

Abstract
The death domain (DD) of the protein kinase Pelle adopts a six-helix bundle fold in the crystal structure of the complex with its dimerization partner, Tube-DD. However, in crystals obtained from a solution of 45% 2-methyl-2,4-pentanediol (MPD), the C-terminal half of Pelle-DD folds into a single helix, and the N-terminal half of the molecule is disordered. The helical segment forms an antiparallel dimer with the corresponding helix of a symmetry-related molecule, and together they form extensive lattice interactions similar in number, composition, and buried surface to those in the six-helix bundle of the native fold. Secondary structure analysis by heteronuclear nuclear magnetic resonance spectroscopy (NMR) demonstrates that Pelle-DD adopts a six-helix bundle fold in aqueous solution. The fold is perturbed by MPD, with the largest chemical shift changes in one helix and two loop regions that encompass the Tube-DD binding site. Pelle-DD is stable to urea denaturation with a folding free energy of 7.9 kcal/mol at 25°C but is destabilized, with loss of urea binding sites, in the presence of MPD. The data are consistent with a cosolvent denaturation model in which MPD denatures the N terminus of Pelle-DD but induces the C terminus to form a more compact structure and aggregate. A similar perturbation in vivo might occur at the plasma membrane and could have consequences for Pelle-mediated regulation. Generally, crystallographers should be aware that high concentrations of MPD or related cosolvents can alter the tertiary structure of susceptible proteins.
Polyhydric alcohols and polyethylene glycols are commonly used as reagents to crystallize proteins. Cosolvents and precipitants of this type are known to promote the preferential hydration of proteins, creating a thermodynamic disequilibrium that can result in precipitation (1, 2) or, in favorable circumstances, crystallization. It is also well established, although not widely appreciated, that the same cosolvents can destabilize the tertiary structures of proteins (3). Here we demonstrate that a commonly used cosolvent, 2-methyl-2,4-pentanediol (MPD), radically transforms the structure of a stable protein domain, the death domain (DD) of Drosophila melanogaster Pelle.
Pelle is a protein kinase that, upon activation of the Toll receptor, is recruited to the plasma membrane where it forms a heterodimer with the protein Tube (4, 5). Similar to Pelle, Tube possesses a DD (6), and it is the interaction between the DDs of the two proteins that mediate their association (7). Formation of the Tube:Pelle heterodimer is believed to initiate a signaling cascade that results in the formation of the dorsal/ventral axis of the Drosophila embryo (8). The same signaling components are used in the Drosophila innate immune response (9).
DDs comprise a diverse family of protein modules that adopt a characteristic six-helix bundle fold (10–12). First discovered in the intracellular domains of receptor proteins involved in apoptotic signaling such as Fas, tumor necrosis factor receptor, and the intracellular interaction partners of these receptors (13), DDs have since been identified in other signaling molecules such as the interleukin-1 receptor and in the structural protein ankyrin (11). The pairwise sequence identities among DDs range from 17 to 32%, and structural analysis of a variety of DDs demonstrates that only a small subset of residues constitutes a conserved structural core (7). DDs belong to a superfamily of folds that include death effector and caspase activator receptor domains.
Here we describe the structure of the Pelle-DD as crystallized from a solution containing MPD. In this state, Pelle-DD is only partly ordered and adopts a tertiary and quaternary structure that differs from the six-helix bundle fold characteristic of DDs. We have investigated further the structure and stability of Pelle-DD in solution in the presence and absence of MPD and consider the mechanism by which MPD transforms the tertiary and quaternary fold of this protein.
Materials and Methods
Crystallization and Characterization of Pelle-DD Crystals.
Recombinant selenomethionine (SeMet) substituted and native Pelle-DD (residues 26–129) were expressed and purified as described (7). Proteins (10 mg/ml) in 20 mM Tris⋅Cl, pH 7.5/100 mM NaCl/1 mM DTT/1 mM ethylenediamine tetraacetic acid (EDTA) were crystallized at 4°C by vapor diffusion against a reservoir solution containing 40–46% MPD, 0.4–0.6 M NaCl, and 0.1 M Tris⋅Cl, pH 8.0. Prism-shaped crystals (Tables 1) grew from precipitates that formed after mixing protein solution with precipitant and grew to a maximum dimension of 0.4 mm after 5 weeks. Two molecules of Pelle-DD are present in the asymmetric unit. Isomorphous crystals of the xenon (AGA specialty gas, OH) derivative were prepared from crystals of native Pelle-DD in a xenon chamber (Hampton Research, Riverside, CA). All crystals were flash-frozen directly in liquid propane for data collection at the Cornell High Energy Synchrotron Source (CHESS) F-1 and F-2 stations (Table 1). The 4-wavelength data set used to derive crystallographic phases by multiple anomalous dispersion (MAD) were measured by using inverse beam geometry. Diffraction data were scaled and merged by using the HKL 2000 software package (14).
Table 1.
X-ray data collection
| Space group: I2I2I2I Unit cell: a = 70.106 Å, b = 95.542 Å, c = 103.196 Å | Dmin, Å | Unique reflections | Redundancy | Completeness, % | 〈I〉/〈σI〉 | Rsym, % |
|---|---|---|---|---|---|---|
| SeMet λ1 (0.9795 Å) | 2.7 | 18,083 | 4.0 | 98.4 (84.2) | 16.6 (3.2) | 8.1 (50.3) |
| SeMet λ2 (0.9791 Å) | 2.7 | 18,041 | 4.0 | 99.5 (99.6) | 16.7 (3.0) | 8.1 (55.0) |
| SeMet λ3 (0.9789 Å) | 2.7 | 18,010 | 4.0 | 99.5 (100) | 14.8 (2.4) | 9.1 (67.0) |
| SeMet λ4 (0.9778 Å) | 2.7 | 17,963 | 4.0 | 99.9 (99.5) | 15.4 (2.6) | 8.9 (60.3) |
| Xenon (0.9470 Å) | 2.7 | 17,499 | 4.5 | 95.5 (99.2) | 16.9 (3.0) | 7.6 (44.8) |
| Native (0.9470 Å) | 2.3 | 15,711 | 5.6 | 99.4 (99.8) | 19.6 (4.5) | 6.3 (25.6) |
Statistics for SeMet and xenon data sets are calculated without merging the Bijvoet mates.
Numbers in parentheses correspond to the last resolution shell.
Rsym = ΣhΣi | Ii(h) − 〈I(h)〉|/ΣhΣ Ii(h), where Ii(h) and 〈I(h)〉 are the ith and mean measurement of the intensity of reflection h, respectively.
Structure Determination and Model Refinement.
The two Se and single Xe positions were located by using SOLVE (15), aided by manual inspection of the Patterson maps; heavy-atom positions and native phases were refined with MLPHARE (16) by using the native data set as the reference data set (Table 2). Density modification using SOLOMON (17) increased the figure of merit from 0.70 to 0.82 at 2.7-Å resolution. A model of Pelle-DD was fit to electron density by using the program O (18), and the model was refined with data extending to 2.3-Å resolution by using CNS 1.0 (ref. 19; Table 3). Noncrystallographic symmetry restraints were not applied in model refinement, because many side chains of the two molecules in the asymmetric unit adopt different conformations. Refinement was monitored by Rfree and inspection of Fo-Fc and 2Fo-Fc maps. The stereochemistry of the model was assessed by the program PROCHECK (16). Solvent-accessible surface areas were calculated by using the program AREAIMOL (16) with a probe radius of 1.4 Å.
Table 2.
Phasing
| Derivative | ΔF/F, % | Sites | RCullis, centric/acentric | PP, centric/acentric | Ranomalous |
| SeMet λ1 | 7.9 | 2 | 0.83/0.71 | 0.91/1.75 | 0.93 |
| SeMet λ2 | 8.7 | 2 | 0.75/0.66 | 1.06/1.91 | 0.85 |
| SeMet λ3 | 9.1 | 2 | 0.78/0.68 | 0.95/1.79 | 0.87 |
| SeMet λ4 | 9.4 | 2 | 0.78/0.68 | 0.97/1.76 | 0.89 |
| Xenon | 14.8 | 1 | 0.90/0.89 | 0.89/0.90 | 0.95 |
ΔF/F = Σh | |FPH| − |FP| |/Σh |FP|, where |FPH| and |FP| are the derivative and native structure amplitudes, respectively.
RCullis = Σh | |FPH ± FP| − FH(calc)|/Σh |FPH± FP|, where |FH| is the calculated scattering amplitude of the heavy-atom structure.
PP (phasing power) = Σh|FH|/Σh[|FPH|(obs)− |FPH|(calc)], where |FH| is the calculated scattering amplitude of the heavy-atom structure, and |FPH|(obs) and |FPH|(calc) are the observed and calculated derivative structure amplitudes, respectively.
Ranomalous = [Σh (|ΔF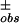 | − |ΔF
| − |ΔF |)2/Σh(ΔF
|)2/Σh(ΔF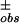 )2]1/2, where ΔF± is the structure factor amplitude difference between Friedel mates. This is calculated for acentric reflections only.
)2]1/2, where ΔF± is the structure factor amplitude difference between Friedel mates. This is calculated for acentric reflections only.
A resolution cutoff of 4.0 Å was applied when these values were calculated.
Table 3.
Refinement statistics
| Model refinement | |
| Resolution range for refinement, Å | 25.0–2.3 |
| No. of protein atoms | 848 |
| No. of water atoms | 19 |
| No. of heteroatoms (Tris and MPD) | 40 |
| rmsd bond lengths, Å | 0.006 |
| rmsd bond angles, ° | 0.840 |
| rmsd bonded mainchain B factors, Å2 | 1.44 |
| rmsd bonded sidechain B factors, Å2 | 2.08 |
| rRwork, % | 23.9 |
| rRfree, % | 25.9 |
| Anisotropic B factor (B11 B22 B33), Å2 | −18.5, 9.8, 8.7 |
| Average B factor, Å2 | 55.9 |
| Wilson B factor, Å2 | 47.7 |
| Ramachandran plot | 100% core |
Rwork = Σh | |Fobs(h)| − |Fcalc(h)| |/Σh|Fobs(h)|, where Fobs(h) and Fcalc(h) are the observed and calculated structure factors, respectively. No I/σ cutoff was applied.
Rfree is the R value obtained for a test set of reflections consisting of a randomly selected 10% subset of the complete data set excluded from refinement.
Analysis of Pelle-DD by NMR.
15N/13C-labeled Pelle-DD was expressed in bacteria grown in M9 medium supplemented with 1 g of 15NH4Cl and 3 g of 13C6-glucose per liter as described (20); labeled proteins were expressed and purified as described (7), but their His-6 tags were not cleaved with thrombin. Protein was dialyzed into sample buffer (20 mM Na+/K+ phosphate, pH 7.2/100 mM NaCl/1 mM DTT/1 mM EDTA) and concentrated to ≈1 mM by using a Centriprep-3 filter unit (Amicon). All NMR experiments were performed on a Varian Inova 500-MHz spectrometer at 25°C. Protein solutions to be analyzed contained 0.5 ml of 1 mM Pelle-DD and 10% 2H2O in sample buffer. 15N 1H heteronuclear single quantum correlation (HSQC) spectra (21) were recorded by using uniformly 15N-labeled protein samples. For MPD titration experiments, solutions of Pelle-DD were prepared with increasing concentrations of MPD, ranging from 5 to 50% (vol/vol). Hydrogen/deuterium exchange experiments were performed with lyophilized 15N-labeled Pelle-DD. The protein was hydrated with 2H2O, and a series of HSQC spectra were recorded to follow exchange. Backbone chemical-shift assignments were obtained by recording three-dimensional HNCO (22, 23), HNCACB (24), and CBCACONH (25, 26) spectra by using uniformly 13C/15N-labeled samples (27). These spectra were processed and analyzed on a Silicon Graphics workstation by using the software packages NMRPIPE (28) and NMRVIEW (29). Backbone φ and ψ dihedral angles were determined from backbone 15N, 13CO, 13Cα, and 13Cβ chemical shifts by using the program TALOS (30).
Urea Denaturation of Pelle-DD and Fas-DD.
The cDNA construct encoding the DD from human Fas (Fas-DD, residues 202–317) was a gift from Steven Fesik of Abbott Laboratories. Fas-DD was expressed and purified as described (10). Urea-induced denaturations of Pelle-DD and Fas-DD were monitored by the change in the wavelength and amplitude of intrinsic tryptophan fluorescence as described by Pace and Shaw (31). Pelle-DD or Fas-DD (1 μM final concentration) was equilibrated overnight at room temperature in a solution containing 30 mM Na+/K+ phosphate, pH 7.0/0.1 mM EDTA and increasing concentrations of urea (0–9 M). Fluorescence spectra were recorded at 25°C with a PTI model A1010 fluorometer with 6-nm excitation and 8-nm-emission slit widths. An excitation wavelength of 280 nm was used, and emission spectra were recorded over the range of 300–380 nm. Spectra were corrected for background, and an intensity-weighted average emission wavelength, 〈λ〉 = Σ(Ii⋅λi)/Σ(Ii), was computed for spectra measured at each urea concentration (32). Free energies of folding were determined by the linear extrapolation method (33); SIGMAPLOT software (Jandel, San Rafael, CA) was used to compute the fit of intensity-weighted emission wavelength with respect to urea concentration to the nonlinear two-state transition model of Santoro and Bolen (31, 34):
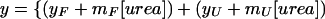 |
 |
 |
where yF and yU are the intercepts, and mF and mU are the slopes of the pre- and posttransition (folded → unfolded) baselines, respectively. The variable y represents intensity-weighted average wavelength, 〈λ〉, at the corresponding urea concentration. ΔG(H2O) is the apparent free energy of folding in the absence of denaturant, and m is the dependence of ΔG on urea concentration:
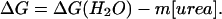 |
To test the reversibility of the denaturation process, native buffer was used to dilute samples in different concentrations of urea, and emission spectra were recorded after the samples were reequilibrated. Denaturation experiments were performed also for Pelle-DD and Fas-DD in the presence of 15% (vol/vol) MPD.
Results
The Structure of Pelle-DD Crystallized from MPD.
Crystals of Pelle-DD (residues 26–129 of D. Melanogaster Pelle) contain two molecules in the asymmetric unit, and they are related by a twofold axis of symmetry. Although no electron density corresponding to the N-terminal ≈50 residues of either Pelle-DD molecule is observed, gel electrophoretic analysis of washed, dissolved crystals demonstrates that intact Pelle-DD is present. Hence, the unobserved residues are thermally or statically disordered. These residues could be accommodated within the large voids present in the crystal lattice (Fig. 1A). Four molecules of Tris buffer and one of MPD (located at the C terminus of molecule A) are visible also in the electron density map. All are within hydrogen-bonding distance of Pelle-DD but possess high temperature factors. The ordered C-terminal segments of the two DDs, residues 78–129 of molecule A and 81–129 of molecule B, each form a continuous α helix with a slight right-handed superhelical twist. Together the two helices form an antiparallel dimer (Fig. 1B), with n, n + 3/n, n + 4 ridge-into-groove packing (35). The twofold symmetrical interface between the two molecules is centered around W90 and is formed mostly by van der Waals interactions. In addition, six hydrogen bonds are observed, four of which are between side chain atoms and main chain carbonyl oxygen atoms. Hydrophobic residues at the dimer (A:B) interface include F86, W90, L101, L104, F105, and L108. Three of these residues, W90, F101, and L105, are among the five most highly conserved residues in the DD family (7). The crystal lattice is built up by contacts between helix dimers (Fig. 1A), at both orthogonal and partly aligned helix–helix interfaces. The former involves only polar residues, whereas the latter, which is more extensive, is largely hydrophobic.
Fig 1.

The structure of Pelle-DD. (A) Packing diagram of Pelle-DD crystallized from MPD. Pelle-DD dimers, depicted as helical ribbons, are shown for three contacting asymmetric units: red/cyan, green/magenta, and blue/yellow. Each asymmetric unit comprises molecules A and B arranged as an antiparallel helix dimer. The red/cyan and green/magenta dimers form orthogonal helix stacks; the green and blue monomers form a partly aligned stack. (B) Molecules A and B of the asymmetric unit, represented as red and cyan ribbons, respectively. The molecules are related by a noncrystallographic twofold axis of symmetry; one half of the symmetrical contact region is shown from opposite sides of the dimer. Side chains are shown as stick models. Carbon atoms in molecule A are magenta, and those in molecule B are green. Oxygen and nitrogen are colored red and blue, respectively. Hydrogen bonds are depicted as yellow dotted lines. (C) The structure of Pelle-DD as determined in the crystal complex with Tube-DD. Helices of the six-helix bundle fold are numbered at their N termini. Only the segment colored red is ordered in crystals of Pelle-DD grown from MPD. Figures were prepared with the programs GL_RENDER (L. Esser, personal communication), BOBSCRIPT (53), MOLSCRIPT (54), and POV-RAY (www.povray.org, ref. 55).
In the complex with Tube-DD, Pelle-DD has a six-helix bundle DD fold (7). In crystals of Pelle-DD obtained by using MPD as precipitant, all of the residues corresponding to the end of helix 3, the succeeding three helices and intervening turns, together with the extended C terminus, are reconfigured as a single α helix (Fig. 1C). In the following discussion, we refer to conformation of Pelle-DD as observed in the complex with Tube-DD as Pelle six-helix bundle (P6hb) and that of Pelle-DD residues 78–128 observed in crystals produced from 45% MPD as pelle helix dimer (Phd). Within the P6hb fold, residues 78–128 of Pelle form a three-helix bundle, to which we refer as ΔNP6hb.
Although the monomer within Phd is an extended helix, ≈1,500 Å2 of its solvent-accessible surface area is buried by lattice contacts, 1,100 Å2 of which are caused by interactions with its dimerization partner. This area is comparable to the 1,660 Å2 of surface of the same residues buried by tertiary contacts in P6hb. In the transformation of ΔNP6hb to a helix, lattice contacts are substituted for tertiary contacts. The global properties of the tertiary and lattice contacts are similar with respect to the fraction of polar and hydrophobic surface buried and in the numbers of van der Waals and hydrogen-bond contacts (887 van der Waals contacts <4 Å and 89 polar/charged contacts for Phd versus 945 and 112, respectively for ΔNP6hb). In contrast, the environments of individual side chains in Phd differ from those of the same residues in P6hb. For example, W90 is located in the hydrophobic core of P6hb, surrounded by V62, F86, L87, H96, L101, and L104. In Phd, W90 is sandwiched between L104 and L108 of its dimerization partner and is in van der Waals contact with the flanking residues F86 and Y94. In similar fashion, most of the residues that form nonbonded contacts in P6hb find alternative partners in the Phd lattice.
Because the N-terminal 50 residues preceding the Phd fold are not visible in the crystal structure, the extent of their exposed surface area cannot be estimated. In the absence of the C-terminal three helices, the remaining helices of P6hb would lose many stabilizing tertiary contacts and must either refold or expose a substantial hydrophobic surface. This segment may possess some degree of residual structure in the crystal lattice, although it must be statically or thermally disordered.
MPD Perturbs the Structure of Pelle-DD in Solution.
It is possible that the structure of Pelle-DD as crystallized from MPD in fact corresponds to the native structure of this protein in solution. To test this hypothesis, we determined the secondary structure of Pelle-DD by using heteronuclear NMR methods. Based on patterns of backbone chemical shifts and protection from deuterium exchange, we identified six stable α helices within Pelle-DD in solution (Fig. 2A). Aside from an apparent shortening of the C terminus of helix α4, the boundaries of these helices agree with those identified in the crystal structure of the Pelle–Tube complex, suggesting that both the complexed and free forms of Pelle-DD have similar structures (7).
Fig 2.

Solution secondary structure and MPD-induced chemical-shift changes of Pelle-DD. (A) Secondary structure of Pelle-DD in solution closely mimics that observed in the Pelle–Tube complex in crystal. The locations of helices in solution are outlined with dashed lines, based on analysis of backbone chemical shifts by the program TALOS (30); plus (+) signs indicate residues with (φ, ψ) values predicted to be within ±20°C of canonical values for α helices). Additional solution evidence regarding the helix locations are provided by residues with significant protection from 2H exchange (filled circles indicate protection factors ≥104; ref. 56). For comparison, these indicators are plotted above the locations of the six α helices in the P6hb conformation of the Pelle–Tube complex structure (PDB entry ; ref. 7). (B) MPD significantly alters the chemical environments of many amides in Pelle-DD. 15N 1H HSQC spectra are shown for 15N-labeled Pelle-DD in the presence of MPD at the following concentrations: 0% (black), 10% (red), 20% (green), and 30% (blue). Assignments for most peaks are shown by residue number, with asterisks indicating peaks that are aliased in the 15N dimension. Signals from side chain NH2 groups are connected by dashed lines. (C) Mapping of the magnitude of shift changes induced by 25% MPD on the tertiary structure of 6hb Pelle-DD. Residues with |Δδ| (see Materials and Methods): greater than 1 standard deviation over the mean, red; less than 1 and greater than 1/2, russet; between +1/2 and −1/2, gray; less than −1 and greater than −1/2, gray-blue; less than −1, blue. Helices of the DD fold are numbered. Tube-DD, as bound to Pelle-DD in the complex of the two proteins, is shown in pale lemon.
We sought to investigate whether MPD induces a structural transition in Pelle-DD in solution by recording a series of 15N 1H HSQC spectra on a sample of 15N-labeled protein titrated with increasing concentrations of MPD (Fig. 2B). At concentrations up to 30%, MPD induced maximal chemical shift changes of 0.3 ppm [1HΔδ2 + (0.17 × 15NΔδ2)]1/2 within these spectra. Although the majority of peaks were perturbed by the addition of MPD, signals from a significant number of sites in Fig. 2B did not shift, suggesting that MPD is not globally unfolding Pelle-DD at these concentrations. We observed a general trend that MPD tended to induce upfield shifts in both the 15N and 1H dimensions, possibly because of bulk changes in solvent properties such as the dielectric constant. This hypothesis was supported by our observation of similar trends in the MPD-induced changes in backbone amide 15N and 1H chemical shifts of the streptococcal protein G β1 domain (data not shown), which has similar structures in solution and in crystals containing up to 70% MPD (36, 37).
To account for the wide range of MPD-induced chemical-shift effects observed for Pelle-DD, we investigated several possible structural causes for these differential changes. Because the magnitude of these shifts failed to correlate with several simple criteria including side chain type, accessible surface area, or mobility in the P6hb fold of Pelle-DD, we surmised that these shifts depended on the surface properties of Pelle-DD. Mapping the magnitude of MPD-induced shifts onto the P6hb fold of the Pelle–Tube complex showed that the largest shifts were observed for residues on the α2-α3 and α4-α5 connecting loops along with the inner surface of α5. Intriguingly, this region of the molecule also forms the binding site for the extended C-terminal polypeptide of Tube-DD as observed in the crystal structure of the Pelle–Tube complex (Fig. 2C). It is possible that MPD preferentially interacts with this surface of Pelle-DD, consistent with prior observations of organic solvents, including alcohols, binding to macromolecular interfaces (38, 39).
In addition to the chemical-shift effects described above, MPD also induced differential peak broadening in the 15N 1H HSQC spectra, particularly at concentrations in excess of 20% (Fig. 2B). At MPD concentrations above 30%, most of the peaks in the 15N 1H HSQC spectra are broadened beyond detection, leaving only a set of intense signals from ≈10 backbone amides and most of the side chain Asn/Gln NH2 groups. Further, the samples became visibly turbid during the short times required to collect these spectra (≈30′, 25°C). All of these observations are consistent with significant MPD-induced destabilization of the folded state of Pelle-DD. Circular dichroism spectra of Pelle-DD recorded at different MPD concentrations are consistent with these results, showing significant helical structure at [MPD] <30% but significant aggregation above this concentration (data not shown). In sum, these data suggest that Pelle-DD retains its native (P6hb) structure at 0 to ≈35% MPD but spontaneously aggregates at higher cosolvent concentration. The structure of the aggregated state cannot be deduced from the NMR data but is not inconsistent with the crystal structure of isolated Pelle-DD, in which partially ordered helical molecules are enmeshed in a three-dimensional network.
Pelle-DD Is a Stable Protein Domain.
In light of the unusual susceptibility of Pelle-DD to MPD, we suspected that its tertiary structure might only be marginally stable. To measure the free energy of folding, the fraction of native structure remaining after titration with urea over a 0–9 M concentration range was monitored by fluorescence-emission spectroscopy. For comparison, we also measured the denaturation profile of a structurally related protein, the DD of Fas. For both Pelle-DD and Fas-DD, denaturation is reversible (Fig. 3). The values of ΔG(H2O) and m for Pelle-DD are 7.9 kcal/mol and 1.3 kcal/mol⋅M, respectively. In comparison, Fas-DD seems less stable, with a ΔG(H2O) and m of 5.0 kcal/mol and 1.20 kcal/mol⋅M, respectively. These parameters fall within the normal range for protein domains of similar mass [for example, thioredoxin has a ΔG(H2O) of 8.7 kcal/mol and m of 1.30, whereas an Src homology 3 domain has a ΔG(H2O) of 2.9 kcal/mol and m of 0.77 kcal/mol⋅M (31)].
Fig 3.

Urea denaturation curves for Pelle-DD and Fas-DD. Pelle-DD, filled circles; Pelle-DD with 15% (vol/vol) MPD, open circles; Fas-DD, filled triangles; Fas-DD with 15% MPD, open triangles.
In the presence of 15% MPD, both Pelle-DD and Fas-DD are destabilized by 1.3 and 0.3 kcal/mol, respectively. MPD at this concentration also reduces the urea dependence of Pelle-DD unfolding (m = 1.05 kcal/mol⋅M), whereas that of Fas-DD is unaffected (Fig. 3).
Discussion
We have demonstrated that the three-dimensional structure of Pelle-DD, which is a well folded protein domain in aqueous buffer near neutral pH, can be transformed radically after exposure to MPD. Moreover, the fold of Pelle-DD is stable, indeed more so than that of the DD of its homolog Fas. MPD, even at moderate concentration, destabilizes the tertiary structures of both proteins, but that of Pelle-DD to a much greater extent.
After titration with MPD, Pelle-DD, as monitored by 15N 1H heteronuclear NMR, undergoes a transition from a cosolvent-perturbed to fully aggregated state. This transition occurs at ≈35% MPD. The upfield shifts observed below this concentration for the majority of amide resonances may reflect nonspecific effects caused by changes in solvent dielectric as well as the amphiphilicity and hydrogen-bonding properties of MPD as a cosolvent. That the largest chemical shift changes are associated with residues that form the interaction surface with Tube-DD suggests that MPD may interact strongly with the Tube-DD binding site, which contains a mix of hydrophobic, polar, and charged amino acids (7). The aggregated state of Pelle-DD that appears at high MPD concentration cannot be observed by NMR but may be similar to the partly ordered crystalline aggregate present in crystals of Pelle-DD obtained at 45% MPD (vol/vol), where the mole fraction of MPD in solvent regions is ≈0.1. In these crystals, Pelle-DD responds to high [MPD] in chimeric fashion. The N-terminal half of the domain is disordered and perhaps partly solvated by MPD, whereas the C-terminal half is fully helical and dimerizes to form extensive lattice interactions.
The behavior of Pelle-DD can be rationalized by the thermodynamic models for the interaction of proteins with cosolvents developed by Tanford (40), Timasheff (41), Shellman (2), and their colleagues. Proteins in aqueous solutions become preferentially hydrated in the presence of cosolvents such as MPD at high concentration, which results in the formation of a boundary layer around the protein from which cosolvent is excluded. The resulting thermodynamic instability can be relieved by two mechanisms. The protein either may undergo a transition that decreases its hydrated surface, or it can unfold, exposing a hydrophobic surface that is capable of binding the cosolvent. For cosolvents such as MPD, the outcome depends on the chemical nature of the protein surface in contact with solvent (3) but not on the inherent stability of the protein fold, as is demonstrated clearly here for Pelle-DD. It is remarkable that Pelle-DD both unfolds (N terminus) and refolds/aggregates (C terminus) in the lattice of crystals obtained in MPD. Hydrated surface within the C terminus is reduced both by extensive lattice interactions and formation of a compact, fully helical conformation, similar to that induced by cosolvents such as trifluoroethanol (42).
In the presence of MPD, a decrease in m, the dependence of the free energy of folding on urea concentration, is observed. In a cosolvent-binding model for denaturation, m is proportional to the protein surface area that becomes accessible to denaturant binding in the unfolded state (31). At 15% MPD, the unfolded state of Pelle-DD may be more compact. Alternatively, the unfolded species may possess fewer urea interaction sites, as might be expected if some of them have equal or greater affinity for MPD in the unfolded state. Residues that show particularly high MPD-induced chemical-shift perturbations might correspond to such sites.
Pelle-DD is not unique in its ability to adopt more than one tertiary fold in the solid state. In certain proteins such transitions can be induced by changes in pH, as in the massive rearrangement of the influenza virus hemagglutinin (43), or by interaction with a regulatory ligand, as in the refolding of the Rac-binding domain of p21 activated kinase (PAK) after activation by Cdc42 (44). Rearrangement of Pelle-DD, however, is irreversible, because protein that has been precipitated by MPD cannot be redissolved in aqueous buffer. Crystals of Pelle-DD obtained from MPD may represent an ordered state of a precipitated aggregate. In some respects, the Pelle-DD lattice is analogous to paracrystalline arrays of amyloid fibrils (45, 46). Although the structure of amyloid is different from that of Pelle-DD in the crystals described here, formation of both requires a refolding event.
Could the refolding and dimerization of Pelle-DD occur in a physiological context? Within the cell, refolding events can accompany the transfer of proteins from the cytosol to the low-dielectric environment of the membrane or cytosol–membrane boundary. In this compartment, proteins such as colicins (47) or cytochrome c (48) and peptides such as mellitin (49) are transformed in tertiary or quaternary structure after insertion or lateral contact with the membrane. Alcohols such as methanol can act as membrane mimetics. Methanol/water mixtures induce the transition of cytochrome c to a molten globule-like state, which possesses native-like secondary structure but only traces of tertiary structure (50). In the case of Pelle, a membrane-induced refolding event might play some role in kinase regulation or dimerization. There is, however, no experimental evidence to suggest that Pelle-DD or its homologs are conformationally labile in vivo.
A more practical concern raised by the present study is the general utility of MPD as a precipitating agent for protein crystallization. After ammonium sulfate and polyethylene glycol (51), MPD is the third most commonly used precipitating agent. Numerous examples of protein crystallization using high concentrations (>45%) of MPD can be found in the Biological Macromolecular Crystallization Database (52). In most if not all of these cases, MPD presumably acts as a precipitant rather than a partial denaturant or refolding reagent. However, as shown here, proteins and protein domains may adopt alternative structures at high MPD concentration in the solid state.
Acknowledgments
We thank Nick Pace for a critical reading of the manuscript, A. Loew, D. E. Coleman, J. Z. Chen, and the staff at the Cornell High Energy Synchrotron Source F-1 and F-2 stations for data collection, C. A. Amezcua, and T. C. Holdeman for assistance with NMR spectra acquisition, Dr. S. W. Fesik for the Fas-DD expression construct, and P. J. Thomas, R. C. Bergstrom, and C. W. Liu for assistance with fluorescence spectroscopy. This research was supported by Welch Foundation Grant I1229 and The John W. and Rhonda K. Pate Professorship (to S.R.S.). K.H.G. acknowledges support from the Searle Scholars Program and the University of Texas Southwestern Endowed Scholars Program as the W. W. Caruth Scholar in Biomedical Research.
Abbreviations
MPD, 2-methyl-2,4-pentanediol
DD, death domain
HSQC, heteronuclear single quantum correlation
P6hb, Pelle six-helix bundle
Phd, Pelle helix dimer
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code ).
References
- 1.Pittz E. P. & Timasheff, S. N. (1978) Biochemistry 17, 615-623. [DOI] [PubMed] [Google Scholar]
- 2.Schellman J. A. (1987) Biopolymers 26, 549-559. [DOI] [PubMed] [Google Scholar]
- 3.Arakawa T., Bhat, R. & Timasheff, S. N. (1990) Biochemistry 29, 1924-1931. [DOI] [PubMed] [Google Scholar]
- 4.Grosshans J., Bergmann, A., Haffter, P. & Nusslein-Volhard, C. (1994) Nature (London) 372, 563-566. [DOI] [PubMed] [Google Scholar]
- 5.Towb P., Galindo, R. L. & Wasserman, S. A. (1998) Development (Cambridge, U.K.) 125, 2443-2450. [DOI] [PubMed] [Google Scholar]
- 6.Feinstein E., Kimchi, A., Wallach, D., Boldin, M. & Varfolomeev, E. (1995) Trends Biochem. Sci. 20, 342-344. [DOI] [PubMed] [Google Scholar]
- 7.Xiao T., Towb, P., Wasserman, S. A. & Sprang, S. R. (1999) Cell 99, 545-555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belvin M. P. & Anderson, K. V. (1996) Annu. Rev. Cell Dev. Biol. 12, 393-416. [DOI] [PubMed] [Google Scholar]
- 9.Imler J. L. & Hoffmann, J. A. (2000) Rev. Immunogenet. 2, 294-304. [PubMed] [Google Scholar]
- 10.Huang B., Eberstadt, M., Olejniczak, E. T., Meadows, R. P. & Fesik, S. W. (1996) Nature (London) 384, 638-641. [DOI] [PubMed] [Google Scholar]
- 11.Aravind L., Dixit, V. M. & Koonin, E. V. (1999) Trends Biochem. Sci. 24, 47-53. [DOI] [PubMed] [Google Scholar]
- 12.Weber C. H. & Vincenz, C. (2001) Trends Biochem. Sci. 26, 475-481. [DOI] [PubMed] [Google Scholar]
- 13.Baker S. J. & Reddy, E. P. (1998) Oncogene 17, 3261-3270. [DOI] [PubMed] [Google Scholar]
- 14.Otwinowski Z. & Minor, W. (1997) Methods Enzymol. 276, 307-326. [DOI] [PubMed] [Google Scholar]
- 15.Terwilliger T. C. & Berendzen, J. (1999) Acta Crystallogr. D 55, 849-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collaborative Computational Project 4 (1994) Acta Crystallogr. D 50, 760-763.15299374 [Google Scholar]
- 17.Abrahams J. P. & Leslie, A. G. W. (1996) Acta Crystallogr. D 52, 30-42. [DOI] [PubMed] [Google Scholar]
- 18.Jones T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991) Acta Crystallogr. A 47, 110-119. [DOI] [PubMed] [Google Scholar]
- 19.Brünger A. T., Adams, P. D., Clore, G. M., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., et al. (1998) Acta Crystallogr. D 54, 905-921. [DOI] [PubMed] [Google Scholar]
- 20.Gardner K. H. & Kay, L. E. (1998) Annu. Rev. Biophys. Biomol. Struct. 27, 357-406. [DOI] [PubMed] [Google Scholar]
- 21.Kay L. E., Keifer, P. & Saarinen, T. (1992) J. Am. Chem. Soc. 114, 10663-10665. [Google Scholar]
- 22.Ikura M., Kay, L. E. & Bax, A. (1990) Biochemistry 29, 4659-4667. [DOI] [PubMed] [Google Scholar]
- 23.Kay L. E., Xu, G. Y. & Yamazaki, T. (1994) J. Magn. Reson. A 109, 129-133. [Google Scholar]
- 24.Wittekind M. & Mueller, M. L. (1993) J. Magn. Reson. B 101, 201-205. [Google Scholar]
- 25.Grzesiek S. & Bax, A. (1992) J. Am. Chem. Soc. 114, 6291-6293. [Google Scholar]
- 26.Kay L. E. (1993) J. Am. Chem. Soc. 115, 2055-2057. [Google Scholar]
- 27.Clore G. M. & Gronenborn, A. M. (1991) Annu. Rev. Biophys. Biophys. Chem. 20, 29-63. [DOI] [PubMed] [Google Scholar]
- 28.Delaglio F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J. & Bax, A. (1995) J. Biomol. NMR 6, 277-293. [DOI] [PubMed] [Google Scholar]
- 29.Johnson B. A. & Blevins, R. A. (1994) J. Biomol. NMR 4, 603-614. [DOI] [PubMed] [Google Scholar]
- 30.Cornilescu G., Delaglio, F. & Bax, A. (1999) J. Biomol. NMR 13, 289-302. [DOI] [PubMed] [Google Scholar]
- 31.Pace C. N. & Shaw, K. L. (2000) Proteins Suppl. 4, 1-7. [DOI] [PubMed] [Google Scholar]
- 32.Royer C. A., Mann, C. J. & Matthews, C. R. (1993) Protein Sci. 2, 1844-1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greene R. F. & Pace, C. N. (1974) J. Biol. Chem. 249, 5388-5393. [PubMed] [Google Scholar]
- 34.Santoro M. M. & Bolen, D. W. (1988) Biochemistry 27, 8063-8068. [DOI] [PubMed] [Google Scholar]
- 35.Chothia C., Levitt, M. & Richardson, D. (1981) J. Mol. Biol. 145, 215-250. [DOI] [PubMed] [Google Scholar]
- 36.Gallagher T., Alexander, P., Bryan, P. & Gilliland, G. L. (1994) Biochemistry 33, 4721-4729. [PubMed] [Google Scholar]
- 37.Kuszewski J., Gronenborn, A. M. & Clore, G. M. (1999) J. Am. Chem. Soc. 121, 2337-2338. [Google Scholar]
- 38.Liepinsh E. & Otting, G. (1997) Nat. Biotechnol. 15, 264-268. [DOI] [PubMed] [Google Scholar]
- 39.Mattos C. & Ringe, D. (2001) Curr. Opin. Struct. Biol. 11, 761-764. [DOI] [PubMed] [Google Scholar]
- 40.Tanford C. (1970) Adv. Protein. Chem. 24, 1-95. [PubMed] [Google Scholar]
- 41.Timasheff S. N. (1993) Annu. Rev. Biophys. Biomol. Struct. 22, 67-97. [DOI] [PubMed] [Google Scholar]
- 42.Buck M. (1998) Q. Rev. Biophys. 31, 297-355. [DOI] [PubMed] [Google Scholar]
- 43.Bullough P. A., Hughson, F. M., Skehel, J. J. & Wiley, D. C. (1994) Nature (London) 371, 37-43. [DOI] [PubMed] [Google Scholar]
- 44.Lei M., Lu, W., Meng, W., Parrini, M. C., Eck, M. J., Mayer, B. J. & Harrison, S. C. (2000) Cell 102, 387-397. [DOI] [PubMed] [Google Scholar]
- 45.Blake C. & Serpell, L. (1996) Structure (London) 4, 989-998. [DOI] [PubMed] [Google Scholar]
- 46.Koo E. H., Lansbury, P. T., Jr. & Kelly, J. W. (1999) Proc. Natl. Acad. Sci. USA 96, 9989-9990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van der Goot F. G., Gonzalez-Manas, J. M., Lakey, J. H. & Pattus, F. (1991) Nature (London) 354, 408-410. [DOI] [PubMed] [Google Scholar]
- 48.Pinheiro T. J., Cheng, H., Seeholzer, S. H. & Roder, H. (2000) J. Mol. Biol. 303, 617-626. [DOI] [PubMed] [Google Scholar]
- 49.Terwilliger T. C., Weissman, L. & Eisenberg, D. (1982) Biophys. J. 37, 353-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bychkova V. E., Dujsekina, A. E., Klenin, S. I., Tiktopulo, E. I., Uversky, V. N. & Ptitsyn, O. B. (1996) Biochemistry 35, 6058-6063. [DOI] [PubMed] [Google Scholar]
- 51.Hennessy D., Buchanan, B., Subramanian, D., Wilkosz, P. A. & Rosenberg, J. M. (2000) Acta Crystallogr. D 56, 817-827. [DOI] [PubMed] [Google Scholar]
- 52.Gilliland G. L. (1988) J. Cryst. Growth 90, 51-59. [Google Scholar]
- 53.Esnouf R. (1997) J. Mol. Graphics 15, 133-138. [DOI] [PubMed] [Google Scholar]
- 54.Kraulis P. J. (1991) J. Appl. Crystallogr. 24, 946-950. [Google Scholar]
- 55.pov-ray Team, (1998) pov-ray (Williamstown, Victoria, Australia).
- 56.Bai Y., Milne, J. S., Mayne, L. & Englander, S. W. (1993) Proteins 17, 75-86. [DOI] [PMC free article] [PubMed] [Google Scholar]