

Abstract
Vascular endothelial growth factor (VEGF) plays a critical role during normal embryonic angiogenesis and also in the pathological angiogenesis that occurs in a number of diseases, including cancer. Initial attempts to block VEGF by using a humanized monoclonal antibody are beginning to show promise in human cancer patients, underscoring the importance of optimizing VEGF blockade. Previous studies have found that one of the most effective ways to block the VEGF-signaling pathway is to prevent VEGF from binding to its normal receptors by administering decoy-soluble receptors. The highest-affinity VEGF blocker described to date is a soluble decoy receptor created by fusing the first three Ig domains of VEGF receptor 1 to an Ig constant region; however, this fusion protein has very poor in vivo pharmacokinetic properties. By determining the requirements to maintain high affinity while extending in vivo half life, we were able to engineer a very potent high-affinity VEGF blocker that has markedly enhanced pharmacokinetic properties. This VEGF-Trap effectively suppresses tumor growth and vascularization in vivo, resulting in stunted and almost completely avascular tumors. VEGF-Trap-mediated blockade may be superior to that achieved by other agents, such as monoclonal antibodies targeted against the VEGF receptor.
The sprouting of new blood vessels, termed angiogenesis, is required to support growth in the embryo and young animal, as well as to allow for repair and remodeling processes in the adult. However, aberrant angiogenesis is also associated with a number of pathological conditions and diseases, including cancer (1, 2). Tumors, like many normal tissues, use the vasculature as a means to obtain oxygen and nutrients and to remove waste products. Although tumors can in part grow by coopting existing host vessels (3–6), most tumors also induce new vessel formation, suggesting that this neovascularization is required for their growth (1, 2, 7). Consequently, much effort has been directed toward discovering antiangiogenic agents and evaluating them as cancer therapeutics. Perhaps the best characterized and most highly validated antiangiogenic approach involves targeting the vascular endothelial growth factor (VEGF) pathway (1, 8–11). Based on numerous animal studies, the VEGF pathway is the only well-defined signaling pathway known to be required for normal development of the vasculature as well as for the pathologic angiogenesis that accompanies cancer and other disease states (8–10).
The VEGF pathway is initiated when VEGF binds to its receptors on endothelial cells. The two best characterized VEGF receptors are termed VEGF receptor 1 (VEGFR1) and VEGF receptor 2 (VEGFR2). VEGFR1 and VEGFR2 are highly related transmembrane tyrosine kinases that use their ectodomains to bind VEGF; this binding in turn activates the intrinsic tyrosine kinase activity of their cytodomains, initiating intracellular signaling. Interestingly, although VEGFR1 binds to VEGF with substantially higher affinity, most of the biologic effects of VEGF seem to be mediated via VEGFR2. In animals, blockade of the VEGF pathway has been achieved by many different means, including blocking antibodies targeted against VEGF (12–14) or its receptors (15), soluble decoy receptors that prevent VEGF from binding to its normal receptors (16–20), as well as small molecule inhibitors of the tyrosine kinase activity of the VEGFRs (21–23). Recently, a study that compared the efficacy of VEGF blockade to other “antiangiogenic” strategies established that this approach is superior to many others (ref. 11). Consistent with predictions from animal studies, blockade of VEGF using a humanized monoclonal antibody has emerged as the first and thus far only antiangiogenesis approach reporting promising results in human cancer patients, based on preliminary reports from early clinical trials.† The hope is that anti-VEGF approaches can be generalized to many different types of cancer, as well as to other diseases in which pathologic angiogenesis contributes, such as diabetic retinopathy and psoriasis.
The clinical promise of initial anti-VEGF approaches highlights the need to optimize blockade of this pathway. Previous studies have found that one of the most effective ways to block the VEGF signaling pathway is to prevent VEGF from binding to its normal receptors by administering decoy VEGF receptors (11, 16, 17, 24). The highest-affinity VEGF blocker described to date is a soluble decoy receptor created by fusing the first three Ig domains of VEGFR1 to the constant region (Fc portion) of human IgG1, resulting in a forced homodimer that has picomolar binding affinity (16, 17). In tumor experiments, this VEGFR1-Fc reagent is efficacious at approximately 500-fold lower concentration than a similar VEGFR-2 construct (11). Despite its high affinity, the VEGFR1-Fc is not a feasible clinical candidate because of its poor pharmacokinetic profile; in rodent studies, this protein has to be administered frequently and at very high levels to achieve efficacious levels (16, 17, 24). In addition, the VEGFR1-Fc exhibits certain toxicological side effects that are not seen with the VEGFR2-Fc (11). These effects appear to be due to nonmechanism-based and nonspecific properties of this agent (see Discussion). By determining the requirements to maintain high affinity while extending in vivo half life, we were able to engineer a very potent high-affinity VEGF blocker that has prolonged in vivo pharmacokinetics and pharmacodynamics, lacks nonspecific toxicities, and can effectively suppress the growth and vascularization of a number of different types of tumors in vivo.
Materials and Methods
Engineering VEGF-Traps.
The parental VEGF-Trap was created by fusing the first three Ig domains of VEGFR1 to the constant region (Fc) of human IgG1. VEGF-TrapΔB1 was created by removing a highly basic 10-aa stretch from the third Ig domain of the parental VEGF-Trap. VEGF-TrapΔB2 was created by removing the entire first Ig domain from VEGF-TrapΔB1. VEGF-TrapR1R2 was created by fusing the second Ig domain of VEGFR1 with the third Ig domain of VEGFR2. All of the VEGF-Trap variants were produced and purified from Chinese hamster ovary cells.
Pharmacokinetic Analysis of VEGF-Traps.
BALB/c mice (25–30 g) were injected s.c. with 4 mg/kg of the various Traps and bled at 1, 2, 4, 6, 24, 48, 72, and 144 hr after injection. Levels of all VEGF-Traps were measured by an ELISA by using human VEGF165 to capture and an antibody to the human Fc region as the reporter.
Extracellular Matrix (ECM)-Binding Assay.
ECM-coated plates (Becton Dickinson no. 35–4607) were incubated with varying concentrations of VEGF-Traps for 1 hr at room temperature. They were washed and incubated with alkaline phosphatase-conjugated anti-human Fc antibody (Promega, 1:4,000 in PBS + 10% BCS) for 1 hr at room temperature. Plates were washed four times with PBS + 0.1%Triton-X 100 and reagent buffer added for color development. Plates were read at 405–570 nm.
VEGF-Trap-Binding Assay.
Binding affinities of VEGF-Traps were measured by using a specific and sensitive ELISA (R&D Systems kit no. DVE00) for detecting free (unbound) human VEGF in mixtures of the VEGF-Traps (ranging in concentration from 0.1 to 160 pM) with human VEGF165 (at 10 pM), incubated overnight at room temperature.
Human Umbilical Vein Endothelial Cell Phosphorylation Assay.
Confluent monolayers of human umbilical vein endothelial cells [Vec Technologies (Rensselaer, NY) passage no. 5] were serum-starved for 2 hr and then challenged for 5 min with vehicle or 40 ng/ml of human VEGF165, alone or preincubated with VEGF-Traps at 1.5-fold molar excess. Cells were then lysed, immunoprecipitated by using a VEGFR2-specific antibody, and immunoblotted with a phosphotyrosine-specific antibody (Upstate Biotechnology, 4G10 mAb).
VEGF-Induced Proliferation Assay.
Cells that proliferate in response to VEGF were generated by stably transfecting NIH 3T3 cells with a VEGFR2/TrkB chimeric receptor (in which the cytodomain of VEGFR2 was replaced with that of TrkB, a receptor for brain-derived neurotrophic factor that effectively drives proliferation in these cells). Five thousand cells were plated per well of a 96-well plate, allowed to settle for 2 hr, incubated for 1 hr with VEGF-Trap variants (titrated from 40 nM to 20 pM), then challenged for 72 hr with human VEGF165 at a concentration of 1.56 nM, followed by addition of [3-(4,5 dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, innersai and spectrophotometric analysis at 450/570 nm.
Acute Hypotension.
Male Wistar–Kyoto rats (180–240 g) from Taconic Laboratories were maintained on a 12:12 light/dark cycle (lights on 0600) with food and water available ad libitum. Before challenge with VEGF, animals were pretreated with VEGF-Traps or PBS as indicated, anesthetized with 1.5–2% isoflurane in oxygen, and the left femoral artery catheterized for direct measurement of systolic blood pressure through a blood pressure transducer (IITC, Woodland Hills, CA) into a chart recorder (Linseis, Princeton Junction, NJ). Animals were then injected in the right jugular vein with a 200-μl bolus containing 10 μg of recombinant human VEGF165. Systolic blood pressure was measured before VEGF injection and every minute thereafter for 20 min. Blood pressures were normalized to baseline preinjection and analyzed by using mixed factorial ANOVAs (see supporting information on the PNAS web site, www.pnas.org).
Tumor Growth Experiments.
C6 glioma cells (1.0 × 106 cells/mouse) and A673 rhabdomyosarcoma cells (2.0 × 106 cells/mouse) were obtained from American Type Culture Collection, and B16F10.9 melanoma cells (5.0 × 105 cells/mouse) were a generous gift from Charles Lin (Duke University, Durham, NC). Cells were suspended in serum-free medium and implanted s.c. on the shaved right flank of male C.B-17 SCID mice at the indicated concentrations. After tumor cell implantation and twice weekly thereafter for the duration of the experiment, mice received a s.c. injection (at the nape of the neck) of vehicle (PBS + 0.5% glycerol), VEGF-Trap, or DC101 (from American Type Culture Collection). After 2–3.0 weeks, animals were killed and tumors were measured ex vivo with calipers (tumor volume = length × width × height). For immunohistochemistry studies, mice were perfused with 4% paraformaldehyde, and tissue was processed as previously described (25).
Results
Reengineering Parental VEGF-Trap to Improve Its Pharmacokinetic Profile.
On the basis of the previously reported high affinity of a soluble decoy receptor in which VEGFR1 is fused to the Fc portion of human IgG1 (16, 17), we produced this fusion protein to study its properties (see parental VEGF-Trap, Fig. 1A). Single s.c. injections of parental VEGF-Trap (4 mg/kg) into mice were performed to confirm that it indeed displayed poor pharmacokinetic properties, with a maximal concentration (Cmax) of only 0.05 μg/ml and total “area under the curve concentration” (AUC) of 0.04 μg × days/ml (Fig. 1C). We postulated that these poor pharmacokinetic properties might be due to the high positive charge of this protein (pI 9.4), which in turn may result in its deposition at the site of s.c. injection because of nonspecific adhesion to highly negatively charged proteoglycans that comprise the extracellular matrix. To test this hypothesis, we next engineered several variants of the parental VEGF-Trap with reduced positive charges. On review of the charge density in the parental molecule, we noted a highly basic stretch of 10 amino acids in the third Ig domain of VEGFR1 (see blue box in Fig. 1A). To reduce the charge, this region was excised, resulting in a decrease in the pI of this VEGF-Trap (termed VEGF-TrapΔB1; see Fig. 1A) from 9.4 to 9.1. It was also noted that the first Ig domain of VEGFR1 had a basic pI, and we thus decided to test removal of this domain as well as the above-noted basic region, resulting in a protein termed VEGF-TrapΔB2 (Fig. 1A), with a further reduced pI of 8.9. Finally, because the third Ig domain of VEGFR2 has a lower pI than the corresponding domain of VEGFR1, we simply switched these domains to make a Trap in which the second Ig domain of VEGFR1 is directly fused to the third Ig domain of VEGFR2; this trap was termed VEGF-TrapR1R2 (Fig. 1A) and had a pI of 8.82. Previous structural analyses indicated that VEGFR1 might make greater use of its second Ig domain in contacting VEGF, whereas VEGFR2 instead makes greater use of its third Ig domain (26), raising the interesting and useful possibility that VEGF-TrapR1R2 might actually bind more tightly to VEGF than the parental versions. Combining the distinct binding regions of two different receptors to create a higher-affinity interactor has previously been used in the creation of a series of interleukin and cytokine blockers also termed Traps (A. Economides, L. Rocco Carpenter, J.S.R., V. Wong, E. Koehler-Stec, C. Hartnett, E. Pyles, T.D., M. Young, J.P.F., Frank Lee, Scott Carver, Jennifer McNay, K.B., S. Ramakanth, R. Hatabarat, C.R., T.H., G.D.Y., and N. Stahl, unpublished results). Using a simple extracellular matrix-binding assay, we then confirmed the hypothesis that decreasing the positive charge of the VEGF-Traps would result in decreased adhesion to extracellular matrix (Fig. 1B). Binding to extracellular matrix in this assay was directly related to the pI of the Traps, with both VEGF-TrapR1R2 and VEGF-TrapΔB2 displaying negligible binding in this assay.
Fig 1.

Engineering of VEGF-Traps with improved pharmacokinetics. (A) Schematics of full-length VEGFR1 (red) and VEGFR2 (blue) are provided, indicating their seven Ig domains, transmembrane regions (black bars), and kinase domains (ovals). The parental VEGF-Trap contains the first three Ig domains of VEGFR1 (including the highly basic 10-aa stretch in Ig3, blue box) fused to the Fc portion of human IgG1. VEGF-TrapΔB1 is identical to the parental VEGF-Trap, except that the basic stretch in Ig3 has been removed. VEGF-TrapΔB2 is the same construct as ΔB1, except that the first Ig domain has been removed. VEGF-TrapR1R2 possesses the second Ig domain of VEGFR1 and the third Ig domain of VEGFR2 fused to the Fc portion of human IgG1. (B) The four indicated VEGF-Traps were assayed in vitro for their capacity to bind to extracellular matrix, with only the parental VEGF-Trap and VEGF-TrapΔB1 demonstrating binding. (C) Pharmacokinetic analysis of the VEGF-Traps reveals that the parental VEGF-Trap has the poorest profile, whereas VEGF-TrapR1R2 showed the best profile.
On the basis of the above results, we next tested these various VEGF-Traps in vivo for their pharmacokinetic behavior. Their in vivo behavior followed the theoretical charge predictions as well as the in vitro adhesion properties. Every reduction in pI was accompanied by a corresponding improvement in Cmax and AUC: VEGF-TrapΔB1 had a Cmax of 1.3 μg/ml and an AUC of 1.36 μg × days/ml; VEGF-TrapΔB2 had a Cmax of 2.65 μg/ml and an AUC of 5.42 μg × days/ml; whereas VEGF-TrapR1R2 revealed the best profile with a Cmax of 16 μg/ml and an AUC of 36.28 μg × days/ml (Fig. 1C). Thus, VEGF-TrapR1R2 had an AUC that was almost 1,000-fold higher than that of the parental VEGF-Trap, raising the possibility that it might be a far superior pharmacologic agent, assuming it retained its ability to bind and block VEGF.
Comparison of Parental VEGF-Trap with VEGF-TrapR1R2 in Binding, Phosphorylation, and Cell Proliferation Assays in Vitro.
Because of the superior pharmacokinetic properties of VEGF-TrapR1R2, we next compared this Trap to its parent for its ability to bind and block VEGF in vitro. To determine binding affinity of the Traps for VEGF, equilibrium binding assays were performed in which different concentrations of the Traps were incubated with VEGF165, and the amount of unbound VEGF165 was measured, revealing that parental VEGF-Trap displays a kD of ≈5 pM, whereas VEGF-TrapR1R2 has a binding affinity of about 1 pM (Fig. 2A). Preliminary analyses show that VEGF-TrapR1R2 has a kD of ≈1–10 pM for VEGF121 and approximately 45 pM for placental growth factor 2 (not shown); other VEGF isoforms and relatives have not been analyzed.
Fig 2.

Binding affinity and inhibitory properties of VEGF-Traps. (A) Affinities of indicated VEGF-Traps for VEGF, as determined by using a binding assay that measures unbound VEGF (ordinate) after incubation of 10 pM of human VEGF165 with varying concentrations of VEGF-Traps (abscissa). (B) Inhibition of VEGF-induced phosphorylation of VEGFR2 in human umbilical vein endothelial cell phosphorylations using indicated VEGF-Traps at 1.5-fold molar excess, as revealed with immunoblotting assay. (C) Inhibition of VEGF-induced proliferation of fibroblasts containing a chimeric VEGFR2/TrkB receptor, using varying concentrations of VEGF-Traps in the presence of 1.56 nM of VEGF.
To determine whether Trap binding of VEGF could potently and effectively block the ability of VEGF to activate its receptor, VEGF and Traps were added to cultured endothelial cells, and the effects on VEGFR2 phosphorylation were examined, revealing that both parental VEGF-Trap as well as VEGF-TrapR1R2 can completely block VEGF-induced VEGFR2 phosphorylation when added at a 1.5-fold molar excess compared with the added VEGF, consistent with very high-affinity binding to VEGF (Fig. 2B). Finally, to assess whether these Traps would also be effective in cell-based proliferation assays, we engineered a cell line containing a chimeric VEGFR2 receptor that mediates a very strong proliferative response to VEGF and found that both parental VEGF-Trap and VEGF-TrapR1R2 potently blocked VEGF-induced proliferation in 3-day growth assays in these cells, with an IC50 at approximately an equimolar concentration of Trap with the added VEGF, once again consistent with very high-affinity binding of the Traps for VEGF (Fig. 2C).
VEGF-TrapR1R2 Provides Long-Term Blockade of Exogenously Administered VEGF-Induced Acute Hypotension.
The above studies indicated that VEGF-TrapR1R2 was at least as impressive a blocker of VEGF as the parental version, but that it had far superior pharmacokinetic properties. To initially explore whether these attributes translated into superior pharmacodynamic performance, we compared these reagents by using an acute readout of VEGF responsiveness in vivo. Administration of a single bolus dose (10 μg) of recombinant VEGF165 to rats results in acute hypotension, with a drop of about 40% from baseline systolic blood pressure; this drop is maximal at 5 min and slowly rectifies to normal by about 30 min (Fig. 3A). To compare the pharmacodynamic efficacy of the VEGF-Traps in blocking this acute response, we preadministered the parental VEGF-Trap or VEGF-TrapR1R2 at 25 mg/kg, 24 hr before VEGF administration (Fig. 3A). Consistent with what would be expected from the above pharmacokinetic studies, this dose of VEGF-TrapR1R2 completely blocked VEGF-induced hypotension, whereas the parental VEGF-Trap had no discernable effect. Thus, although the parental VEGF-Trap and its VEGF-TrapR1R2 derivative are quite comparable in vitro (see above), the VEGF-TrapR1R2 performs much better in vivo, presumably because of its dramatically enhanced pharmacokinetic profile.
Fig 3.
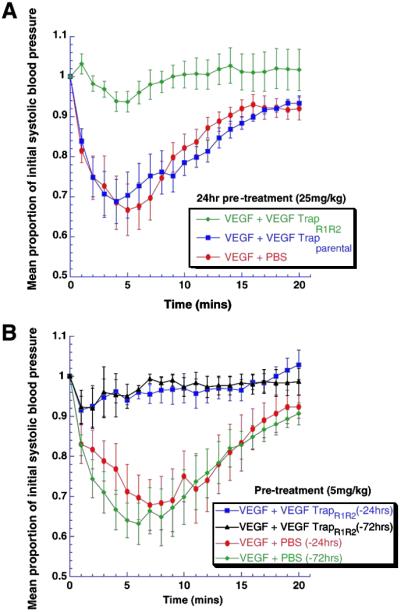
Using blockade of VEGF-induced acute hypotension to pharmacodynamically compare VEGF-Traps. (A) When rats were treated with VEGF-Traps at 25 mg/kg at 1 day before VEGF challenge, VEGF-TrapR1R2 (n = 8) completely blocked VEGF-induced hypotension, whereas PBS (n = 6) and parental VEGF-Trap (n = 6) were ineffective. ANOVA shows treatment effect, P < 0.007. (B) At a 5-fold lower dose (5 mg/kg), VEGF-TrapR1R2 was still effective at 1 day (n = 4) or 3 days (n = 3) before the VEGF challenge. ANOVA shows treatment effect, P < 0.03.
To further characterize the length of time in which VEGF-TrapR1R2 remained efficacious, we waited 1, 3, and 7 days after injection of the Trap at 5 mg/kg before inducing hypotension. At this dose, VEGF-TrapR1R2 was completely effective in blocking VEGF-induced acute hypotension at 1 and 3 days after a single bolus (Fig. 3B) but was not significantly different from controls at 7 days (data not shown).
VEGF-TrapRiR2 Dramatically Blocks Tumor Growth in Vivo.
Altogether, the above pharmacokinetic and pharmacodynamic studies indicated that VEGF-TrapR1R2 has the potential to be a long-term and potent pharmacologic blocker of VEGF-mediated activities in vivo, far superior to that of parental VEGF-Trap. To begin to explore the value of VEGF-TrapR1R2 as an anticancer therapeutic and to compare it to other effective agents targeting the VEGF pathway, we evaluated its ability to block the growth of a variety of tumor cell lines in s.c. tumor models in mice. Tumor cells were derived from diverse tissue origins and different species (mouse B16F10.9 melanoma, human A673 rhabdomyosarcoma, and rat C6 glioma). After implantation of tumor cells, mice were allowed a brief recovery period and then received s.c. injections of VEGF-TrapR1R2 (25 mg/kg) or vehicle twice weekly for the duration of the experiment (2–3.0 weeks), after which the animals were killed and tumors excised and measured. VEGF-TrapR1R2 significantly inhibited the growth of all three types of tumors (Fig. 4A). In the study using C6 glioma cells, a 10-fold lower dose of VEGF-TrapR1R2 (2.5 mg/kg) was tested and found to be equally effective at inhibiting tumor growth.
Fig 4.

VEGF-TrapR1R2 dramatically inhibits the s.c. growth and vascularity of implanted tumors from diverse tissues and species. (A) VEGF-Trap R1R2 substantially blocked the growth of the indicated s.c. implanted tumors, at the indicated doses twice weekly for 2 weeks (C6 and B16F10.9) or 3.0 weeks (A673). Error bars represent standard error of mean, n = five mice/treatment group. The differences between control tumor volumes and VEGF-TrapR1R2–treated tumor volumes were analyzed by using Student's t tests and found to be significant at the following levels: B16F10 P = 0.01; A673 P = 0.06; C6 P < 0.0001. (B–D) Histological analysis reveals that VEGF-TrapR1R2 can effectively block blood vessel growth in these implanted tumors. Sections of C6 tumors stained with antibodies to platelet–endothelial cell adhesion molecule reveal that vehicle-treated animals had large tumors that were highly vascularized (B), whereas animals treated with 25 mg/kg VEGF-TrapR1R2 (C) had tumors that were largely avascular with large areas of necrosis (N). Viable tumor appeared to be vascularized because of cooption of preexisting host vessels (white arrowheads) associated with hypodermal musculature (M) and dermis. Treatment with 2.5 mg/kg VEGF-TrapR1R2 greatly stunted tumor growth (C) and resulted in large necrotic regions (N), although small pockets of vessels were occasionally apparent (black arrows). (Bar = 100 μm.)
To evaluate the effects of VEGF-TrapR1R2 on tumor-associated angiogenesis, the tumors from the above studies were sectioned and immunostained with antibodies to platelet–endothelial cell adhesion molecule, so that the vasculature could be visualized (Fig. 4 B–D). This analysis revealed that the higher dose of VEGF-TrapR1R2 almost completely blocked tumor-associated angiogenesis, with the stunted tumors being largely avascular, save for regions in which preexisting host vessels appeared to be coopted by surrounding tumor (see open arrowheads, Fig. 4C). The lower dose of VEGF-TrapR1R2, which was quite comparable at inhibiting tumor growth (see above), appeared to be slightly less effective at completely blocking tumor-associated angiogenesis, allowing for small pockets of tumor-associated vessels in otherwise avascular tumors (see black arrowheads in Fig. 4D). In contrast to the VEGF-Trap-treated tumors, control tumors in vehicle-treated mice not only were much larger (see above) but also had a very high vascular density (Fig. 4B).
VEGF-TrapR1R2 Compares Favorably with Antibodies Targeting VEGFR2.
After establishing that VEGF-TrapR1R2 was effective at blocking s.c. tumor growth, we undertook studies to compare its efficacy with other known VEGF blockers. One particularly effective and well-characterized blocker is a monoclonal antibody, termed DC101, that targets VEGFR2 (15). When equimolar doses of VEGF-TrapR1R2 and DC101 were compared in the B16F10 melanoma model, it was apparent that much higher doses of DC101 are required to inhibit tumor growth (Fig. 5). Furthermore, because antibodies have longer circulation times in mice than simple Fc fusion proteins, the highly efficacious dose of DC101 accumulates to approximately 60-fold higher serum levels than that of the equally efficacious low dose of VEGF-Trap: circulating levels of DC101 in animals treated with the 40-mg/kg dose were 2,442 ± 272 μg/ml, in contrast to the circulating levels of VEGF-Trap in animals treated with 3.2 mg/kg, which were 40 ± 8 μg/ml. Thus, circulating levels of VEGF-Trap that were approximately 60-fold lower than those of DC101 were equally efficacious in inhibiting tumor growth. Importantly, the favorable allometric scaling of Fc fusion proteins relative to antibodies (27, 28) suggests that in humans the circulation time for the VEGF-Trap will be much more comparable to that of antibodies, which in turn suggests that in humans the difference in efficacious doses would be further magnified and may be as great as 60-fold.
Fig 5.

VEGF-TrapR1R2 blocks tumor growth (of subcutaneously implanted B16F10.9 cells) at far lower concentrations than DC101, a monoclonal antibody directed to VEGFR2. Mice were treated twice weekly with the indicated dose of VEGF-TrapR1R2, DC101, or vehicle. After 2.5 weeks, mice were killed, and tumors were excised and measured. Individual tumor volumes are shown (colored bars), as are average tumor volumes for each treatment (black bars) ± SEM, n = six mice/treatment group. Differences between treatment groups were analyzed by using a one-way ANOVA followed by Fisher's protected least significant difference test. Average volume of tumors in all treatment groups is significantly smaller than control tumor volume (P < 0.01). Differences in tumor volume between the high-dose VEGF-Trap, low-dose VEGF-Trap, and high-dose DC101 treatment groups are not significantly different, but they are significantly different from those of the low-dose DC101 treatment group (P < 0.02).
As described in an accompanying manuscript (29), when used at the same dose, VEGF-Trap shows efficacy equal to or better than a monoclonal antibody to VEGF (30). As noted above, because Fc fusion proteins have much shorter circulating half-lives than antibodies in mice, but comparable half-lives in humans, the finding that the VEGF-TrapR1R2 is at least as potent as the monoclonal antibody in mice suggests that the efficacious dose of VEGF-Trap will be much lower than that of the monoclonal antibody in humans.
Discussion
Validation of VEGF as an important new target in the war against cancer comes from pioneering clinical studies using a humanized monoclonal antibody that binds and blocks VEGF.† Because anti-VEGF approaches act by blocking tumor-associated angiogenesis, which appears to be widely required by many different types of tumors, these approaches may prove to be generally useful against a wide assortment of cancers. In addition, pathological angiogenesis seems to contribute to a number of non-neoplastic diseases, such as diabetic retinopathy (31) and psoriasis (32), extending the potential utility of anti-VEGF therapeutics. All this promise highlights the need to optimize anti-VEGF approaches. Herein we describe the engineering of an anti-VEGF agent, termed VEGF-TrapR1R2. VEGF-TrapR1R2 is a derivative of perhaps the most potent VEGF binder known, VEGFR1. Soluble forms of VEGFR1 suffer from poor pharmacokinetic properties, which seem to correlate with their nonspecific interactions with extracellular matrix. VEGF-TrapR1R2 was engineered to have minimal interactions with extracellular matrix, and this property apparently accounts for its satisfying pharmacokinetic profile. The combination of high-affinity and improved pharmacokinetics apparently contributes toward making VEGF-TrapR1R2 one of the most, if not the most, potent and efficacious VEGF blocker available. An additional advantage is that VEGF-TrapR1R2 is composed of entirely human sequences, hopefully minimizing the possibility that it might prove immunogenic in human patients. Despite its wholly human nature, VEGF-TrapR1R2 binds all species of VEGF tested, from human to chicken VEGF (not shown), making it a very versatile reagent that can be used in almost any experimental animal models.
A recent study comparing numerous antiangiogenesis approaches concluded that anti-VEGF approaches were the most efficacious (11). The particular anti-VEGF agent used for these studies was essentially equivalent to our parental VEGF-Trap but was delivered in an adenoviral system in which it was highly expressed in the livers of infected animals. In contrast to other anti-VEGF approaches that seem to be well-tolerated, the adenovirally delivered VEGF-Trap caused severe liver toxicity and ascites, raising the possibility that it might have some unique mechanism-based side effects compared with other anti-VEGF approaches. To explore this possibility, we made adenoviral versions of both the parental VEGF-Trap as well as VEGF-TrapR1R2 and found that, whereas adenoviral delivery of parental VEGF-Trap reproduces the previously reported toxicities (11), adenoviral delivery of VEGF-TrapR1R2 did not cause these side effects even though much higher levels were achieved in the circulation. Our conclusion is that the nonspecific interactions of the parental VEGF-Trap with extracellular matrix contribute to its increased toxicity after adenoviral administration, and that comparable toxicity is not noted with adenoviral administration of the engineered VEGF-TrapR1R2.
In addition to the anticancer findings reported here, recent studies have shown that various versions of the VEGF-Trap can efficaciously treat a cancer-associated condition in mice similar to liver peliosis (33), as well as noncancer-associated disease models, such as of diabetic retinopathy (34–36) and psoriasis (Y.-P. Xia, M. Detmar, G.D.Y., and J.S.R., unpublished results). The accompanying manuscript (29) compares the efficacy of the VEGF-Trap to that of several other VEGF blockers, including a humanized monoclonal antibody to VEGF, in a model of kidney cancer. Among the several VEGF blockers tested, the VEGF-Trap shows the best overall efficacy. In this manuscript, we compare the efficacy of the VEGF-Trap to that of a monoclonal antibody to VEGFR2 in cancer models and find that far lower circulating levels of VEGF-TrapR1R2 are required for similar efficacy. Tumors treated with highest doses of the VEGF-Trap are not only stunted but also strikingly avascular. Our description of a VEGF blocker with such superior blocking and pharmacologic properties seems to demand that it be tested in human patients suffering from diseases involving neoangiogenesis. Toward this end, the safety of the VEGF-Trap has recently been confirmed in toxicological studies in cynomologus monkeys (data not shown). Consequently, the VEGF-Trap is currently in human clinical trials for several different types of cancer.
Supplementary Material
Acknowledgments
We thank the following for their support: A. Rafique, B. Luan, S. Keerthy, A. Polotskaia, S. Mahon, S. Xu, M. Fezoui, S. Jiang, L. Pasnikowski, V. Mosher, H. Collins, J. Kintner, L. Kasselman, and Q. Zhang. We also thank S. Staton and V. Lan for image processing.
Abbreviations
VEGF, vascular endothelial growth factor
VEGFR1, VEGF receptor 1
VEGFR2, VEGF receptor 2
AUC, area under the curve
Yang, J., Haworth, L., Steinberg, S., Rosenberg, S. & Novotny, W. (2002) Am. Soc. Clin. Oncol. (abstr. 15).
References
- 1.Ferrara N. & Alitalo, K. (1999) Nat. Med. 5, 1359-1364. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J. (1995) Nat. Med. 1, 27-31. [DOI] [PubMed] [Google Scholar]
- 3.Holash J., Wiegand, S. J. & Yancopoulos, G. D. (1999) Oncogene 18, 5356-5362. [DOI] [PubMed] [Google Scholar]
- 4.Pezzella F., Pastorino, U., Tagliabue, E., Andreola, S., Sozzi, G., Gasparini, G., Menard, S., Gatter, K. C., Harris, A. L., Fox, S., et al. (1997) Am. J. Pathol. 151, 1417-1423. [PMC free article] [PubMed] [Google Scholar]
- 5.Zagzag D., Friedlander, D. R., Margolis, B., Grumet, M., Semenza, G. L., Zhong, H., Simons, J. W., Holash, J., Wiegand, S. J. & Yancopoulos, G. D. (2000) Pediatr. Neurosurg. 33, 49-55. [DOI] [PubMed] [Google Scholar]
- 6.Zagzag D., Amirnovin, R., Greco, M. A., Yee, H., Holash, J., Wiegand, S. J., Zabski, S., Yancopoulos, G. D. & Grumet, M. (2000) Lab. Invest. 80, 837-849. [DOI] [PubMed] [Google Scholar]
- 7.van Hinsbergh V. W., Collen, A. & Koolwijk, P. (1999) Ann. Oncol. 10, 60-63. [PubMed] [Google Scholar]
- 8.Eriksson U. & Alitalo, K. (1999) Curr. Top. Microbiol. Immunol. 237, 41-57. [DOI] [PubMed] [Google Scholar]
- 9.Ferrara N. (1999) Curr. Top. Microbiol. Immunol. 237, 1-30. [DOI] [PubMed] [Google Scholar]
- 10.Yancopoulos G. D., Davis, S., Gale, N. W., Rudge, J. S., Wiegand, S. J. & Holash, J. (2000) Nature (London) 407, 242-248. [DOI] [PubMed] [Google Scholar]
- 11.Kuo C. J., Farnebo, F., Yu, E. Y., Christofferson, R., Swearingen, R. A., Carter, R., von Recum, H. A., Yuan, J., Kamihara, J., Flynn, E., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 4605-4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim K. J., Li, B., Winer, J., Armanini, M., Gillett, N., Phillips, H. S. & Ferrara, N. (1993) Nature (London) 362, 841-844. [DOI] [PubMed] [Google Scholar]
- 13.Asano M., Yukita, A., Matsumoto, T., Hanatani, M. & Suzuki, H. (1998) Hybridoma 17, 185-190. [DOI] [PubMed] [Google Scholar]
- 14.Kamiya K., Konno, H., Tanaka, T., Baba, M., Matsumoto, K., Sakaguchi, T., Yukita, A., Asano, M., Suzuki, H., Arai, T. & Nakamura, S. (1999) Jpn. J. Cancer Res. 90, 794-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prewett M., Huber, J., Li, Y., Santiago, A., O'Connor, W., King, K., Overholser, J., Hooper, A., Pytowski, B., Witte, L., Bohlen, P. & Hicklin, D. J. (1999) Cancer Res. 59, 5209-5218. [PubMed] [Google Scholar]
- 16.Ferrara N., Chen, H., Davis-Smyth, T., Gerber, H. P., Nguyen, T. N., Peers, D., Chisholm, V., Hillan, K. J. & Schwall, R. H. (1998) Nat. Med. 4, 336-340. [DOI] [PubMed] [Google Scholar]
- 17.Gerber H. P., Vu, T. H., Ryan, A. M., Kowalski, J., Werb, Z. & Ferrara, N. (1999) Nat. Med. 5, 623-628. [DOI] [PubMed] [Google Scholar]
- 18.Gerber H. P., Hillan, K. J., Ryan, A. M., Kowalski, J., Keller, G. A., Rangell, L., Wright, B. D., Radtke, F., Aguet, M. & Ferrara, N. (1999) Development (Cambridge, U.K.) 126, 1149-1159. [DOI] [PubMed] [Google Scholar]
- 19.Goldman C. K., Kendall, R. L., Cabrera, G., Soroceanu, L., Heike, Y., Gillespie, G. Y., Siegal, G. P., Mao, X., Bett, A. J., Huckle, W. R., et al. (1998) Proc. Natl. Acad. Sci. USA 95, 8795-8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Millauer B., Longhi, M. P., Plate, K. H., Shawver, L. K., Risau, W., Ullrich, A. & Strawn, L. M. (1996) Cancer Res. 56, 1615-1620. [PubMed] [Google Scholar]
- 21.Fong T. A., Shawver, L. K., Sun, L., Tang, C., App, H., Powell, T. J., Kim, Y. H., Schreck, R., Wang, X., Risau, W., et al. (1999) Cancer Res. 59, 99-106. [PubMed] [Google Scholar]
- 22.Drevs J., Hofmann, I., Hugenschmidt, H., Wittig, C., Madjar, H., Muller, M., Wood, J., Martiny-Baron, G., Unger, C. & Marme, D. (2000) Cancer Res. 60, 4819-4824. [PubMed] [Google Scholar]
- 23.Wedge S. R., Ogilvie, D. J., Dukes, M., Kendrew, J., Curwen, J. O., Hennequin, L. F., Thomas, A. P., Stokes, E. S., Curry, B., Richmond, G. H., et al. (2000) Cancer Res. 60, 970-975. [PubMed] [Google Scholar]
- 24.Gerber H. P., Kowalski, J., Sherman, D., Eberhard, D. A. & Ferrara, N. (2000) Cancer Res. 60, 6253-6258. [PubMed] [Google Scholar]
- 25.Holash J., Maisonpierre, P. C., Compton, D., Boland, P., Alexander, C. R., Zagzag, D., Yancopoulos, G. D. & Wiegand, S. J. (1999) Science 284, 1994-1998. [DOI] [PubMed] [Google Scholar]
- 26.Lu D., Kussie, P., Pytowski, B., Persaud, K., Bohlen, P., Witte, L. & Zhu, Z. (2000) J. Biol. Chem. 275, 14321-14330. [DOI] [PubMed] [Google Scholar]
- 27.Mordenti J. & Green, J. D. (1991) in New Trends in Pharmacokinetics, eds. Rescigno, A. & Thakur, A. K. (Plenum, New York), pp. 411–424.
- 28.Lin Y. S., Nguyen, C., Mendoza, J. L., Escandon, E., Fei, D., Meng, Y. G. & Modi, N. B. (1999) J. Pharmacol. Exp. Ther. 288, 371-378. [PubMed] [Google Scholar]
- 29.Kim E. S., Serur, A., Huang, J., Manley, C. A., McCrudden, K. W., Frischer, J. S., Soffer, S. Z., Ring, L., New, T., Zabski, S., et al. (2002) Proc. Natl. Acad. Sci. USA 99, 11399-11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mordenti J., Thomsen, K., Licko, V., Chen, H., Meng, Y. G. & Ferrara, N. (1999) Toxicol. Pathol. 27, 14-21. [DOI] [PubMed] [Google Scholar]
- 31.Adamis A. P., Miller, J. W., Bernal, M. T., D'Amico, D. J., Folkman, J., Yeo, T. K. & Yeo, K. T. (1994) Am. J. Ophthalmol. 118, 445-450. [DOI] [PubMed] [Google Scholar]
- 32.Detmar M., Brown, L. F., Claffey, K. P., Yeo, K. T., Kocher, O., Jackman, R. W., Berse, B. & Dvorak, H. F. (1994) J. Exp. Med. 180, 1141-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong A. K., Alfert, M., Castrillon, D. H., Shen, Q., Holash, J., Yancopoulos, G. D. & Chin, L. (2001) Proc. Natl. Acad. Sci. USA 98, 7481-7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joussen A. M., Poulaki, V., Qin, W., Kirchhof, B., Mitsiades, N., Wiegand, S. J., Rudge, J., Yancopoulos, G. D. & Adamis, A. P. (2002) Am. J. Pathol. 160, 501-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poulaki V., Qin, W., Joussen, A. M., Hurlbut, P., Wiegand, S. J., Rudge, J., Yancopoulos, G. D. & Adamis, A. P. (2002) J. Clin. Invest. 109, 805-815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qaum T., Xu, Q., Joussen, A. M., Clemens, M. W., Qin, W., Miyamoto, K., Hassessian, H., Wiegand, S. J., Rudge, J., Yancopoulos, G. D. & Adamis, A. P. (2001) Invest. Ophthalmol. Visual Sci. 42, 2408-2413. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.