

Abstract
The impact of nitric oxide (NO) synthesis on different biological cascades can rapidly change dependent on the rate of NO formation and composition of the surrounding milieu. With this perspective, we used diaminonaphthalene (DAN) and diaminofluorescein (DAF) to examine the nitrosative chemistry derived from NO and superoxide (O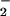 ) simultaneously generated at nanomolar to low micromolar per minute rates by spermine/NO decomposition and xanthine oxidase-catalyzed oxidation of hypoxanthine, respectively. Fluorescent triazole product formation from DAN and DAF increased as the ratio of O
) simultaneously generated at nanomolar to low micromolar per minute rates by spermine/NO decomposition and xanthine oxidase-catalyzed oxidation of hypoxanthine, respectively. Fluorescent triazole product formation from DAN and DAF increased as the ratio of O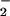 to NO approached equimolar, then decreased precipitously as O
to NO approached equimolar, then decreased precipitously as O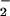 exceeded NO. This pattern was also evident in DAF-loaded MCF-7 carcinoma cells and when stimulated macrophages were used as the NO source. Cyclic voltammetry analysis and inhibition studies by using the N2O3 scavenger azide indicated that DAN- and DAF-triazole could be derived from both oxidative nitrosylation (e.g., DAF radical + NO) and nitrosation (NO+ addition). The latter mechanism predominated with higher rates of NO formation relative to O
exceeded NO. This pattern was also evident in DAF-loaded MCF-7 carcinoma cells and when stimulated macrophages were used as the NO source. Cyclic voltammetry analysis and inhibition studies by using the N2O3 scavenger azide indicated that DAN- and DAF-triazole could be derived from both oxidative nitrosylation (e.g., DAF radical + NO) and nitrosation (NO+ addition). The latter mechanism predominated with higher rates of NO formation relative to O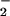 . The effects of oxymyoglobin, superoxide dismutase, and carbon dioxide were examined as potential modulators of reactant availability for the O
. The effects of oxymyoglobin, superoxide dismutase, and carbon dioxide were examined as potential modulators of reactant availability for the O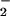 + NO pathway in vivo. The findings suggest that the outcome of NO biosynthesis in a scavenger milieu can be focused by O
+ NO pathway in vivo. The findings suggest that the outcome of NO biosynthesis in a scavenger milieu can be focused by O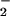 toward formation of NO adducts on nucleophilic residues (e.g., amines, thiols, hydroxyl) through convergent mechanisms involving the intermediacy of nitrogen dioxide. These modifications may be favored in microenvironments where the rate of O
toward formation of NO adducts on nucleophilic residues (e.g., amines, thiols, hydroxyl) through convergent mechanisms involving the intermediacy of nitrogen dioxide. These modifications may be favored in microenvironments where the rate of O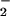 production is temporally and spatially contemporaneous with nitric oxide synthase activity, but not in excess of NO generation.
production is temporally and spatially contemporaneous with nitric oxide synthase activity, but not in excess of NO generation.
Biosynthesis of nitrogen monoxide (NO) from nitric-oxide synthase (NOS)-catalyzed oxidation of l-arginine can lead to selective formation of NO adducts on protein residues, which modulates homeostatic metabolism and affects numerous pathophysiologic responses. N-nitrosamines and S-nitrosothiols are typically formed via donation of a nitrosonium equivalent (NO+) from dinitrogen trioxide (N2O3) to the nucleophilic residue (1). The function of many proteins has been altered by nitrosation in vitro including caspases (2, 3), glyceraldehyde-3-phosphate dehydrogenase (4), the N-methyl-d-aspartate receptor (5, 6), O6-methylguanine-DNA-methyltransferase (7, 8), ras (9) and the ryanodine receptor (10).
The mechanism through which these modifications may occur in biological systems is a subject of debate. Many have questioned the relevance of nitrosation via NO autoxidation, reasoning that sufficient levels of NO cannot be achieved in vivo to satisfy the rate-limiting step for N2O3 formation, which is second order in NO (11–20). In contrast to NO autoxidation, the reaction between NO and superoxide (O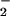 ) is first order in both reactants and occurs at near diffusion control (21–24). Much emphasis has been placed on the product of this reaction, peroxynitrite (ONOO−), as an important mediator of oxidation and nitration (23–28). In this study, we tested the hypothesis that ONOO− may also serve as an intermediary in a pathway leading to formation of NO adducts (e.g., N-nitrosamines, S-nitrosothiols) in vivo. The results suggest that these moieties may be produced through both nitrosation and oxidative nitrosylation dependent mechanisms, which are strongly influenced by the relative rates of NO and O
) is first order in both reactants and occurs at near diffusion control (21–24). Much emphasis has been placed on the product of this reaction, peroxynitrite (ONOO−), as an important mediator of oxidation and nitration (23–28). In this study, we tested the hypothesis that ONOO− may also serve as an intermediary in a pathway leading to formation of NO adducts (e.g., N-nitrosamines, S-nitrosothiols) in vivo. The results suggest that these moieties may be produced through both nitrosation and oxidative nitrosylation dependent mechanisms, which are strongly influenced by the relative rates of NO and O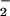 formation.
formation.
Methods
Generation of O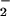 and NO.
and NO.
Formation of O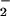 from conversion of hypoxanthine (HX, 500 μM; Sigma) to xanthine was catalyzed by xanthine oxidase (XO; Roche; ref. 29) in PBS (pH 7.4, 37°C) containing the metal chelator diethylenetriaminepentaacetic acid (DTPA, 50 μM; Sigma). The rate of O
from conversion of hypoxanthine (HX, 500 μM; Sigma) to xanthine was catalyzed by xanthine oxidase (XO; Roche; ref. 29) in PBS (pH 7.4, 37°C) containing the metal chelator diethylenetriaminepentaacetic acid (DTPA, 50 μM; Sigma). The rate of O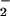 formation by stock enzyme was assessed by cytochrome c reduction (Sigma; 570 nm, ɛ = 21,000 M−1⋅cm−1; ref. 30). NO was produced by decomposition of spermine/NO (a generous gift from J. A. Hrabie, National Cancer Institute-Frederick Cancer Research and Development Center, Frederick, MD; ref. 31). Buffer pH (7.4) and temperature (37°C) were confirmed before each experiment because of the major influence these parameters have on the rate of spermine/NO decomposition (t1/2 = 42 min). Rates of NO release were determined by measuring oxymyoglobin oxidation (582 nm, ɛ = 9,100 M−1⋅cm−1) in PBS solution containing DTPA and HX (HX buffer, ref. 32). Oxymyoglobin was prepared by reducing myoglobin (500 μM; Sigma) in water with excess sodium dithionite (Fluka) followed by passage through a Sephadex G-25 column (PD-10; Amersham Pharmacia). Steady-state NO concentrations were also assessed by using a NO selective electrode (World Precision Instruments, Sarasota, FL) controlled by a DUO18 amplifier. Peak amplitude was calibrated by using standard curves generated with argon-purged PBS solutions of saturated NO (Matheson) after determination of NO concentration with 2-2′-azinobis(3-ethylbenzthiazoline-6-sulfonic acid) (660 nm, ɛ = 12,000 M−1⋅cm−1; Sigma; refs. 32 and 33). Murine ANA-1 macrophages were stimulated to generate NO by treatment overnight with IFN-γ (100 units/ml; R&D Systems) followed by 4 h incubation with lipopolysaccharide (LPS; E. coli, 0111:B4; Sigma; ref. 34).
formation by stock enzyme was assessed by cytochrome c reduction (Sigma; 570 nm, ɛ = 21,000 M−1⋅cm−1; ref. 30). NO was produced by decomposition of spermine/NO (a generous gift from J. A. Hrabie, National Cancer Institute-Frederick Cancer Research and Development Center, Frederick, MD; ref. 31). Buffer pH (7.4) and temperature (37°C) were confirmed before each experiment because of the major influence these parameters have on the rate of spermine/NO decomposition (t1/2 = 42 min). Rates of NO release were determined by measuring oxymyoglobin oxidation (582 nm, ɛ = 9,100 M−1⋅cm−1) in PBS solution containing DTPA and HX (HX buffer, ref. 32). Oxymyoglobin was prepared by reducing myoglobin (500 μM; Sigma) in water with excess sodium dithionite (Fluka) followed by passage through a Sephadex G-25 column (PD-10; Amersham Pharmacia). Steady-state NO concentrations were also assessed by using a NO selective electrode (World Precision Instruments, Sarasota, FL) controlled by a DUO18 amplifier. Peak amplitude was calibrated by using standard curves generated with argon-purged PBS solutions of saturated NO (Matheson) after determination of NO concentration with 2-2′-azinobis(3-ethylbenzthiazoline-6-sulfonic acid) (660 nm, ɛ = 12,000 M−1⋅cm−1; Sigma; refs. 32 and 33). Murine ANA-1 macrophages were stimulated to generate NO by treatment overnight with IFN-γ (100 units/ml; R&D Systems) followed by 4 h incubation with lipopolysaccharide (LPS; E. coli, 0111:B4; Sigma; ref. 34).
Experimental Paradigms.
Assays were conducted in either black microtiter plates (200 μl, nonstirring; Dynex, Manasas, VA) or fluorometric cuvettes (2 ml, stirring; Spectrocell, Oreland, PA) containing XO in HX buffer and either 2,3-diaminonaphthalene (DAN; Fluka), 4,5-diaminofluorescence (DAF, Calbiochem) with copper/zinc superoxide dismutase (SOD, Sigma) or oxymyoglobin (Sigma) as indicated. Sodium azide (Sigma) concentration was 1 mM.
Experiments commenced upon addition of spermine/NO, with the exception of macrophage experiments, where XO was added secondary to equilibration of cells into HX buffer (pH 7.4, 37°C) supplemented with l-arginine (1 mM, Sigma) and d-glucose (20 mM, Sigma). For CO2 experiments, 25 mM NaHCO3 was added to the buffer (final pH 7.4), and the reaction was carried out in an atmosphere of 5% CO2, 95% air. Incubation times were 1 h at 37°C unless otherwise indicated. Intracellular triazole formation was examined with 85–90% confluent adherent human MCF-7 breast carcinoma cells [American Type Culture Collection (ATCC), Manassas, VA] incubated with RPMI media 1640 containing DAF diacetate (5 μM; Calbiochem) in black-wall microtiter plates for 30 min. After several washes with PBS solution, the cells were gently covered with HX buffer (200 μl, pH 7.4) supplemented with d-glucose (20 mM) and then were exposed to spermine/NO and XO while maintained at 37°C in an atmosphere of 5% CO2, 95% air. Intracellular DAF was of sufficient quantity to detect nitrosation at NO concentrations ≥100-fold the maximum concentration used (35). End-point MCF-7 viability, assessed by trypan blue dye exclusion, was found to be >95% (35). DAF fluorescence in the supernatant was <2% of the total, indicating that DAF release from cells under experimental conditions was negligible.
Spectroscopy.
UV-visible spectroscopy was performed with a Hewlett–Packard 8452A diode-array spectrophotometer. Fluorescence measurements were obtained on either a Perkin-Elmer HTS 7100 plate reader or an LS50B fluorometer. Formation of the fluorescent products DAN- and DAF-triazole was linear from 15–90 min (data not shown). The fluorescent products from DAN and DAF derived from either NO autoxidation or the NO + O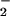 paradigms had identical fluorescent excitation and emission spectra (data not shown).
paradigms had identical fluorescent excitation and emission spectra (data not shown).
Cyclic Voltammetry.
Cyclic voltammetry was performed with an EG & G Princeton Applied Research potentiostat/galvanostat (273/PAR 270 software). The working electrode was a glassy carbon disk with a Pt counter electrode and Ag/AgCl reference electrode [+0.222 V vs. normal hydrogen electrode (NHE)]. Compounds were examined from 0 to +1.8 V at a rate of 100 mV/s in acetonitrile (Baker) containing tetraethylammonium perchlorate (0.1 M, Fluka) as the electrolyte. The electrode surface was cleaned between runs because of interference from electroplating, which contributed to the lack of signal reversibility. Fluoresceinamine (AF; Sigma) and 5,6-carboxyfluorescein (CF; Molecular Probes) served as fluorescein derivative controls.
Results
Reactions with DAN.
N-nitrosation of DAN results in the formation of fluorescent product 2,3-naphthotriazole (DAN-triazole; refs. 34 and 36). Consistent with previous experimental findings on aqueous NO autoxidation, decomposition of spermine/NO (6 μM, ≈140 nM NO/min) in HX buffer containing DAN resulted in formation of DAN-triazole (Fig. 1, 0 O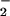 ). Addition of XO to this system augmented fluorescence incrementally, with maximal signal observed at an XO activity of 60 nM/min O
). Addition of XO to this system augmented fluorescence incrementally, with maximal signal observed at an XO activity of 60 nM/min O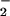 , which was ≈175% relative to spermine/NO alone. At higher XO activities, DAN-triazole fluorescence decreased markedly, with complete quenching at ≥1 μM O
, which was ≈175% relative to spermine/NO alone. At higher XO activities, DAN-triazole fluorescence decreased markedly, with complete quenching at ≥1 μM O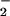 /min. XO alone did not result in an increase in fluorescence above background (data not shown).
/min. XO alone did not result in an increase in fluorescence above background (data not shown).
Fig 1.

Reacitivy of DAN with NO + O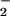 . Reactions were carried out in HX buffer as described in Methods containing DAN (30 μM) and XO to give the indicated O
. Reactions were carried out in HX buffer as described in Methods containing DAN (30 μM) and XO to give the indicated O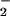 flux for 1 h after the addition of spermine/NO (6 μM). Shown are representative data (n = three trials) of fluorescence (triplicate mean ± SEM) monitored at λex/em of 360/465 nm (gain 75) minus background.
flux for 1 h after the addition of spermine/NO (6 μM). Shown are representative data (n = three trials) of fluorescence (triplicate mean ± SEM) monitored at λex/em of 360/465 nm (gain 75) minus background.
Double reciprocal plots of DAN-triazole fluorescence as a function of DAN concentration (Fig. 4, which is published as supporting information on the PNAS web site, www.pnas.org) indicate that the species produced by NO autoxidation and by the NO + O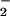 reactions have different affinities for DAN. The −X
reactions have different affinities for DAN. The −X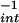 is an indication of the relative reactivity of species formed by either pathway. Exposure to spermine/NO (6 μM, ≈140 nM NO/min; 90 min) resulted in a −X
is an indication of the relative reactivity of species formed by either pathway. Exposure to spermine/NO (6 μM, ≈140 nM NO/min; 90 min) resulted in a −X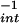 of 95 ± 15 μM, whereas addition of 125 nM O
of 95 ± 15 μM, whereas addition of 125 nM O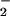 /min decreased the −X
/min decreased the −X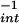 to 62 ± 12 μM. At infinite concentrations of DAN, the projected maximal fluorescence values (Y
to 62 ± 12 μM. At infinite concentrations of DAN, the projected maximal fluorescence values (Y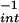 ) were similar, indicating that the maximal amounts of reactive species formed were comparable. The difference in relative reactivity for DAN (−X
) were similar, indicating that the maximal amounts of reactive species formed were comparable. The difference in relative reactivity for DAN (−X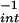 ) suggests distinct mechanisms for DAN-triazole formation via NO autoxidation and a NO + O
) suggests distinct mechanisms for DAN-triazole formation via NO autoxidation and a NO + O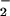 reaction pathway.
reaction pathway.
Reactions with DAF.
Studies have shown that N-nitrosation of DAF results in formation of fluorescent 4,5-naphthotriazole (DAF-triazole; refs. 35 and 37). NO generated in tandem with varied rates of O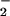 resulted in DAF-triazole fluorescence changes that were similar to those observed with DAN (data not shown). DAF-triazole formation as a function of NO at several different rates of O
resulted in DAF-triazole fluorescence changes that were similar to those observed with DAN (data not shown). DAF-triazole formation as a function of NO at several different rates of O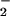 formation is shown in Fig. 2A. The enhancement of this signal in the presence of O
formation is shown in Fig. 2A. The enhancement of this signal in the presence of O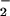 vs. NO alone (NO autoxidation) was determined by subtracting the fluorescence values derived from NO in the absence of O
vs. NO alone (NO autoxidation) was determined by subtracting the fluorescence values derived from NO in the absence of O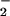 (Fig. 2A, open symbols) from values obtained with simultaneous NO and O
(Fig. 2A, open symbols) from values obtained with simultaneous NO and O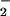 production (solid symbols). The actual data are a combination of these values (open symbols + solid symbols). Doubling O
production (solid symbols). The actual data are a combination of these values (open symbols + solid symbols). Doubling O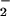 formation increased the fluorescence maxima in a staircase pattern. For each data set, peaks occurred at points where the rate of O
formation increased the fluorescence maxima in a staircase pattern. For each data set, peaks occurred at points where the rate of O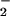 formation approached the rate of NO production, then decreased as the rate of O
formation approached the rate of NO production, then decreased as the rate of O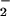 formation exceeded that of NO.
formation exceeded that of NO.
Fig 2.

Reacitivy of DAF with NO + O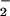 . (A) XO was added to HX buffer containing DAF (1 μM) to give O
. (A) XO was added to HX buffer containing DAF (1 μM) to give O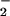 flux as indicated. Spermine/NO was subsequently added forming NO as indicated. Shown are representative data (triplicate mean ± SEM, n = three trials) of fluorescence monitored at λex/em of 485/535 nm (gain = 75) after 1 h incubation at 37°C. Fluorescence values obtained with spermine/NO in the absence of XO (□) have been subtracted from the data acquired in the presence of XO (filled symbols). (B) ANA-1 murine macrophages (1 × 106) stimulated to express iNOS as described in Methods. Arrow indicates time of XO addition to HX buffer to give O
flux as indicated. Spermine/NO was subsequently added forming NO as indicated. Shown are representative data (triplicate mean ± SEM, n = three trials) of fluorescence monitored at λex/em of 485/535 nm (gain = 75) after 1 h incubation at 37°C. Fluorescence values obtained with spermine/NO in the absence of XO (□) have been subtracted from the data acquired in the presence of XO (filled symbols). (B) ANA-1 murine macrophages (1 × 106) stimulated to express iNOS as described in Methods. Arrow indicates time of XO addition to HX buffer to give O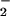 flux as indicated. (C) XO was added to HX buffer covering adherent MCF-7 cells loaded with DAF-diacetate as described in Methods to give the indicated O
flux as indicated. (C) XO was added to HX buffer covering adherent MCF-7 cells loaded with DAF-diacetate as described in Methods to give the indicated O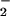 fluxes. Fluorescence values were obtained (as in A) 1 h after addition of spermine/NO to give the indicated NO fluxes.
fluxes. Fluorescence values were obtained (as in A) 1 h after addition of spermine/NO to give the indicated NO fluxes.
Macrophages stimulated to express inducible NOS (iNOS) with IFN-γ and lipopolysaccharide (LPS) have been shown to elicit nitrosative reactions (34). We examined whether XO-catalyzed formation of O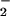 influences macrophage-mediated reactivity with DAF in solution (Fig. 2B). The burst of DAF-triazole formation visible immediately after addition of XO may be attributed to O
influences macrophage-mediated reactivity with DAF in solution (Fig. 2B). The burst of DAF-triazole formation visible immediately after addition of XO may be attributed to O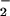 reacting with the steady-state concentration of NO available in the cuvette. Within minutes, a new ratio between NO generated by iNOS and each concentration of XO-derived O
reacting with the steady-state concentration of NO available in the cuvette. Within minutes, a new ratio between NO generated by iNOS and each concentration of XO-derived O could be distinguished as varying rates of fluorescence production. Analogous to the pattern observed by using the synthetic NO source spermine/NO, a bell-shaped concentration dependence for O
could be distinguished as varying rates of fluorescence production. Analogous to the pattern observed by using the synthetic NO source spermine/NO, a bell-shaped concentration dependence for O in macrophage-mediated augmentation of DAF-triazole formation was evident.
in macrophage-mediated augmentation of DAF-triazole formation was evident.
To determine how simultaneously generated O and NO and their subsequent reaction products interact with the complex milieu of viable cells, MCF-7 carcinoma cells were incubated with DAF-diacetate, which was taken up from the media and retained as DAF in the cytoplasm after esterase-mediated cleavage (35). A 2- to 4-fold increase in the concentrations of both O
and NO and their subsequent reaction products interact with the complex milieu of viable cells, MCF-7 carcinoma cells were incubated with DAF-diacetate, which was taken up from the media and retained as DAF in the cytoplasm after esterase-mediated cleavage (35). A 2- to 4-fold increase in the concentrations of both O and NO was required to achieve a relative fluorescence signal inside cells comparable to that observed with DAF in buffer (Fig. 2C). Important issues to consider when comparing reactions in homogeneous buffer to a heterogeneous cellular system are fluorophore compartmentalization and cellular consumptive processes that do not exist in buffer. The shape of profiles derived from intracellular DAF closely resembled those generated with either DAN or DAF in buffer (e.g., Fig. 1). Peak intracellular DAF-triazole fluorescence was augmented ≈4- to 5-fold by the combination of O
and NO was required to achieve a relative fluorescence signal inside cells comparable to that observed with DAF in buffer (Fig. 2C). Important issues to consider when comparing reactions in homogeneous buffer to a heterogeneous cellular system are fluorophore compartmentalization and cellular consumptive processes that do not exist in buffer. The shape of profiles derived from intracellular DAF closely resembled those generated with either DAN or DAF in buffer (e.g., Fig. 1). Peak intracellular DAF-triazole fluorescence was augmented ≈4- to 5-fold by the combination of O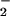 and NO relative to the signal generated from NO autoxidation in the absence of O
and NO relative to the signal generated from NO autoxidation in the absence of O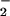 .
.
Specificity of Diamine Fluorophores for Nitrosation.
We and others have observed that DAF has the potential to yield fluorescent product after exposure to nitrogen oxide oxidants (38, †). The ability of DAF and DAN to donate electrons was measured by cyclic voltammetry vs. Ag/AgCl (Fig. 5A, which is published as supporting information on the PNAS web site). These data show that the oxidation potential for DAN is 0.88 V (1.11 V vs. NHE), whereas DAF is oxidized between 0.85 to 1.25 V (1.07 to 1.47 V vs. NHE). Fluoresceinamine has a single oxidation potential of 1.2 V (1.42 V vs. NHE), whereas 5,6-carboxyfluorescein was essentially resistant to oxidation (E > 2.0 V; 2.2 V vs. NHE), suggesting that oxidation was localized to the amino moieties of DAF with some facilitation by the fluorescein ring. Under these conditions, an oxidation potential of 1.0 V (1.22 V vs. NHE) was observed with dihydrorhodamine, a commonly used indicator of oxidation (data not shown). These data suggest that oxidation of DAN and DAF may be feasible under biological conditions.
The contribution of N2O3 to DAN-triazole and DAF-triazole formation was assessed by azide scavenging studies (12). Addition of azide subsequent to triazole formation did not affect fluorescence (data not shown). Azide (1 mM) quenched >90% of DAN-triazole formation (30 to 200 μM DAN) during NO autoxidation derived from relatively high concentrations of NO (≥50 μM spermine/NO, ≥1.2 μM NO/min; Fig. 5B). The combination of XO (125 nM O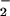 /min) and spermine/NO (6 μM, 140 nM/min) was tested in the presence and absence of azide to determine the level of nitrosation via N2O3 at the optimal ratio of O
/min) and spermine/NO (6 μM, 140 nM/min) was tested in the presence and absence of azide to determine the level of nitrosation via N2O3 at the optimal ratio of O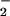 to NO for maximal fluorescent product formation. Under these conditions, the presence of azide inhibited DAN- and DAF-triazole formation 23 ± 5% and 18 ± 3%, respectively (Fig. 5B). These data indicate that ≈20% of the fluorescence signal derived from the diamine fluorophores at low nM per minute rates of simultaneous O
to NO for maximal fluorescent product formation. Under these conditions, the presence of azide inhibited DAN- and DAF-triazole formation 23 ± 5% and 18 ± 3%, respectively (Fig. 5B). These data indicate that ≈20% of the fluorescence signal derived from the diamine fluorophores at low nM per minute rates of simultaneous O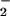 and NO generation was due to N2O3.
and NO generation was due to N2O3.
Modulators of the O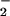 + NO Reaction.
+ NO Reaction.
The influence of several biologically relevant scavengers for O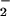 , NO, and their related species on nitrosation was examined by using DAF as an indicator. The ubiquitous metabolic by-product CO2 can readily react with ONOO− (39–44). The effect of CO2 on DAF-triazole formation by simultaneous production of O
, NO, and their related species on nitrosation was examined by using DAF as an indicator. The ubiquitous metabolic by-product CO2 can readily react with ONOO− (39–44). The effect of CO2 on DAF-triazole formation by simultaneous production of O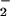 (varied XO) and NO (spermine/NO 3 μM, ≈70 nM NO/min) is shown in Fig. 3A. Reaction in buffer equilibrated with CO2 at physiologic concentration resulted in an approximate 2-fold increase in fluorescence maximum. The effect was lessened with each point away from the peak.
(varied XO) and NO (spermine/NO 3 μM, ≈70 nM NO/min) is shown in Fig. 3A. Reaction in buffer equilibrated with CO2 at physiologic concentration resulted in an approximate 2-fold increase in fluorescence maximum. The effect was lessened with each point away from the peak.
Fig 3.

Modulators of NO + O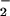 reaction. (A) Spermine/NO (3 μM) was added to HX buffer containing DAF (1 μM) and XO to give O
reaction. (A) Spermine/NO (3 μM) was added to HX buffer containing DAF (1 μM) and XO to give O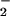 flux as indicated in either the presence (▴) or absence (□) of 25 mM NaHCO3 with a 5% CO2, 95% air atmosphere. Fluorescence was measured (λex/em of 485/535 nm) after incubation at 37°C for 1 h. (B) DAF-triazole formation was monitored (as in A) in HX buffer containing DAF (1 μM) under conditions as indicated. (C) Spermine/NO (6 μM) was added to HX buffer containing DAF (1 μM) and XO to give O
flux as indicated in either the presence (▴) or absence (□) of 25 mM NaHCO3 with a 5% CO2, 95% air atmosphere. Fluorescence was measured (λex/em of 485/535 nm) after incubation at 37°C for 1 h. (B) DAF-triazole formation was monitored (as in A) in HX buffer containing DAF (1 μM) under conditions as indicated. (C) Spermine/NO (6 μM) was added to HX buffer containing DAF (1 μM) and XO to give O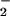 flux as indicated in the presence of SOD as indicated. Fluorescence was measured (as in A) after incubation at 37°C for 1 h.
flux as indicated in the presence of SOD as indicated. Fluorescence was measured (as in A) after incubation at 37°C for 1 h.
The capacity of the O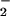 + NO pathway to form DAF-triazole in the presence of oxymyoglobin is shown in Fig. 3B. Initial experiments showed that addition of 10 μM oxymyoglobin to DAF-triazole (1 μM) decreased fluorescence 23% because of an interference filter effect (data not shown). Although quenching was significant, sufficient fluorescence formation was observed under the assay conditions to allow real-time analysis. The presence of 10 μM oxymyoglobin was adequate to competitively scavenge NO (4 μM spermine/NO, ≈90 nM NO/min) evidenced by a lack of fluorescence production from DAF. Under these conditions, an emergence of fluorescence was observed on introduction of O
+ NO pathway to form DAF-triazole in the presence of oxymyoglobin is shown in Fig. 3B. Initial experiments showed that addition of 10 μM oxymyoglobin to DAF-triazole (1 μM) decreased fluorescence 23% because of an interference filter effect (data not shown). Although quenching was significant, sufficient fluorescence formation was observed under the assay conditions to allow real-time analysis. The presence of 10 μM oxymyoglobin was adequate to competitively scavenge NO (4 μM spermine/NO, ≈90 nM NO/min) evidenced by a lack of fluorescence production from DAF. Under these conditions, an emergence of fluorescence was observed on introduction of O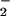 generated by XO/HX at a rate of 2 μM/min. These results indicate that this level of O
generated by XO/HX at a rate of 2 μM/min. These results indicate that this level of O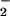 was sufficient to outcompete oxymyoglobin for reaction with NO subsequently leading to DAF-triazole formation.
was sufficient to outcompete oxymyoglobin for reaction with NO subsequently leading to DAF-triazole formation.
Superoxide dismutase (SOD), which catalytically converts O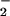 to hydrogen peroxide (30, 45), did not significantly effect DAF-triazole formation via NO autoxidation during decomposition of spermine/NO (6 μM, ≈140 nM NO/min; data not shown). In contrast, relatively small amounts of SOD (≥3.1 μg/ml; equivalent to ≈10 units/ml) significantly influenced the development of DAF-triazole fluorescence by this level of NO when produced in combination with O
to hydrogen peroxide (30, 45), did not significantly effect DAF-triazole formation via NO autoxidation during decomposition of spermine/NO (6 μM, ≈140 nM NO/min; data not shown). In contrast, relatively small amounts of SOD (≥3.1 μg/ml; equivalent to ≈10 units/ml) significantly influenced the development of DAF-triazole fluorescence by this level of NO when produced in combination with O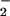 (Fig. 3C). The presence of SOD shifted the initial peak for fluorescence to a higher O
(Fig. 3C). The presence of SOD shifted the initial peak for fluorescence to a higher O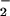 rate, indicating that a 2-fold greater concentration of O
rate, indicating that a 2-fold greater concentration of O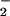 was required to achieve a comparable initial level of DAF-triazole formation. In addition, SOD broadened the range of XO concentrations in which DAF-triazole was formed in a dose-dependent fashion. The highest level of SOD tested (25 μg/ml) increased DAF-triazole production derived from spermine/NO (140 nM NO/min) and XO (1 μM O
was required to achieve a comparable initial level of DAF-triazole formation. In addition, SOD broadened the range of XO concentrations in which DAF-triazole was formed in a dose-dependent fashion. The highest level of SOD tested (25 μg/ml) increased DAF-triazole production derived from spermine/NO (140 nM NO/min) and XO (1 μM O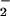 /min) over 17-fold. Inclusion of azide under these conditions quenched DAF-triazole formation ≈12% (data not shown), suggesting a limited, but measurable, contribution from N2O3. Experiments conducted with both SOD and catalase showed a decrease in DAF-triazole formation only when catalase levels were sufficient to competitively react with NO to form a stable a catalase-nitrosylheme complex, thereby altering the NO balance with O
/min) over 17-fold. Inclusion of azide under these conditions quenched DAF-triazole formation ≈12% (data not shown), suggesting a limited, but measurable, contribution from N2O3. Experiments conducted with both SOD and catalase showed a decrease in DAF-triazole formation only when catalase levels were sufficient to competitively react with NO to form a stable a catalase-nitrosylheme complex, thereby altering the NO balance with O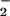 (data not shown). This finding suggests that H2O2 was inconsequential under our experimental conditions.
(data not shown). This finding suggests that H2O2 was inconsequential under our experimental conditions.
Discussion
XO in conjunction with synthetic and macrophage iNOS sources of NO has been used as a model to study the O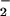 /NO reaction. Beyond this simple system, XO-mediated generation of O
/NO reaction. Beyond this simple system, XO-mediated generation of O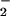 through purine catalysis plays a role in inflammatory or ischemia and reperfusion injury (46, 47). During these events, XO from distal sources can be enriched on endothelial cell surfaces or the interstitial matrix by binding glycosaminoglycan residues, which can result in aberrant O
through purine catalysis plays a role in inflammatory or ischemia and reperfusion injury (46, 47). During these events, XO from distal sources can be enriched on endothelial cell surfaces or the interstitial matrix by binding glycosaminoglycan residues, which can result in aberrant O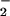 formation at these sites (48, 49). These conditions can also elicit changes in NO formation from the various NOS isoforms expressed by the endothelia, associated neurons, and resident or recruited immune cells, thereby altering the dynamic for NO participation in signaling cascades.
formation at these sites (48, 49). These conditions can also elicit changes in NO formation from the various NOS isoforms expressed by the endothelia, associated neurons, and resident or recruited immune cells, thereby altering the dynamic for NO participation in signaling cascades.
The rapid reaction between O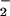 and NO (21–24) has prompted numerous investigators to focus on ONOO− and the roles it may play in oxidation and nitration of susceptible molecules (23–28). Data have been largely lacking, however, on whether the putative damage elicited by ONOO− may be modulated or counteracted by the additional reactions of ONOO− occurring when NO is produced at a variable rate compared with O
and NO (21–24) has prompted numerous investigators to focus on ONOO− and the roles it may play in oxidation and nitration of susceptible molecules (23–28). Data have been largely lacking, however, on whether the putative damage elicited by ONOO− may be modulated or counteracted by the additional reactions of ONOO− occurring when NO is produced at a variable rate compared with O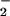 . Therefore, we examined the potential for nitrosation during simultaneous, but varied generation of O
. Therefore, we examined the potential for nitrosation during simultaneous, but varied generation of O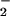 and NO under conditions relevant to their formation at basal levels (nanomolar) and levels that may disrupt regulatory processes (low micromolar).
and NO under conditions relevant to their formation at basal levels (nanomolar) and levels that may disrupt regulatory processes (low micromolar).
A characteristic reaction pattern based on relative O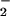 and NO fluxes was evident in experiments with the N-nitrosation indicators (34–37) DAN (Fig. 1) and DAF (Fig. 2). In both cases, product formation was maximal when the ratio of O
and NO fluxes was evident in experiments with the N-nitrosation indicators (34–37) DAN (Fig. 1) and DAF (Fig. 2). In both cases, product formation was maximal when the ratio of O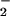 to NO was nearly equivalent, but with NO still in slight excess. These findings were consistent with studies that have shown that the balance between O
to NO was nearly equivalent, but with NO still in slight excess. These findings were consistent with studies that have shown that the balance between O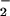 and NO rates of formation is a critical determinant in the type of chemistries they bring to bear. Under conditions of intracellular O
and NO rates of formation is a critical determinant in the type of chemistries they bring to bear. Under conditions of intracellular O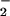 formation from menadione and mitomycin C redox cycling within tumor cells, subtle increases in NO exposure (from 250–500 nM/min) resulted in switching from amplification of dihydrorhodamine oxidation to abatement (50). These data were consistent with the patterns of dihydrorhodamine oxidation observed in solution during covariation of NO and O
formation from menadione and mitomycin C redox cycling within tumor cells, subtle increases in NO exposure (from 250–500 nM/min) resulted in switching from amplification of dihydrorhodamine oxidation to abatement (50). These data were consistent with the patterns of dihydrorhodamine oxidation observed in solution during covariation of NO and O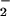 rates of formation (44, 51). Activity measurements from purified alcohol dehydrogenase after incubation with XO/HX and spermine/NO showed that inhibitory oxidation was maximal at a correspondence between NO and O
rates of formation (44, 51). Activity measurements from purified alcohol dehydrogenase after incubation with XO/HX and spermine/NO showed that inhibitory oxidation was maximal at a correspondence between NO and O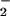 rates of formation (52). Relief of inhibition in favor of nitrosative chemistry was observed as NO was increased ≥2-fold relative to O
rates of formation (52). Relief of inhibition in favor of nitrosative chemistry was observed as NO was increased ≥2-fold relative to O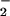 . The importance of stoichiometry between O
. The importance of stoichiometry between O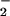 and NO has also been noted in the oxidation of lipids (53), hydroxylation of benzoate (51), nitration of tyrosine (54) and various peptides (unpublished observation) as well as in the formation of disulfides vs. S-nitrosothiols (refs. 55–57; X. Wang, M. T. Gladwin, and M.G.E., unpublished observation). These examples typify how contemporaneous production of O
and NO has also been noted in the oxidation of lipids (53), hydroxylation of benzoate (51), nitration of tyrosine (54) and various peptides (unpublished observation) as well as in the formation of disulfides vs. S-nitrosothiols (refs. 55–57; X. Wang, M. T. Gladwin, and M.G.E., unpublished observation). These examples typify how contemporaneous production of O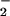 and NO may result in a variety of functional outcomes dependent on their relative rates of formation, the target characteristics, and the composition of the surrounding milieu.
and NO may result in a variety of functional outcomes dependent on their relative rates of formation, the target characteristics, and the composition of the surrounding milieu.
The pivotal importance of balance in the biochemistry of O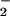 and NO generation cannot be fully appreciated in experimental paradigms that use bolus application of synthetic ONOO−. Peroxynitrite anion can be protonated to form peroxynitrous acid (ONOOH, pKa = 6.8; ref. 23). NO can react with intermediates formed in the decomposition of ONOO−/ONOOH in either the absence or presence of CO2 (44, 55). Although these reactions generally lead to an abatement of oxidation, nitrosation reactions can still occur under conditions of excess NO via formation of N2O3 (Eq. 1–3).
and NO generation cannot be fully appreciated in experimental paradigms that use bolus application of synthetic ONOO−. Peroxynitrite anion can be protonated to form peroxynitrous acid (ONOOH, pKa = 6.8; ref. 23). NO can react with intermediates formed in the decomposition of ONOO−/ONOOH in either the absence or presence of CO2 (44, 55). Although these reactions generally lead to an abatement of oxidation, nitrosation reactions can still occur under conditions of excess NO via formation of N2O3 (Eq. 1–3).
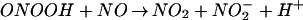 |
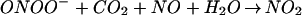 |
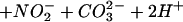 |
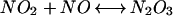 |
DAF was chosen as a probe of N-nitrosation for its sensitivity and ability to be sequestered within cells (35, 37). However, we and others† have observed that DAF-triazole formation is not strictly related to reaction with N2O3. Cyclic voltammetry analysis indicated that the oxidation potentials of DAF and DAN are in the range of 0.85–1.2 V (Fig. 5A; 1.07 to 1.42 V vs. NHE) suggesting the possibility of an oxidative pathway for nitrosation rather than addition of nitrosonium by N2O3. At the ratio of O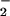 to NO required for optimal fluorescent product formation from either DAN or DAF, inclusion of the specific N2O3 scavenger azide (12) resulted in only ≈20% inhibition of triazole generation (Fig. 5B). These data signify that triazole product may be formed predominantly by oxidative nitrosylation under these conditions, a process that involves oxidation of the compound followed by direct reaction with NO (Eqs. 4 and 5; refs. 58 and 59).
to NO required for optimal fluorescent product formation from either DAN or DAF, inclusion of the specific N2O3 scavenger azide (12) resulted in only ≈20% inhibition of triazole generation (Fig. 5B). These data signify that triazole product may be formed predominantly by oxidative nitrosylation under these conditions, a process that involves oxidation of the compound followed by direct reaction with NO (Eqs. 4 and 5; refs. 58 and 59).
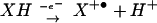 |
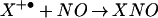 |
From a mechanistic viewpoint, the reactions of DAN+• and DAF+• intermediates with NO must be competitive with respect to a variety of reactive species produced during simultaneous generation of O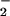 and NO. Of note, azide inhibition studies indicated that oxidative nitrosylation did not completely preclude nitrosation via N2O3 (Eq. 6) during O
and NO. Of note, azide inhibition studies indicated that oxidative nitrosylation did not completely preclude nitrosation via N2O3 (Eq. 6) during O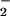 and NO cogeneration, despite only nanomolar rates of NO formation in our system (Fig. 5B).
and NO cogeneration, despite only nanomolar rates of NO formation in our system (Fig. 5B).
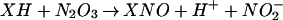 |
This observation suggests that, during the 1-h experimental time frame, a small portion of NO could react by Eq. 1–3 to form N2O3 despite competing with the reaction in Eq. 5.
From these observations, it becomes evident that formation of nitrosated triazole complexes can be produced by either nitrosation (intermediacy of N2O3) or through oxidative nitrosylation. The factors that determine which pathway leads to formation of nitrosated product include differences in relative rates of O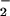 and NO formation and qualities inherent to the substrate nucleophilicity, oxidation potential, residue location, and interaction with metals. Daiber et al. exposed phenol to relatively high levels of NO (spermine/NO, 100 μM) in combination with SIN-1 (200 μM), which simultaneously decomposes to NO and O
and NO formation and qualities inherent to the substrate nucleophilicity, oxidation potential, residue location, and interaction with metals. Daiber et al. exposed phenol to relatively high levels of NO (spermine/NO, 100 μM) in combination with SIN-1 (200 μM), which simultaneously decomposes to NO and O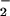 , and observed that azide inhibited 4-nitrosophenol formation by 64% (52). This finding suggests that, under these conditions (25), nitrosation of phenol proceeded mostly through nitrosation rather than oxidative nitrosylation. These data support earlier studies that suggest that a susceptibility hierarchy of amine, hydroxyl, and thiol constituents likely exist for formation of NO adducts in vivo (12, 25, 35, 51, 55).
, and observed that azide inhibited 4-nitrosophenol formation by 64% (52). This finding suggests that, under these conditions (25), nitrosation of phenol proceeded mostly through nitrosation rather than oxidative nitrosylation. These data support earlier studies that suggest that a susceptibility hierarchy of amine, hydroxyl, and thiol constituents likely exist for formation of NO adducts in vivo (12, 25, 35, 51, 55).
An important observation in this study was that the pattern of DAF-triazole formation in solution was comparable to that within intact cells (Fig. 2 A and C). These data suggest formation of similar reactive intermediates, which may readily cross the plasma membrane. Data showing incomplete inhibition by azide coupled with the relatively high oxidation potentials observed for DAN and DAF (Fig. 5A) indicate formation of a reactive intermediate capable of transmembrane diffusion during simultaneous O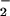 and NO generation. On the basis of the near diffusion control rate constant for the O
and NO generation. On the basis of the near diffusion control rate constant for the O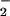 + NO reaction (21–24) and the relative membrane impermeability of O
+ NO reaction (21–24) and the relative membrane impermeability of O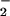 (pKa = 4.8; refs. 30 and 45), the ingressive species most likely is a reactive nitrogen oxide. Under our experimental conditions, initial formation of ONOO− on the outside of the cell is predicted (21–24). Once protonated, this species can decompose leading to secondary formation of other oxidants, chiefly NO2 and hydroxyl radical (•OH; refs. 21, 23, and 60–62). It is unlikely that the highly reactive •OH molecule would have sufficient lifetime for diffusion into the cell. A more likely candidate is NO2, a sufficient oxidant (63) that can be produced via Eqs. 1 and 2 that is capable of permeating cells (35, 40, 50) for reactivity with cytoplasmic DAF (35).
(pKa = 4.8; refs. 30 and 45), the ingressive species most likely is a reactive nitrogen oxide. Under our experimental conditions, initial formation of ONOO− on the outside of the cell is predicted (21–24). Once protonated, this species can decompose leading to secondary formation of other oxidants, chiefly NO2 and hydroxyl radical (•OH; refs. 21, 23, and 60–62). It is unlikely that the highly reactive •OH molecule would have sufficient lifetime for diffusion into the cell. A more likely candidate is NO2, a sufficient oxidant (63) that can be produced via Eqs. 1 and 2 that is capable of permeating cells (35, 40, 50) for reactivity with cytoplasmic DAF (35).
An important facet to consider in the biochemistry of NO and O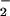 is the influence of CO2 on this system. Reports on the ingress of ONOO−/ONOOH into cells (41, 64) suggest that these species could potentially react with intracellular DAF. Cell experiments were performed under standard culture conditions in which buffer was equilibrated with physiologic CO2 levels. Several groups have demonstrated that ONOO− can be rapidly consumed by CO2 to form the CO2 adduct nitrosoperoxocarbonate (ONOOCO
is the influence of CO2 on this system. Reports on the ingress of ONOO−/ONOOH into cells (41, 64) suggest that these species could potentially react with intracellular DAF. Cell experiments were performed under standard culture conditions in which buffer was equilibrated with physiologic CO2 levels. Several groups have demonstrated that ONOO− can be rapidly consumed by CO2 to form the CO2 adduct nitrosoperoxocarbonate (ONOOCO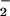 ; refs. 39–44). This reaction pathway would decrease the lifetime, thus diffusion distance of ONOO− circumventing the putative homolysis of ONOOH to NO2 and •OH in favor of ONOOCO
; refs. 39–44). This reaction pathway would decrease the lifetime, thus diffusion distance of ONOO− circumventing the putative homolysis of ONOOH to NO2 and •OH in favor of ONOOCO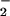 decomposition into NO2 and carbonate radical (CO
decomposition into NO2 and carbonate radical (CO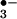 ; Eqs. 7 and 8).
; Eqs. 7 and 8).
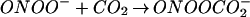 |
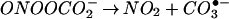 |
In our cell-free system, CO2 slightly enhanced peak DAF-triazole formation (2-fold, Fig. 3A), but did not alter the overall NO to O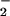 relationship, suggesting that the CO2 reaction pathway may result in a modestly enhanced level of NO2 formation relative to that produced by alternate routes of ONOO− catabolism. Taken together with the intracellular DAF results, these data argue against ONOO−, ONOOH or ONOOCO
relationship, suggesting that the CO2 reaction pathway may result in a modestly enhanced level of NO2 formation relative to that produced by alternate routes of ONOO− catabolism. Taken together with the intracellular DAF results, these data argue against ONOO−, ONOOH or ONOOCO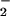 as a significant cell permeable species relative to NO2 (39, 49).
as a significant cell permeable species relative to NO2 (39, 49).
In vivo, CO2 may modulate NO2 formation through the intermediacy of ONOOCO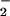 during contemporaneous NO and O
during contemporaneous NO and O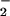 generation, resulting in selective and focal zones of oxidation, nitration, and nitrosation/oxidative nitrosylation dependent on the inter- and intracellular compartmentalization of the target substrate (40, 43, 44, 49, 50, 64). Oxidation of dihydrorhodamine by either the ONOO−/ONOOH or ONOOCO
generation, resulting in selective and focal zones of oxidation, nitration, and nitrosation/oxidative nitrosylation dependent on the inter- and intracellular compartmentalization of the target substrate (40, 43, 44, 49, 50, 64). Oxidation of dihydrorhodamine by either the ONOO−/ONOOH or ONOOCO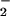 route in aqueous solution was found to be comparable (44). An elegant study by the Kalyanaraman group showed that ONOO− exposure in the presence of CO2 augmented oxidation and nitration of a tyrosine probe in solution, but did not enhance these reactions in membranes (43). Using vesicular-trapped Fe(CN)
route in aqueous solution was found to be comparable (44). An elegant study by the Kalyanaraman group showed that ONOO− exposure in the presence of CO2 augmented oxidation and nitration of a tyrosine probe in solution, but did not enhance these reactions in membranes (43). Using vesicular-trapped Fe(CN)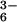 , Hurst and colleagues showed that, in the presence of CO2, the vast majority of extracellular ONOOH was converted via the intermediacy of ONOOCO
, Hurst and colleagues showed that, in the presence of CO2, the vast majority of extracellular ONOOH was converted via the intermediacy of ONOOCO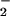 to NO2 before transmembrane diffusion could occur (40). The commonality of NO2 as a putative product of the ONOOH or ONOOCO
to NO2 before transmembrane diffusion could occur (40). The commonality of NO2 as a putative product of the ONOOH or ONOOCO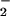 decomposition pathways in combination with the ability of NO2 to diffuse into cells and mediate oxidation (40, 49) suggest it to be a key intermediary in the process of NO adduct formation via the oxidative nitrosylation pathway. The findings advocate that CO2 would not block formation of NO adducts under physiologic conditions (65) and suggest how O
decomposition pathways in combination with the ability of NO2 to diffuse into cells and mediate oxidation (40, 49) suggest it to be a key intermediary in the process of NO adduct formation via the oxidative nitrosylation pathway. The findings advocate that CO2 would not block formation of NO adducts under physiologic conditions (65) and suggest how O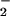 may facilitate this process.
may facilitate this process.
We observed that DAN had a greater affinity for the reactive species formed by the O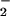 + NO pathway relative to that produced during NO autoxidation (Fig. 4). This finding was consistent with our earlier analysis of DAF, which showed similar differences in relative reactivity of species produced by NO + NO2 vs. aqueous NO autoxidation (35). We promote the viewpoint that the cell may provide an environment, such as within membranes (66), that is favorable for NO autoxidation to produce NO2 (35) and in the current study suggest that NO2 formation through the intermediacy of ONOO− is a key agent leading to formation of NO adducts on biological molecules. An examination of nitration-induced quenching of green fluorescent protein within cells showed that hemeperoxidase-catalyzed oxidation of nitrite was a more effective generator of NO2 than balanced NO and O
+ NO pathway relative to that produced during NO autoxidation (Fig. 4). This finding was consistent with our earlier analysis of DAF, which showed similar differences in relative reactivity of species produced by NO + NO2 vs. aqueous NO autoxidation (35). We promote the viewpoint that the cell may provide an environment, such as within membranes (66), that is favorable for NO autoxidation to produce NO2 (35) and in the current study suggest that NO2 formation through the intermediacy of ONOO− is a key agent leading to formation of NO adducts on biological molecules. An examination of nitration-induced quenching of green fluorescent protein within cells showed that hemeperoxidase-catalyzed oxidation of nitrite was a more effective generator of NO2 than balanced NO and O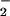 generation (50). These data suggest that the presence of peroxidase and H2O2 may also affect formation of NO adducts through the intermediacy of NO2 when present in conjunction with NOS activity.
generation (50). These data suggest that the presence of peroxidase and H2O2 may also affect formation of NO adducts through the intermediacy of NO2 when present in conjunction with NOS activity.
Regardless of the reactive intermediates involved, the pathways for generation of NO adducts differ in the context of the relative rate of NO formation and the presence of O , which may form the basis for divergent reaction mechanisms and functional outcomes from NOS activity. Nitrosation via NO autoxidation may be the dominant route when relatively high rates of NO formation occur under aerobic conditions without significant O
, which may form the basis for divergent reaction mechanisms and functional outcomes from NOS activity. Nitrosation via NO autoxidation may be the dominant route when relatively high rates of NO formation occur under aerobic conditions without significant O generation, which would favor nitrosation through N2O3 formation subsequent to reaction of NO2 with an additional NO molecule (Eq. 3; Scheme 1, which is published as supporting information on the PNAS web site; refs. 11–20, 35, 36, and 66). Triazole formation (e.g., Fig. 5A) and azide inhibition studies (Fig. 5B) showed that >600 nM NO/min (spermine/NO 24 μM) was sufficient to foster significant formation of the nitrosating species N2O3 (<250 nM O
generation, which would favor nitrosation through N2O3 formation subsequent to reaction of NO2 with an additional NO molecule (Eq. 3; Scheme 1, which is published as supporting information on the PNAS web site; refs. 11–20, 35, 36, and 66). Triazole formation (e.g., Fig. 5A) and azide inhibition studies (Fig. 5B) showed that >600 nM NO/min (spermine/NO 24 μM) was sufficient to foster significant formation of the nitrosating species N2O3 (<250 nM O /min). This condition may be most applicable to recruitment of immune cells expressing iNOS to sites either spatially or temporally dissociated from O
/min). This condition may be most applicable to recruitment of immune cells expressing iNOS to sites either spatially or temporally dissociated from O sources, for instance during inflammatory events secondary to the acute phase response.
sources, for instance during inflammatory events secondary to the acute phase response.
In contrast, oxidative nitrosylation and nitrosation mediated by an ONOO− derived NO2 pathway (Eqs. 1-3) may chiefly prevail under conditions where O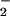 was present, but not in great excess of NO. Basal formation of nitrosated compounds may for the most part occur on a subtle scale, in discrete zones that favor such equivalence between nanomolar levels of O
was present, but not in great excess of NO. Basal formation of nitrosated compounds may for the most part occur on a subtle scale, in discrete zones that favor such equivalence between nanomolar levels of O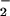 and NO by virtue of their reactivity with each other vs. consumption by respective scavenger substances and competing reactions. This result was illustrated in Fig. 3B, where the addition of sufficient O
and NO by virtue of their reactivity with each other vs. consumption by respective scavenger substances and competing reactions. This result was illustrated in Fig. 3B, where the addition of sufficient O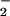 successfully diverted NO from reaction with oxymyglobin toward ONOO− formation and subsequent reactions to promote DAF-triazole generation. SOD in our system consumed excess O
successfully diverted NO from reaction with oxymyglobin toward ONOO− formation and subsequent reactions to promote DAF-triazole generation. SOD in our system consumed excess O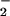 with respect to NO, thereby maintaining the appropriate alignment between O
with respect to NO, thereby maintaining the appropriate alignment between O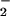 and NO rates, effectively augmenting reactivity with DAF over a broader range of O
and NO rates, effectively augmenting reactivity with DAF over a broader range of O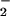 (Fig. 3C; refs. 54 and 67). The intermediacy of ONOO− in the formation of NO adducts on nucleophiles may be a relevant regulatory mechanism where simultaneous, but low-level, generation of both O
(Fig. 3C; refs. 54 and 67). The intermediacy of ONOO− in the formation of NO adducts on nucleophiles may be a relevant regulatory mechanism where simultaneous, but low-level, generation of both O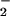 and NO may episodically occur; for instance, membrane proteins on leukocyte and endothelial cell surfaces (68, 69) and within organelles such as mitochondria (3, 70).
and NO may episodically occur; for instance, membrane proteins on leukocyte and endothelial cell surfaces (68, 69) and within organelles such as mitochondria (3, 70).
This process may affect a different set of signal cascades if high levels of O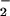 and NO are produced in tandem. Fig. 2 A and C shows that a balanced spatial and temporal relationship between of O
and NO are produced in tandem. Fig. 2 A and C shows that a balanced spatial and temporal relationship between of O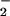 and NO may focus this chemistry, typified by a staircase pattern for peak formation of DAF-triazole as each set of increased rates of O
and NO may focus this chemistry, typified by a staircase pattern for peak formation of DAF-triazole as each set of increased rates of O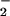 and NO came into concordance. In lesion sites involving XO deposition and leukocytic infiltration under pathological conditions, for instance, higher rates of O
and NO came into concordance. In lesion sites involving XO deposition and leukocytic infiltration under pathological conditions, for instance, higher rates of O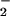 and NO formation may coincide accordingly to foster a greater magnitude and duration of oxidative nitrosylation/nitrosation, a possibility supported by the ability of stimulated macrophages to produce this chemistry during XO catalysis of hypoxanthine (Fig. 2B). Under pathogenic conditions, formation of NO adducts via these oxidative nitrosylation and nitrosation mechanisms may serve to abate and resolve deleterious actions derived from ONOO− formation (<2 NO to 1 O
and NO formation may coincide accordingly to foster a greater magnitude and duration of oxidative nitrosylation/nitrosation, a possibility supported by the ability of stimulated macrophages to produce this chemistry during XO catalysis of hypoxanthine (Fig. 2B). Under pathogenic conditions, formation of NO adducts via these oxidative nitrosylation and nitrosation mechanisms may serve to abate and resolve deleterious actions derived from ONOO− formation (<2 NO to 1 O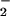 ) such as oxidation (refs. 53, 55, and 68–72; Scheme 1).
) such as oxidation (refs. 53, 55, and 68–72; Scheme 1).
In summary, this study suggests that small differences in the location and rates of O and NO generation in vivo will have a dramatic influence on the ultimate functional outcomes these species mediate. In a biological system, these factors are constantly changing. Therefore, the sphere of influence for a particular chemistry resulting from NO biosynthesis (e.g., oxidation, nitration, oxidative nitrosylation, nitrosation) may be delineated and dynamically focused by O
and NO generation in vivo will have a dramatic influence on the ultimate functional outcomes these species mediate. In a biological system, these factors are constantly changing. Therefore, the sphere of influence for a particular chemistry resulting from NO biosynthesis (e.g., oxidation, nitration, oxidative nitrosylation, nitrosation) may be delineated and dynamically focused by O .
.
Supplementary Material
Abbreviations
NOS, nitric-oxide synthase
iNOS, inducible NOS
HX, hypoxanthine
XO, xanthine oxidase
DTPA, diethylenetriaminepentaacetic acid
DAN, diaminonaphthalene
DAF, diaminofluorescein
NHE, normal hydrogen electrode
SOD, superoxide dismutase
This paper was submitted directly (Track II) to the PNAS office.
Jourd'heuil, D. (2001) Free Radical Biol. Med. 31, S80 (abstr.).
References
- 1.Williams D. L. H., (1988) Nitrosation (Cambridge Univ. Press, Cambridge, U.K.).
- 2.Rossig L., Fichtlscherer, B., Breitschopf, K., Haendeler, J., Zeiher, A. M., Mulsch, A. & Dimmeler, S. (1999) J. Biol. Chem. 274, 6823-6826. [DOI] [PubMed] [Google Scholar]
- 3.Mannick J. B., Schonhoff, C., Papeta, N., Ghafourifar, P., Szibor, M., Fang, K. & Gaston, B. (2001) J. Cell Biol. 154, 1111-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Molina y Vedia L., McDonald, B., Reep, B., Brune, B., DiSilvio, M., Billiar, T. R. & Lapetina, E. G. (1992) J. Biol. Chem. 267, 24929-24932. [PubMed] [Google Scholar]
- 5.Gbadegesin M., Vicini, S., Hewett, S. J., Wink, D. A., Espey, M., Pluta, R. M. & Colton, C. A. (1999) Am. J. Physiol. 277, C673-C683. [DOI] [PubMed] [Google Scholar]
- 6.Espey M. G., Colton, C. A., Pluta, R. M., Miranda, K. M., Hewett, S. J. & Wink, D. A., (2000) Free Radicals in Brain Pathophysiology (Dekker, New York), pp. 523–540.
- 7.Laval F. & Wink, D. A. (1994) Carcinogenesis 15, 443-447. [DOI] [PubMed] [Google Scholar]
- 8.Wink D. A. & Laval, J. (1994) Carcinogenesis 15, 2125-2129. [DOI] [PubMed] [Google Scholar]
- 9.Lander H. M., Hajjar, D. P., Hempstead, B. L., Mirza, U. A., Chait, B. T., Campbell, S. & Quilliam, L. A. (1997) J. Biol. Chem. 272, 4323-4326. [DOI] [PubMed] [Google Scholar]
- 10.Sun J., Xin, C., Eu, J. P., Stamler, J. S. & Meissner, G. (2001) Proc. Natl. Acad. Sci. USA 98, 11158-11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wink D. A., Darbyshire, J. F., Nims, R. W., Saavedra, J. E. & Ford, P. C. (1993) Chem. Res. Toxicol. 6, 23-27. [DOI] [PubMed] [Google Scholar]
- 12.Wink D. A., Nims, R. W., Darbyshire, J. F., Christodoulou, D., Hanbauer, I., Cox, G. W., Laval, F., Laval, J., Cook, J. A., Krishna, M. C., et al. (1994) Chem. Res. Toxicol. 7, 519-525. [DOI] [PubMed] [Google Scholar]
- 13.Wink D. A., Ford, P. C. & Stanbury, D. M. (1993) FEBS Lett. 326, 1-3. [DOI] [PubMed] [Google Scholar]
- 14.Fukuto J. (1995) Adv. Pharmacol. 34, 1-15. [DOI] [PubMed] [Google Scholar]
- 15.Lewis R. S. & Dean, W. M. (1994) Chem. Res. Toxicol. 7, 568-574. [DOI] [PubMed] [Google Scholar]
- 16.Bonner F. T. & Stedman, G., (1996) Methods in Nitric Oxide Research (Wiley, New York), pp. 1–18.
- 17.Pires M., Ross, D. S. & Rossi, M. J. (1994) Int. J. Chem. Kinet. 26, 1207-1227. [Google Scholar]
- 18.Challis, B. C. & Kyrtopoulos, S. A. (1978) J. Chem. Soc. Perkin Trans. 1, 1296–1302.
- 19.Goldstein S. & Czapski, G. (1995) J. Am. Chem. Soc. 117, 12078-12084. [Google Scholar]
- 20.Goldstein S. & Czapski, G. (1996) J. Am. Chem. Soc. 118, 3419-3425. [Google Scholar]
- 21.Kissner R., Nauser, T., Bugnon, P., Lye, P. G. & Koppenol, W. H. (1997) Chem. Res. Toxicol. 10, 1285-1292. [DOI] [PubMed] [Google Scholar]
- 22.Huie R. E. & Padmaja, S. (1993) Free Radical Res. Commun. 18, 195-199. [DOI] [PubMed] [Google Scholar]
- 23.Koppenol W. H., Moreno, J. J., Pryor, W. A., Ischiropoulos, H. & Beckman, J. S. (1992) Chem. Res. Toxicol. 5, 834-842. [DOI] [PubMed] [Google Scholar]
- 24.Goldstein S. & Czapski, G. (1995) Free Radical Res. Commun. 19, 505-510. [DOI] [PubMed] [Google Scholar]
- 25.Beckman J. S. & Crow, J. P. (1995) Adv. Pharmacol. 34, 17-43. [DOI] [PubMed] [Google Scholar]
- 26.Pryor W. A. & Squadrito, G. L. (1995) Am. J. Physiol. 268, L699-L722. [DOI] [PubMed] [Google Scholar]
- 27.Ischiropoulos H. (1998) Arch. Biochem. Biophys. 356, 1-11. [DOI] [PubMed] [Google Scholar]
- 28.Radi R., Peluffo, G., Alvarez, M. N., Naviliat, M. & Cayota, A. (2001) Free Radical Biol. Med. 30, 463-488. [DOI] [PubMed] [Google Scholar]
- 29.Olson J. S., Ballou, D. P., Palmer, G. & Massey, V. (1974) J. Biol. Chem. 249, 4350-4362. [PubMed] [Google Scholar]
- 30.McCord J. M. & Fridovich, I. (1969) J. Biol. Chem. 244, 6049-6055. [PubMed] [Google Scholar]
- 31.Hrabie J. A., Klose, J. R., Wink, D. A. & Keefer, L. K. (1993) J. Org. Chem. 58, 1472-1476. [Google Scholar]
- 32.Nims R. W., Darbyshire, J. F., Saavedra, J. E., Christodoulou, D., Hanbauer, I., Cox, G. W., Grisham, M. B., Laval, F., Cook, J. A., Krishna, M. C. & Wink, D. A. (1995) Methods 7, 48-54. [Google Scholar]
- 33.Christodoulou D., Kudo, S., Cook, J. A., Krishna, M. C., Miles, A., Grisham, M. B., Murugesan, M., Ford, P. C. & Wink, D. A. (1996) Methods Enzymol. 268, 69-83. [DOI] [PubMed] [Google Scholar]
- 34.Espey M. G., Miranda, K. M., Pluta, R. M. & Wink, D. A. (2000) J. Biol. Chem. 275, 11341-11347. [DOI] [PubMed] [Google Scholar]
- 35.Espey M. G., Miranda, K. M., Thomas, D. D. & Wink, D. A. (2001) J. Biol. Chem. 276, 30085-30091. [DOI] [PubMed] [Google Scholar]
- 36.Miles A. M., Wink, D. A., Cook, J. C. & Grisham, M. B. (1996) Methods Enzymol. 268, 105-120. [DOI] [PubMed] [Google Scholar]
- 37.Kojima H., Nakatsubo, N., Kikuchi, K., Kawahara, S., Kirino, Y., Nagoshi, H., Hirata, Y. & Nagano, T. (1998) Anal. Chem. 70, 2446-2453. [DOI] [PubMed] [Google Scholar]
- 38.Espey, M. G., Miranda, K. M., Thomas, D. D. & Wink, D. A. (2002) Free Radical Biol. Med., in press. [DOI] [PubMed]
- 39.Lymar S. V., Jiang, Q. & Hurst, J. K. (1996) Biochemistry 35, 7855-7861. [DOI] [PubMed] [Google Scholar]
- 40.Khairutdinov R. F., Coddington, J. W. & Hurst, J. K. (2000) Biochemistry 39, 14238-14249. [DOI] [PubMed] [Google Scholar]
- 41.Denicola A., Freeman, B. A., Trujillo, M. & Radi, R. (1996) Arch. Biochem. Biophys. 333, 49-58. [DOI] [PubMed] [Google Scholar]
- 42.Romero N., Denicola, A., Souza, J. M. & Radi, R. (1999) Arch. Biochem. Biophys. 368, 23-30. [DOI] [PubMed] [Google Scholar]
- 43.Zhang H., Joseph, J., Feix, J., Hogg, N. & Kalyanaraman, B. (2001) Biochemistry 40, 7675-7686. [DOI] [PubMed] [Google Scholar]
- 44.Jourd'heuil D., Miranda, K. M., Kim, S. M., Espey, M. G., Vodovotz, Y., Laroux, S., Mai, C. T., Miles, A. M., Grisham, M. B. & Wink, D. A. (1999) Arch. Biochem. Biophys. 365, 92-100. [DOI] [PubMed] [Google Scholar]
- 45.Fridovich I. (1997) J. Biol. Chem. 272, 18515-18517. [DOI] [PubMed] [Google Scholar]
- 46.Granger D. N. (1988) Am. J. Physiol. 255, H1269-H1275. [DOI] [PubMed] [Google Scholar]
- 47.Yokoyama Y., Beckman, J. S., Beckman, T. K., Wheat, J. K., Cash, T. G., Freeman, B. A. & Parks, D. A. (1990) Am. J. Physiol. 258, G564-G570. [DOI] [PubMed] [Google Scholar]
- 48.Radi R., Rubbo, H., Bush, K. & Freeman, B. A. (1997) Arch. Biochem. Biophys. 339, 125-135. [DOI] [PubMed] [Google Scholar]
- 49.Houston M., Estevez, A., Chumley, P., Aslan, M., Marklund, S., Parks, D. A. & Freeman, B. (1999) J. Biol. Chem. 274, 4985-4994. [DOI] [PubMed] [Google Scholar]
- 50.Espey M. G., Xavier, S., Thomas, D. D., Miranda, K. M. & Wink, D. A. (2002) Proc. Natl. Acad. Sci. USA 99, 3481-3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miles A. M., Bohle, D. S., Glassbrenner, P. A., Hansert, B., Wink, D. A. & Grisham, M. B. (1996) J. Biol. Chem. 271, 40-47. [DOI] [PubMed] [Google Scholar]
- 52.Daiber, A., Frein, D., Namgaladze, D. & Ullrich, V. (2002) J. Biol. Chem. 11882–11888. [DOI] [PubMed]
- 53.Rubbo H., Radi, R., Trujillo, M., Telleri, R., Kalyanaraman, B., Barnes, S., Kirk, M. & Freeman, B. A. (1994) J. Biol. Chem. 269, 26066-26075. [PubMed] [Google Scholar]
- 54.Pfeiffer S. & Mayer, B. (1998) J. Biol. Chem. 273, 27280-27285. [DOI] [PubMed] [Google Scholar]
- 55.Wink D. A., Cook, J. A., Kim, S., Vodovotz, Y., Pacelli, R., Krishna, M. C., Russo, A., Mitchell, J. B., Jourd'heuil, D., Miles, A. M. & Grisham, M. B. (1997) J. Biol. Chem. 272, 11147-11151. [DOI] [PubMed] [Google Scholar]
- 56.Radi R., Beckman, J. S., Bush, K. M. & Freeman, B. A. (1991) J. Biol. Chem. 266, 4244-4250. [PubMed] [Google Scholar]
- 57.Moro M. A., Darley-Usmar, V. M., Goodwin, D. A., Read, N. G., Zamora-Pino, R., Feelisch, M., Radomski, M. W. & Moncada, S. (1994) Proc. Natl. Acad. Sci. USA 91, 6702-6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Challis, B. C. & Outram, J. R. (1978) J. Chem. Soc. Chem. Commun., 707–708.
- 59.Williams D. L. H. (1997) Nitric Oxide 1, 522-527. [DOI] [PubMed] [Google Scholar]
- 60.Goldstein S., Czapski, G., Lind, J. & Merenyi, G. (1998) Inorg. Chem. 37, 3943-3947. [DOI] [PubMed] [Google Scholar]
- 61.Gerasimov O. V. & Lymar, S. V. (1999) Inorg. Chem. 37, 4317-4321. [Google Scholar]
- 62.Koppenol W. H. & Kissner, R. (1998) Chem. Res. Toxicol. 11, 87-90. [DOI] [PubMed] [Google Scholar]
- 63.Stanbury D. M. (1989) Adv. Inorg. Chem. 33, 69-139. [Google Scholar]
- 64.Denicola A., Souza, J. M. & Radi, R. (1998) Proc. Natl. Acad. Sci. USA 95, 3566-3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Caulfield J. L., Singh, S. P., Wishnok, J. S., Deen, W. M. & Tannenbaum, S. R. (1996) J. Biol. Chem. 271, 25859-25863. [DOI] [PubMed] [Google Scholar]
- 66.Liu X., Miller, M. J., Joshi, M. S., Thomas, D. D. & Lancaster, J. R., Jr. (1998) Proc. Natl. Acad. Sci. USA 95, 2175-2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miles A. M., Gibson, M. F., Kirshina, M., Cook, J. C., Pacelli, R., Wink, D. A. & Grisham, M. B. (1995) Free Radical Res. 23, 379-390. [DOI] [PubMed] [Google Scholar]
- 68.Wung B. S., Cheng, J. J., Shyue, S. K. & Wang, D. L. (2001) Arterioscler. Thromb. Vasc. Biol. 21, 1941-1947. [DOI] [PubMed] [Google Scholar]
- 69.Chiu J. J., Wung, B. S., Hsieh, H. J., Lo, L. W. & Wang, D. L. (1999) Circ. Res. 85, 238-246. [DOI] [PubMed] [Google Scholar]
- 70.Ghafourifar P., Schenk, U., Klein, S. D. & Richter, C. (1999) J. Biol. Chem. 274, 31185-31188. [DOI] [PubMed] [Google Scholar]
- 71.Wink D. A., Hanbauer, I., Krishna, M. C., DeGraff, W., Gamson, J. & Mitchell, J. B. (1993) Proc. Natl. Acad. Sci. USA 90, 9813-9817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wink D. A., Miranda, K. M., Espey, M. G., Pluta, R. M., Colton, C., Vitek, M., Feelisch, M. & Grisham, M. B. (2001) Antiox. Redox Signal. 3, 203-213. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.