correlation of topologically local residues (Fig. 10). In summary, our analyses indicate a strong correlation of protein residues with respect to specific compound binding during the simulation process.
Fig. 10.

The dynamic cross correlation matrix of the protein complexes to find the positive correlation in protein residues. A The DCCM of VL-1 complex. B The DCCM of VL-2 complex
MM/GBSA
The MM/GBSA method was used to calculate the total binding free energy (ΔG_total) of both protein–ligand complexes. The energy calculation considered van der Waals (ΔE_vdW), electrostatic (ΔE_ele), and solvation (ΔG_GB) contributions. For the complexes, the Van der Waals and and electrostatic components were similar; however GB contributions were greater for the VL-1 ligand compared to VL-2. The calculated ΔG_total values were − 37.55 ± 0.50 for VL-1 and − 28.85 ± 0.33 for VL-2, indicating a greater stability of the former complex (Table 4). Figure 11 displays the energy component contributions.
Table 4.
The Binding free energies of the complexes calculated by implying MM/GBSA module
| Energy components | GSK-3β-VL-1 | GSK-3β-VL-2 |
|---|---|---|
| ΔEvdW | − 49.13 ± 0.34 | − 48.06 ± 0.30 |
| ΔEele | − 50.70 ± 0.84 | − 5.52 ± 0.24 |
| ΔEGB | 69.09 ± 0.62 | 30.87 ± 0.30 |
| ΔEsurf | − 6.81 ± 0.01 | − 6.13 ± 0.02 |
| ΔGgas | − 99.83 ± 0.86 | − 53.59 ± 0.36 |
| ΔGsolv | 62.27 ± 0.62 | 24.73 ± 0.29 |
| ΔGtotal | − 37.55 ± 0.50 | − 28.85 ± 0.33 |
Fig. 11.

The contribution of binding energy components in total binding free energy
Free Energy Landscape (FEL) Analysis
FEL analysis was performed for both VL-1 and VL-2 complexes for provide further validation of the stability of ligand–protein complexes. The FEL of VL-1 (Fig. 12A) confirmed the presence of a single deep minimum at x = − 0.56445, y = − 5.79882, z = 0.0, suggesting that the complex remained very stable during the entire simulation. The existence of only one deep basin points to the presence of little conformational fluctuations with regards to the binding stability of VL-1 within the GSK-3β active site. In the case of VL-2 (Fig. 12B), two minima were observed in the FEL; at x = 2.27375, y = − 2.22768, z = 0.931, and at x = 3.40904, y = − 2.22768, z = 0.931, indicating the presence of two distinct stable conformations. The first minimum seems to suggest a dominant stable state, while the presence of the second minimum indicates the existence of possible conformational flexibility, which may permit VL-2 to adapt its binding mode inside the active site. Thus, the overall evidence from the FEL analysis confirms that both VL-1 and VL-2 are thermodynamically stable, with VL-1 exhibiting a more rigid binding mode, while VL-2 exhibits some flexibility in its binding conformation. This, in turn, supports the viability of both compounds as GSK-3β inhibitors, while also suggesting that VL-1 may have an advantage in stability for therapeutic application.
Fig. 12.

Free Energy Landscape (FEL) analysis of VL-1 (A) and VL-2 (B) complexes. Minima points indicate stable conformations, with VL-1 showing a single deep basin and VL-2 displaying two stable states
Discussion
In recent years, drug discovery strategies have become essential in initial identification of potentially therapeutic drugs for different diseases. Among the various approaches, high-throughput screening and advanced computational techniques such as molecular docking, network pharmacology, and virtual screening of large chemical databases have emerged as key tools. One particularly promising method is pharmacophore modeling-aided virtual screening, which focuses on defining the pharmacophoric characteristics of ligands. A pharmacophore model captures the essential structural features that a ligand must possess in order to bind effectively to the binding site of a biological macromolecule, thus maximizing binding affinity and optimizing the pharmacological response. Structure-based pharmacophore models are typically developed by utilizing the structural coordinates of a target protein and its corresponding ligand, leading to the prediction of the specific biological activity expected from interactions with that protein. After the construction of the pharmacophore model, hits are extracted from large databases of compounds by considering them compatible with the model. Exploitation of natural resources to discover drugs has been a preferable advantage for centuries, since in most cases natural compounds offer quite unique chemical structures and show a diverse range of biological activities that may not be easily acquired by synthetic compounds. In recent years, several groups have employed this strategy in order to identify inhibitors against key biotargets implicated in AD; such as amyloid-beta (Purgatorio et al. 2020), β-secretase (Pinhiero et al. 2015), tau (Pradeepkiran et al. 2019), acetylcholinesterase and butyrylcholinesterase (Gao et al. 2021), adenosine A2A receptor (Ajala et al. 2025), glutamatergic NMDA receptors (El Fadili et al. 2024), dual-specificity tyrosine phosphorylation-regulated kinase (Dyrk1A; Ajala et al. 2024a, b), and monoamine oxidase-B (MAO-B; Ajala et al. 2024a). Studies have also been directed at identification of GSK-3β inhibitors in the recent past with varying successes (Czeleń 2017; Shukla and Singh 2021; Galati et al. 2023; Ahmad et al. 2024). However, in absence of an approved GSK-3β inhibitor, and following discontinuation of tideglusib as an FDA-approved anti-AD agent (Alzforum 2019), there is an urgent need for identification of lead compounds to target GSK-3β for a plethora of pathologies (Shri et al. 2023).
In the present study, virtual screening was carried out on a small molecule database by using a pharmacophore model of the target protein GSK-3β, which appears to be a primary kinase linking amyloid and tau pathologies in AD. The database used in this study includes 20 co-crystal structures of GSK-3β obtained from PDB. The ligands of these co-crystals were aligned, decoupled from their parent proteins, and compared for similarity. Then, common features of these ligands were used to prepare pharmacophore model queries. A co-crystal ligand with PDB ID: 6LQ was selected as the template for the generation of the pharmacophore query. Before virtual screening, queries were validated through AUC values. Pharmacophore-based virtual screening was performed on small molecule databases, and the hits (four in total; VL-1 to 4) were docked onto the active site of GSK-3β to select compounds with the best binding profiles. VL-1 and VL-2 were chosen for MDS because they have better binding affinity (Glide GScores: − 8.63, − 8.49 kcal/mol) and more intense molecular interactions with the important GSK-3β residues. VL-3 and VL-4, even with good ADME characteristics, possessed weaker docking scores and less stable interactions and hence were less desirable candidate. The selected hits (VL-1 and 2) were shown to have strong and stable bindings within the GSK-3β pocket, and their stabilities were further assessed via MDS, which calculated RMSD, RMSF, and changes in protein–ligand contacts. PCA also supported our results. The binding free energy analysis from MM/GBSA showed an enormous contribution of energy from interactions between the selected hit molecules and GSK-3β. FEL analysis not only confirmed binding stability, but also pointed to significant differences in the dynamical behavior of VL-1 and VL-2. The single deep minimum of VL-1 suggests that a rigid binding conformation, while the two minima of VL-2 speak of structural adaptability occurring in the GSK-3β active site. Thus, our analyses identified VL-1 and VL-2 as potential ATP-competitive inhibitors of GSK-3β. Their interaction with major ATP-site residues, such as Val135, Asp181, and Lys183, further attested to their competitive inhibition over allosteric modulation.
Our work under this study builds on existing literature focused on identification of GSK-3β inhibitors. It employs pharmacophore-based screening, molecular docking, ADMET filtering, and MDS across a multi-level filtration scheme toward identification of its high-affinity inhibitors. We also compared our results with well-established GSK-3β inhibitors (CHEMBL410072 and ChEMBL474807) as reference compounds to evaluate the efficacy of VL-1 and VL-2. Comparison of our results with previous studies suggests probable advantages of VL-1 and VL-2 in GSK-3β inhibition over CHEMBL410072 (Czeleń 2017) and CHEMBL474807 (Czeleń and Szefler 2015) in binding affinity and stability. VL-1 appears to have exhibited the best protein–ligand interactions compared with other compounds by maintaining stable hydrogen bonding with Val135 and Asp181, therefore preserving low RMSD fluctuation levels. Also, VL-2 appears to show a higher degree of pharmacokinetic suitability compared to CHEMBL410072 and CHEMBL474807, which lose stability in GSK-3β binding due to enhanced ligand mobility and relatively weaker stacking interactions (detailed comparison of the results are presented in Supplementary Table S1). Hence, VL-1 and VL-2 may be considered as better prospects for experimental validation toward the selective inhibition of GSK-3β for possible therapeutic applications. Thus, our in silico analyses demonstrated that VL-1 and VL-2 exhibit strong binding affinities and stable binding properties, compared to other known GSK-3β ligands. Nonetheless, further studies are needed to confirm the relevance of these compounds as anti-AD agents capable of specifically targeting GSK-3β under in vivo pathophysiological conditions.
Conclusion
This study aimed to discover novel GSK-3β-mediated ATP-competitive inhibitors for AD and other CNS disorders. GSK-3β phosphorylates tau and promotes neurodegeneration, so selectively inhibiting GSK-3β may aid in modifying and retarding disease outcomes. Identification of GSK-3β inhibitors relied on a computational drug discovery approach, which included pharmacophore-based virtual screening, molecular docking, ADMET, and MDS to screen for high-affinity inhibitors. A pharmacophore model was developed using the crystal structure of the target protein (6LQ). Initial screening of the VITAS-M Lab database identified 174 chemicals with a phase score > 1.7 for further considerations. Subsequent selection was based upon the Glide GScores; and among these, VL-1 and VL-2 were observed to exhibit most favorable binding affinities and interactions with active residues of GSK-3β. MDS for a duration of 100 ns confirmed their binding stability under low RMSD and RMSF fluctuations, indicating high stability and selectivity of VL-1 and VL-2 for GSK-3β-targeted anti-AD therapeutics. Indeed, our results have important implications for drug discovery as VL-1 and VL-2 may be researched as viable lead compounds for further medicinal chemistry refinement, in vitro validation, and preclinical studies. In conclusion, while this work provides a valuable platform for development of novel therapies aimed at controlling pathophysiology of GSK-3β, further experimental validation is warrantied.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgement
The authors gratefully acknowledge the funding of the Deanship of Graduate Studies and Scientific Research, Jazan University, Saudi Arabia, through Project Number: RG24-L06. The authors are grateful to Dr. Giuseppe Ermondi, Group Leader, CASSMedChem, Department of Molecular Biotechnology and Health Sciences—University of Turin, Italy, for allowing us the use of his group’s computers and various software programs installed on them. Also, the authors are grateful to Dr. Md Tabish Rehman, King Saud University, Riyadh, Saudi Arabia for providing help in MDS studies.
Author Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by SH, DMM, MW, HS, and FA. The first draft of the manuscript was written by SH and FA, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Funding
Deanship of Graduate Studies and Scientific Research, Jazan University, Saudi Arabia, Project # RG24-L06.
Data Availability
All data generated in this study are presented in the main manuscript and supplementary files.
Declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical Approval
Not applicable, as no human or animal subjects were used in the study.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Ahmad F, Gupta A, Marzook H et al (2024) Natural compound screening predicts novel GSK-3 isoform-specific inhibitors. Biochimie 225:68–80. 10.1016/j.biochi.2024.05.002 [DOI] [PubMed] [Google Scholar]
- Ajala A, Eltayb WA, Abatyough TM et al (2024a) In-silico screening and ADMET evaluation of therapeutic MAO-B inhibitors against Parkinson disease. Intell Pharm 2:554–564. 10.1016/j.ipha.2023.12.008 [Google Scholar]
- Ajala A, Uzairu A, Shallangwa GA et al (2024b) QSAR application of natural therapeutics inhibitors against Alzheimer’s disease through in-silico virtual-screening, docking-simulation, molecular dynamics, and pharmacokinetic prediction analysis. Intell Pharm 2:505–515. 10.1016/j.ipha.2023.12.004 [Google Scholar]
- Ajala A, Asipita OH, Michael AT et al (2025) Therapeutic exploration potential of adenosine receptor antagonists through pharmacophore ligand-based modelling and pharmacokinetics studies against Parkinson disease. Silico Pharmacol 13:17. 10.1007/s40203-025-00305-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzforum (2019) Therapeutics: Tideglusib
- Alzheimer’s Association Report (2024) 2024 Alzheimer’s disease facts and figures. Alzheimer’s Dement 20:3708–3821. 10.1002/alz.13809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I-J, Foloppe N (2010) Drug-like bioactive structures and conformational coverage with the LigPrep/ConfGen suite: comparison to programs MOE and catalyst. J Chem Inf Model 50:822–839. 10.1021/ci100026x [DOI] [PubMed] [Google Scholar]
- Chow E, Rendleman CA, Bowers KJ et al (2008) Desmond performance on a cluster of multicore processors. SIMULATION 1:1–14 [Google Scholar]
- Czeleń P (2017) Inhibition mechanism of CDK-2 and GSK-3β by a sulfamoylphenyl derivative of indoline: a molecular dynamics study. J Mol Model 23:230. 10.1007/s00894-017-3395-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czeleń P, Szefler B (2015) Molecular dynamics study of the inhibitory effects of ChEMBL474807 on the enzymes GSK-3β and CDK-2. J Mol Model 21:74. 10.1007/s00894-015-2627-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SL, Smondyrev AM, Rao SN (2006) PHASE: a novel approach to pharmacophore modeling and 3D database searching. Chem Biol Drug des 67:370–372. 10.1111/j.1747-0285.2006.00384.x [DOI] [PubMed] [Google Scholar]
- El Fadili M, Er-Rajy M, Mujwar S et al (2024) In silico insights into the design of novel NR2B-selective NMDA receptor antagonists: QSAR modeling, ADME-toxicity predictions, molecular docking, and molecular dynamics investigations. BMC Chem 18:142. 10.1186/s13065-024-01248-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari C, Sorbi S (2021) The complexity of Alzheimer’s disease: an evolving puzzle. Physiol Rev 101:1047–1081. 10.1152/physrev.00015.2020 [DOI] [PubMed] [Google Scholar]
- Ferreira LLG, Andricopulo AD (2019) ADMET modeling approaches in drug discovery. Drug Discov Today 24:1157–1165. 10.1016/j.drudis.2019.03.015 [DOI] [PubMed] [Google Scholar]
- Galande AK, Rohane SH (2021) Insilico molecular docking analysis in maestro software. Asian J Res Chem 14:1–4. 10.5958/0974-4150.2021.00017.1 [Google Scholar]
- Galati S, Di Stefano M, Bertini S et al (2023) Identification of new GSK3β inhibitors through a consensus machine learning-based virtual screening. Int J Mol Sci 24:17233. 10.3390/ijms242417233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo G, Holtzman DM (2019) Amyloid-β and Tau at the Crossroads of Alzheimer’s Disease. In: Takashima A, Wolozin B, Buee L (eds) Tau Biology. Springer, Singapore, pp 187–203 [Google Scholar]
- Gao H, Jiang Y, Zhan J, Sun Y (2021) Pharmacophore-based drug design of AChE and BChE dual inhibitors as potential anti-Alzheimer’s disease agents. Bioorg Chem 114:105149. 10.1016/j.bioorg.2021.105149 [DOI] [PubMed] [Google Scholar]
- Genheden S, Ryde U (2015) The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov 10:449–461. 10.1517/17460441.2015.1032936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BJ, Skjærven L, Yao X (2021) The Bio3D packages for structural bioinformatics. Protein Sci 30:20–30. 10.1002/pro.3923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren TA, Murphy RB, Friesner RA et al (2004) Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 47:1750–1759. 10.1021/jm030644s [DOI] [PubMed] [Google Scholar]
- Hernandez F, Lucas JJ, Avila J (2012) GSK3 and Tau: two convergence points in Alzheimer’s disease. J Alzheimer’s Dis 33:S141–S144. 10.3233/JAD-2012-129025 [DOI] [PubMed] [Google Scholar]
- Hu R, Xiao Z, Qiao M et al (2024) Construction and validation of a bioinformatics-based screen for cuproptosis-related genes and risk model for Alzheimer’s disease. Mol Med Rep 30:194. 10.3892/mmr.2024.13318 [DOI] [PubMed] [Google Scholar]
- Ioakimidis L, Thoukydidis L, Mirza A et al (2008) Benchmarking the reliability of QikProp. correlation between experimental and predicted values. QSAR Comb Sci 27:445–456. 10.1002/qsar.200730051 [Google Scholar]
- Jellinger KA (2022) Recent update on the heterogeneity of the Alzheimer’s disease spectrum. J Neural Transm 129:1–24. 10.1007/s00702-021-02449-2 [DOI] [PubMed] [Google Scholar]
- Laoui A, Polyakov VR (2011) Web services as applications’ integration tool: QikProp case study. J Comput Chem 32:1944–1951. 10.1002/jcc.21778 [DOI] [PubMed] [Google Scholar]
- Lauretti E, Dincer O, Praticò D (2020) Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim Biophys Acta - Mol Cell Res 1867:118664. 10.1016/j.bbamcr.2020.118664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandlik DS, Mandlik SK (2024) Therapeutic implications of glycogen synthase kinase-3β in Alzheimer’s disease: a novel therapeutic target. Int J Neurosci 134:603–619. 10.1080/00207454.2022.2130297 [DOI] [PubMed] [Google Scholar]
- Mehta SL, Kim T, Chelluboina B, Vemuganti R (2023) Tau and GSK-3β are critical contributors to α-synuclein-mediated post-stroke brain damage. NeuroMolecular Med 25:94–101. 10.1007/s12017-022-08731-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro AR, Barbosa DJ, Remião F, Silva R (2023) Alzheimer’s disease: Insights and new prospects in disease pathophysiology, biomarkers and disease-modifying drugs. Biochem Pharmacol 211:115522. 10.1016/j.bcp.2023.115522 [DOI] [PubMed] [Google Scholar]
- Onufriev AV, Izadi S (2018) Water models for biomolecular simulations. Wires Comput Mol Sci 8:e1347. 10.1002/wcms.1347 [Google Scholar]
- Onufriev A, Bashford D, Case DA (2004) Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins Struct Funct Bioinforma 55:383–394. 10.1002/prot.20033 [DOI] [PubMed] [Google Scholar]
- Pinhiero A, Silva K, Silva A et al (2015) In silico identification of novel potential BACE-1 inhibitors for Alzheimer’s disease treatment: molecular docking, pharmacophore Modeling and activity and synthetic accessibility predictions. Br J Pharm Res 7:217–229. 10.9734/BJPR/2015/18013 [Google Scholar]
- Pollastri MP (2010) Overview on the rule of five. Curr Protoc Pharmacol 49:9–12. 10.1002/0471141755.ph0912s49 [DOI] [PubMed] [Google Scholar]
- Pradeepkiran JA, Reddy AP, Reddy PH (2019) Pharmacophore-based models for therapeutic drugs against phosphorylated tau in Alzheimer’s disease. Drug Discov Today 24:616–623. 10.1016/j.drudis.2018.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purgatorio R, Gambacorta N, Catto M et al (2020) Pharmacophore modeling and 3D-QSAR study of indole and isatin derivatives as antiamyloidogenic agents targeting Alzheimer’s disease. Molecules 25:5773. 10.3390/molecules25235773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostkowski M, Olsson MH, Søndergaard CR, Jensen JH (2011) Graphical analysis of pH-dependent properties of proteins predicted using PROPKA. BMC Struct Biol 11:6. 10.1186/1472-6807-11-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelley JC, Cholleti A, Frye LL et al (2007) Epik: a software program for pK a prediction and protonation state generation for drug-like molecules. J Comput Aided Mol des 21:681–691. 10.1007/s10822-007-9133-z [DOI] [PubMed] [Google Scholar]
- Shivakumar D, Williams J, Wu Y et al (2010) Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J Chem Theory Comput 6:1509–1519. 10.1021/ct900587b [DOI] [PubMed] [Google Scholar]
- Shri SR, Manandhar S, Nayak Y, Pai KSR (2023) Role of GSK-3β inhibitors: new promises and opportunities for Alzheimer’s disease. Adv Pharm Bull 13:688–700. 10.34172/apb.2023.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla R, Singh TR (2021) High-throughput screening of natural compounds and inhibition of a major therapeutic target HsGSK-3β for Alzheimer’s disease using computational approaches. J Genet Eng Biotechnol 19:61. 10.1186/s43141-021-00163-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein SAM, Loccisano AE, Firestine SM, Evanseck JD (2006) Chapter principal components analysis: a review of its application on molecular dynamics data. Annu Rep Comput Chem 22:233–261 [Google Scholar]
- Tuccinardi T (2021) What is the current value of MM/PBSA and MM/GBSA methods in drug discovery? Expert Opin Drug Discov 16:1233–1237. 10.1080/17460441.2021.1942836 [DOI] [PubMed] [Google Scholar]
- Wang E, Sun H, Wang J et al (2019) End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem Rev 119:9478–9508. 10.1021/acs.chemrev.9b00055 [DOI] [PubMed] [Google Scholar]
- Yuan S, Chan HCS, Hu Z (2017) Using PyMOL as a platform for computational drug design. Wires Comput Mol Sci 7:e1298. 10.1002/wcms.1298 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated in this study are presented in the main manuscript and supplementary files.