

Abstract
Background
A typical microarray experiment has many sources of variation which can be attributed to biological and technical causes. Identifying sources of variation and assessing their magnitude, among other factors, are important for optimal experimental design. The objectives of this study were: (1) to estimate relative magnitudes of different sources of variation and (2) to evaluate agreement between biological and technical replicates.
Results
We performed a microarray experiment using a total of 24 Affymetrix GeneChip® arrays. The study included 4th mammary gland samples from eight 21-day-old Sprague Dawley CD female rats exposed to genistein (soy isoflavone). RNA samples from each rat were split to assess variation arising at labeling and hybridization steps. A general linear model was used to estimate variance components. Pearson correlations were computed to evaluate agreement between technical and biological replicates.
Conclusion
The greatest source of variation was biological variation, followed by residual error, and finally variation due to labeling when *.cel files were processed with dChip and RMA image processing algorithms. When MAS 5.0 or GCRMA-EB were used, the greatest source of variation was residual error, followed by biology and labeling. Correlations between technical replicates were consistently higher than between biological replicates.
Background
Microarray chips are a powerful technology capable of measuring expression levels of thousands of genes simultaneously. Expression profiling has led to dramatic advances in the understanding of cellular processes at the molecular level, which may lead to improvements in molecular diagnostics and personalized medicine [1]. The number of experiments involving microarrays grows nearly exponentially each year [2]. Several platforms are currently available, including the commonly used short oligonucleotide-based Affymetrix GeneChip® arrays, which utilize multiple probes for each gene and automated control of the experimental process from hybridization to quantification. Although microarrays have tremendous potential, great effort and care is required in planning and designing microarray experiments, analyzing gene expression data, and interpreting results [3-6].
A typical microarray experiment has many different sources of variation which can be attributed to biological and technical causes [4]. Biological variation results from tissue heterogeneity, genetic polymorphism, and changes in mRNA levels within cells and among individuals due to sex, age, race, genotype-environment interactions and other factors [7-10]. Biological variation reflects true variation among experimental units (i.e. individual mice, rats, tissue samples, etc.) and is of interest to investigators. However, preparation of samples, labeling, hybridization, and other steps of microarray experiment can contribute to technical variation, which can significantly impact the quality of array data [11-16]. To ensure highly reproducible microarray data, technical variation should be minimized by controlling the quality of the RNA samples, and by efficient labeling and hybridization [17].
Identifying sources of experimental variation and assessing their magnitude are important for optimal experimental design, as for example, in the planning of mRNA pooling in microarray experiments [18]. Similarly, this information is useful for estimating the optimal number of required technical replicates because measurement accuracy and reliability affect researchers' power to identify differentially expressed genes [19]. However, other considerations, such as the goals of the study, the features of a particular microarray platform, or the cost of arrays and samples may influence experimental design [4-6]. Several studies have been conducted to examine the relative contributions of various factors in different experimental settings [7-15]. Here, we estimated the relative magnitudes of sources of variation in experiments involving Affymetrix GeneChip® arrays and evaluated agreement between biological and technical replicates.
Results
Experimental design
The experiment was set up as described in Materials and Methods (see Figure 1). Source *.cel data files from 24 GeneChip® arrays were subjected to image processing by four popular methods for probe-level data implemented in BioConductor [20]: DNA Chip Analyzer (dChip) [21], MAS 5.0 [22], RMA [23], and GCRMA-EB [24].
Figure 1.

Experimental design. The scheme of hierarchical unbalanced design used in our experiment is shown. A total of 8 rats and 24 chips were used.
Variance components estimation
For each probe set, expression levels were modeled as follows: yg = μg + Bg + L(B)g + εg, where Bg ~ N(0, 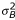 ) is the effect of biological variation among experimental units; L(B)g ~ N(0,
) is the effect of biological variation among experimental units; L(B)g ~ N(0, 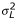 ) is the effect of labeling nested within biological replications; and, εg ~ N(0,
) is the effect of labeling nested within biological replications; and, εg ~ N(0, 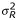 ) is the residual error. It should be noted that in our case biological variation could be confounded by technical variation arising during tissue isolation and preparation of mRNA samples. Dobbin et al., 2005, found that variation at this stage of microarray processing was small compared to variation at the hybridization step [25]. The model was fit separately on the gene expression measurements of each of dChip, MAS 5.0, RMA and GCRMA-EB probe set summaries. Both the effects of biological replication and the labeling effect nested within biological cases were treated as random. We estimated variance components and applied shrinkage variance estimators to them. These shrunken estimators borrow information across genes and have been shown to improve statistical tests [26]. Figure 2 shows the density plots of the distributions of relative magnitudes of different sources of variation. The results indicated that for most of the genes, the biggest source of variation was biological when using dChip and RMA, whereas the biggest source of variation was residual error when using GCRMA-EB or MAS 5.0. For all algorithms, a significant number of probe sets had biological and labeling variance components estimates equal or very close to zero. The findings are summarized in Table 1.
) is the residual error. It should be noted that in our case biological variation could be confounded by technical variation arising during tissue isolation and preparation of mRNA samples. Dobbin et al., 2005, found that variation at this stage of microarray processing was small compared to variation at the hybridization step [25]. The model was fit separately on the gene expression measurements of each of dChip, MAS 5.0, RMA and GCRMA-EB probe set summaries. Both the effects of biological replication and the labeling effect nested within biological cases were treated as random. We estimated variance components and applied shrinkage variance estimators to them. These shrunken estimators borrow information across genes and have been shown to improve statistical tests [26]. Figure 2 shows the density plots of the distributions of relative magnitudes of different sources of variation. The results indicated that for most of the genes, the biggest source of variation was biological when using dChip and RMA, whereas the biggest source of variation was residual error when using GCRMA-EB or MAS 5.0. For all algorithms, a significant number of probe sets had biological and labeling variance components estimates equal or very close to zero. The findings are summarized in Table 1.
Figure 2.

Density plots of different sources of variation. Density plots of relative magnitudes of different sources of variation are shown for data analyzed with four image processing algorithms. The proportions of different variance components are shown on x-axis and frequencies of probe sets are shown on y-axis.
Table 1.
Proportions of different sources of variation
| Source | dChip | MAS 5.0 | RMA | GCRMA-EB | ||||
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| Biological variation | 0.431 | 0.304 | 0.292 | 0.300 | 0.393 | 0.306 | 0.310 | 0.292 |
| Labeling variation | 0.206 | 0.230 | 0.136 | 0.198 | 0.221 | 0.224 | 0.147 | 0.192 |
| Residual error | 0.363 | 0.274 | 0.572 | 0.311 | 0.386 | 0.272 | 0.543 | 0.298 |
Assessment of reproducibility
We investigated agreement between technical replicates and biological replicates using Pearson correlations between chips. The correlations for the following three groups were compared: (1) Correlations between two technical replicates at the hybridization stage within a biological replicate (i.e., chips i_2A vs. i_2B; total of 8 correlations); (2) Correlations between two technical replicates at the labeling stage within a biological replicate (i.e. chips i_1 vs. i_2A and i_1 vs. i_2B; total of 16 correlations); (3) Correlations between different biological replicates (all possible pairwise comparisons; total of 252 correlations). Results indicated that technical replicates at the hybridization step agree more closely (i.e. have consistently higher correlations) either than technical replicates at the labeling stage or than different biological replicates (Figure 3). This finding can be illustrated using scatter plots: regardless of the image processing method, technical replicates of the same biological replicate (Figure 4) show less dispersion than data from different animals (Figure 5).
Figure 3.

Boxplots of pairwise correlations between chips. Box plots of Pearson correlations between technical replicates at the hybridization step (Hybr; i_2A vs. i_2B chips, where i is biological replicate), labeling step (Label; i_1 vs. i_2A and i_1 vs i_2B chips), and between different biological replicates (Bio; all pairwise combinations) are shown for four image processing algorithms (dChip, MAS 5.0, RMA, GCRMA-EB). Technical replicates have consistently higher correlations than different biological replicates.
Figure 4.

Comparison of two technical replicates of the same biological replicate using different image processing techniques. Expression levels detected on the 1_2A chip (x-axis) are plotted against levels detected on the 1_2B chip (y-axis). Results obtained with different image processing algorithms are shown. dChip and MAS 5.0 are shown on the log scale for compatibility with RMA and GCRMA-EB. Good agreement between two chips will result in data grouped along the identity line, while lack of agreement will lead to dispersion.
Figure 5.

Comparison of two different biological replicates using different image processing techniques. Expression levels detected on the 1_2A chip (x-axis) are plotted against levels detected on the 6_1 chip (B) (y-axis). Results obtained with different image processing algorithms are shown. dChip and MAS 5.0 are shown on the log scale for compatibility with RMA and GCRMA-EB. Good agreement between two chips will result in data grouped along the identity line, while lack of agreement will lead to dispersion.
The reproducibility at the hybridization stage was assessed by testing the significance of the differences between expression levels of technical replicates at the hybridization step using a paired t-test analysis as described in Material and Methods. Briefly, for each probe set we tested the hypothesis that a difference in expression levels between two technical replicates (i.e., between i_2A and i_2B chips) is equal to zero. A total of 15,923 paired t-tests were conducted and 15,923 p-values obtained for each image processing algorithm. The distribution of p-values was modeled using a mixture model approach [27]. Under a global null hypothesis, there are no differentially expressed genes and distribution of p-values is expected to be uniform on [0, 1]. If some genes are truly differentially expressed, we expect an increased number of small p-values (near 0). Distributions of p-values for the data obtained by four image processing methods are presented on Figure 5. By fitting the mixture of two beta distributions, one can estimate proportion of differentially expressed genes. We obtained the following estimates: dChip – 10.8%; MAS 5.0 – 4.8%; RMA – 2.3%, and GCRMA-EB – 13.6%. Thus, at the nominal α-level 0.05, the number of differentially expressed genes was smaller than expected by chance when data were processed with MAS 5.0 or RMA, but above the nominal α-level when data was processed with dChip or GCRMA-EB.
Discussion
Using Affymetrix GeneArray® chips, we examined the relative magnitudes of different sources of variation in microarray experiment. Analysis of variance using mixed-effects linear models is a common way to account for and test the significance of various factors contributing to overall variation [3]. Due to limitations of our hierarchical unbalanced experimental design and relatively small number of degrees of freedom, we did not include factors that can potentially contribute to variation such as day of processing, scanning order, mRNA preparation, etc. We assume that such factors were not significant. However, to formally test this assumption, another experiment is needed.
We used a general linear model to partition variance for each probe set into three components. The first source was biological (i.e. animal-to-animal) variation. The biological variation may be confounded by technical variation at the mRNA preparation step, but this variation is probably relatively small compared to variation at the hybridization step [25]. Thus, we assume that most of the variation for this effect was due to true biological differences among animals. The second source of variation was the effect of labeling. Although our experiments were carried out by the same person, using the same equipment, under the same experimental conditions as much as realistically possible, there is always some variation caused by minor environmental differences in temperature, duration, pipetting etc., which influences labeling efficiency. The third source of variation other than animal-to-animal variation and labeling-effect variation was residual error caused by differences in hybridization, scanning and other factors. To compare the relative magnitudes of different sources of variation, we estimated variance components and applied shrunken variance estimators that borrow information across genes. We constructed these shrunken variance estimators by shrinking a group of individual variance estimators toward their common corrected geometric mean [26]. The amount of shrinkage depends on the variation on the individual variance components estimators. These estimators were shown to be robust in respect to variance heterogeneity in gene expression data among groups [26].
We found that our results depend on the image processing algorithm used: biological variation was the largest source when dChip or RMA were used, but when *.cel files were processed with GCRMA-EB or MAS 5.0, the largest source was residual error. Bakay et al., 2002, found that biological variation presumably caused by tissue heterogeneity and genetic polymorphism was a major source of variation while technical variation was minor [12]. Han et al., 2004, found that biological variation was about of the same size as other sources combined [14]. Whitney et al., 2003, found that inter-individual variation in gene expression profiles was correlated with gender, age, and the time of day at which the sample was taken. These intrinsic differences in expression patterns were likely caused by differences in genotype, although they might also reflect epigenetic or environmental factors [9]. Oleksiak et al., 2002, in their studies of teleost fish have observed significant differences in gene expression levels between individuals from the same population and between different populations. These differences could be caused by genetic variation as well as other factors, including maternal effects and genotype-environment interactions [10]. On the contrary, Dumur et al., 2004, found that day-to day variation was the main source of variation [17]. Woo et al., 2004, in studies of inbred mice strains, detected that most of the genes had small biological variance, but about 10% of genes showed large variation between individuals [28].
We found that technical replicates within a biological replicate had higher and more consistent correlations with each other than with other biological replicates. Generally, our correlations were higher than those observed by Dobbin et al., 2005, for interlaboratory correlations between tumor samples [25] and were compatible with values for in-lab correlations obtained in another study [29].
The consistency of the hybridization step was evaluated using paired t-tests following by modeling of distribution of resulting p-values. The significance depends on the image processing algorithm used: the hybridization effect was not significant for MAS 5.0 (4.8% of genes were differentially expressed between two technical replicates) and RMA (2.3% of genes), but the proportion of differentially expressed genes was higher than expected by chance for dChip (10.8% of genes) and GCRMA-EB (13.6% of genes).
The low-level data were analyzed using four popular methods implemented in the BioConductor [20] package: dChip [21], MAS 5.0 [22], RMA [23], and GCRMA-EB [24]. We found that different low-level data processing algorithms produced different results. We provide comparisons mainly to illustrate the compatibility of several algorithms. Evaluation of the strengths and weaknesses of different image processing algorithms may require other experimental settings, such as spike-in data. Shedden et al., 2005, performed a comprehensive comparison of seven image processing methods for Affymetrix arrays and demonstrated that the choice of image processing algorithm has a major impact on the results of microarray data analysis [30]. The authors found that the dChip method operates consistently well, while MAS 5.0 and GCRMA-EB consistently performed poorly. GCRMA-EB had a particular disagreement with other methods when a t-test was used for group comparison, presumably because it might be more sensitive to the underlying statistical assumptions of a test (e.g. independence of genes). Similarly, we observed that estimates of the proportion of differentially expressed genes between two technical replicates at the hybridization stage were different than those for data processed with GCRMA-EB compared to other methods, which is consistent with finding of Shedden et al. [30].
The results presented here are specific for the systems being studied, and other experimental conditions may yield different estimates. For example, we used an outbred strain of rats, which had greater inherent biological variation than inbred strains. In cell cultures of inbred mice strains under otherwise equal conditions, the relative magnitude of biological variation presumably would be smaller. Different steps in microarray data analysis, such as normalization, transformation, and gene filtering, may affect results as well [31-35]. A microarray platform and microarray facility can also have a significant impact, as was demonstrated in several recent studies [25,36-38]. Testing the influence of these various factors could be an interesting topic of future research.
Conclusion
Identification of sources of variation and their relative magnitudes, among other factors, is important for optimal experimental design and the development of quality control procedures. In this study, we evaluated the relative magnitudes of different sources of variation in Affymetrix microarray experiments. Different image processing algorithms gave different variance components estimates: the greatest source was animal-to-animal (i.e. biological) variation when dChip and RMA were used, and residual error when MAS 5.0 or GCRMA-EB were used. We observed that correlations between technical replicates within one biological replicate were consistently higher than between different biological replicates. It should be noted that estimates obtained here were specific for our experimental system, and results would probably change if we used another organism or tissue, or another microarray platform.
Methods
Samples and microarrays
This study included samples taken from eight 21-day-old Sprague Dawley CD female rats exposed to genistein (a soy isoflavone) via their mother's milk. The mothers were fed AIN-76A diet supplemented with 200 mg genistein / kg chow. Young rats were sacrificed at day 21 and the 4th mammary glands extracted and flash-frozen in liquid nitrogen within 3 minutes of ex-sanguination. Samples were frozen at -70°C for approximately 90 days, at which point the extraneous fat was dissected off and samples processed in Trizol RNA extraction buffer. Total RNA was generated using Affymetrix RNA extraction and labeling kits according to manufacturer's protocols, and each of the RNA samples was split in half. The first half was labeled and run on a RAE 230A Affymetrix GeneChip®, and the other half was labeled, split, and run across two RAE 230A chips (see Figure 1). Affymetrix arrays were run in the Genomics Core facility of the Heflin Center for Human Genetics at the University of Alabama at Birmingham. Images were scanned on a HP 2500 scanner.
Image processing
Each of the low-level *.cel data files was processed using four popular image analysis algorithms: DNA Chip Analyzer (dChip) [21], MAS 5.0 [22], RMA [23], and GCRMA-EB [24]. The processing was done in R 1.8.1 / R 1.9.1 [39]. The default settings for all normalization procedures were used as implemented in the BioConductor [20]; in particular, the scale normalization for MAS 5.0; the quantile-quantile normalization for RMA; the invariant-set normalization for dChip; and the quantile-quantile normalization for GCRMA-EB (see [35] for the details of the different normalization methods). The default implementation of dChip, RMA, and GCRMA-EB used only the PM (perfect match) intensity matrix, while MAS 5.0 by default used both PM and MM (mismatch) matrices.
Evaluation of relative magnitudes of different sources of variation
The relative magnitudes of different sources of variation were estimated using a general linear model in PROC VARCOMP procedure of SAS 9.1 (SAS Institute Inc., Cary, NC) using REML option. The expression levels of each probe set, yg, were modeled as follows: yg = μg + Bg + L(B)g + εg, where Bg ~ N(0, ) is the effect of biological variation among experimental units; L(B)g ~ N(0, ) is the effect of labeling variation nested within biological replications; and εg ~ N(0, ) is the residual error, i.e. technical variation caused by factors other than labeling. Biological effect could be confounded by technical variation arising during mRNA sample preparation. For each probe set, variance components were estimated. We applied shrinkage variance estimators that borrow information across probe sets and improve individual variance estimators by shrinking them toward their corrected geometric mean [26]. The total variance was assumed to be the sum of three components: VARTot = VARBio + VARLabel + VARResidual, where VARBio is the shrunken estimate of biological variance; VARLabel is the shrunken estimate of variance due to labeling; and VARResidual is the shrunken variance estimate of residual error. The relative proportion of each source of variation was calculated as a ratio of the shrunken variance estimate to the sum of all three shrunken variance estimates:, i.e. 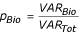 calculates the proportion of biological variation,
calculates the proportion of biological variation, 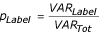 calculates the proportion of variation due to labeling within biological replicates, and
calculates the proportion of variation due to labeling within biological replicates, and 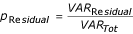 calculates the proportion of variation due to unaccounted technical variation (residual error).
calculates the proportion of variation due to unaccounted technical variation (residual error).
Assessment of reproducibility across different replicates
Pearson correlations between chips were calculated for the following three groups: (1) Correlations between two technical replicates at the hybridization step (i.e., chips i_2A vs. i_2B; total of 8 correlations); (2) Correlations between two technical replicates at the labeling step (i.e. chips i_1 vs. i_2A and i_1 vs. i_2B; total of 16 correlations); (3) Correlations between different biological replicates (all possible pairwise comparisons; total of 252 correlations).
To evaluate the significance of variation introduced at the hybridization step, paired t-tests were performed on 16 chips (i_2A and i_2B chips from each of 8 separate rats). For each probe set, the null hypothesis was that the difference between the expression levels of two replicates was equal to zero. A total of 15,923 t-tests were performed and 15,923 p-values were generated for each image processing algorithm. The distribution of resulting p-values was modeled using a mixture of two beta distributions [24]. If the global null hypothesis is true, there are no differentially expressed genes and the distribution of p-values is expected to be uniform [0, 1]. We expect an increased number of p-values close to 0 if some genes are truly differentially expressed. By fitting the mixture of two beta distributions, one can estimate a proportion of differentially expressed genes. At the nominal α-level 0.05, one expects 5% of genes to be differentially expressed just by chance. Thus, the differences between replicates were considered significant only if the proportion of differentially expressed genes was > 5%.
Authors' contributions
SOZ performed analysis of the data, drafted and finalized the manuscript. KK and RP helped with analysis and contributed to discussion. KES performed microarray experiment that generated the data. SB provided support for microarray experiment and contributed to discussion. TM and LC analyzed the data with four image-analysis algorithms. GPP planned and designed the experiment. GPP and DBA supervised and coordinated the project and assisted with the interpretation. All authors have read and approved the manuscript.
Figure 6.

Distributions of p-values for the paired t-test for hybridization effect. Histograms of p-values for four image processing algorithms. If the global null hypothesis is true, the distribution of p-values would be uniform from 0 to 1 (dotted line). If differentially expressed genes are present, the number of small p-values will be increased.
Acknowledgments
Acknowledgements
This work was supported in part by NIH grants U54CA100949 and T32HL072757, and NSF grants 0217651 and 0090286. We thank Dr. Xiangqin Cui for critical reading of the manuscript and for making valuable suggestions.
Contributor Information
Stanislav O Zakharkin, Email: szakharkin@ms.soph.uab.edu.
Kyoungmi Kim, Email: KKim@ms.soph.uab.edu.
Tapan Mehta, Email: TMehta@ms.soph.uab.edu.
Lang Chen, Email: LChen@ms.soph.uab.edu.
Stephen Barnes, Email: sbarnes@uab.edu.
Katherine E Scheirer, Email: kscheire@uab.edu.
Rudolph S Parrish, Email: rudy.parrish@louisville.edu.
David B Allison, Email: dallison@uab.edu.
Grier P Page, Email: gpage@uab.edu.
References
- Jain KK. Applications of biochips: from diagnostics to personalized medicine. Curr Opin Drug Discov Devel. 2004;7:285–289. [PubMed] [Google Scholar]
- Gracey AY, Cossins AR. Application of microarray technology in environmental and comparative physiology. Annu Rev Physiol. 2003;65:231–259. doi: 10.1146/annurev.physiol.65.092101.142716. [DOI] [PubMed] [Google Scholar]
- Kerr MK, Martin M, Churchill GA. Analysis of variance for gene expression microarray data. J Comput Biol. 2000;7:819–837. doi: 10.1089/10665270050514954. [DOI] [PubMed] [Google Scholar]
- Churchill GA. Fundamentals of experimental design for cDNA microarrays. Nat Genet. 2002;32 Suppl:490–495. doi: 10.1038/ng1031. [DOI] [PubMed] [Google Scholar]
- Yang YH, Speed T. Design issues for cDNA microarray experiments. Nat Rev Genet. 2002;3:579–88. doi: 10.1038/nrg863. [DOI] [PubMed] [Google Scholar]
- Kerr MK, Churchill GA. Experimental design for gene expression microarrays. Biostatistics. 2001;2:183–201. doi: 10.1093/biostatistics/2.2.183. [DOI] [PubMed] [Google Scholar]
- Molloy MP, Brzezinski EE, Hang J, McDowell MT, VanBogelen RA. Overcoming technical variation and biological variation in quantitative proteomics. Proteomics. 2003;3:1912–1919. doi: 10.1002/pmic.200300534. [DOI] [PubMed] [Google Scholar]
- Spruill SE, Lu J, Hardy S, Weir B. Assessing sources of variability in microarray gene expression data. BioTechniques. 2002;33:916–923. doi: 10.2144/02334mt05. [DOI] [PubMed] [Google Scholar]
- Whitney AR, Diehn M, Popper SJ, Alizadeh AA, Boldrick JC, Relman DA, Brown PO. Individuality and variation in gene expression patterns in human blood. Proc Natl Acad Sci. 2003;100:1896–1901. doi: 10.1073/pnas.252784499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleksiak MF, Churchill GA, Crawford DL. Variation in gene expression within and among natural populations. Nat Genet. 2002;32:261–266. doi: 10.1038/ng983. [DOI] [PubMed] [Google Scholar]
- Brown JS, Kuhn D, Wisser R, Power E, Schnell R. Quantification of sources of variation and accuracy of sequence discrimination in a replicated microarray experiment. BioTechniques. 2004;36:324–332. doi: 10.2144/04362MT04. [DOI] [PubMed] [Google Scholar]
- Bakay M, Chen YW, Borup R, Zhao P, Nagaraju K, Hoffman EP. Sources of variability and effect of experimental approach on expression profiling data interpretation. BMC Bioinformatics. 2002;3:4. doi: 10.1186/1471-2105-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breit S, Nees M, Schaefer U, Pfoersich M, Hagemeier C, Muckenthaler M, Kulozik AE. Impact of pre-analytical handling on bone marrow mRNA gene expression. Br J Haematol. 2004;126:231–243. doi: 10.1111/j.1365-2141.2004.05017.x. [DOI] [PubMed] [Google Scholar]
- Han ES, Wu Y, McCarter R, Nelson JF, Richardson A, Hilsenbeck SG. Reproducibility, sources of variability, pooling, and sample size: important considerations for the design of high-density oligonucleotide array experiments. J Gerontol A Biol Sci Med Sci. 2004;59:306–315. doi: 10.1093/gerona/59.4.b306. [DOI] [PubMed] [Google Scholar]
- Liu J, Walker N, Waalkes MP. Hybridization buffer systems impact the quality of filter array data. J Pharmacol Toxicol Methods. 2004;50:67–71. doi: 10.1016/j.vascn.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Page GP, Edwards JW, Barnes S, Weindruch R, Allison DB. A design and statistical perspective on microarray gene expression studies in nutrition: the need for playful creativity and scientific hard-mindedness. Nutrition. 2003;19:997–1000. doi: 10.1016/j.nut.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Dumur CI, Nasim S, Best AM, Archer KJ, Ladd AC, Mas VR, Wilkinson DS, Garrett CT, Ferreira-Gonzalez A. Evaluation of quality-control criteria for microarray gene expression analysis. Clin Chem. 2004;50:1994–2002. doi: 10.1373/clinchem.2004.033225. [DOI] [PubMed] [Google Scholar]
- Kendziorski CM, Zhang Y, Lan H, Attie AD. The efficiency of pooling mRNA in microarray experiments. Biostatistics. 2003;4:465–477. doi: 10.1093/biostatistics/4.3.465. [DOI] [PubMed] [Google Scholar]
- Allison DB, Allison RL, Faith MS, Paultre F, Pi-Sunyer FX. Power and money: designing statistically powerful studies while minimizing financial costs. Psychological Methods. 1997;2:20–33. doi: 10.1037//1082-989X.2.1.20. [DOI] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. BioConductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci. 2001;98:31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbell E, Liu WM, Mei R. Robust estimators for expression analysis. Bioinformatics. 2002;18:1585–92. doi: 10.1093/bioinformatics/18.12.1585. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Wu Z, Irizarry R, Gentleman R, Murillo F, Spencer F. Technical Report. John Hopkins University, Department of Biostatistics Working Papers, Baltimore, MD; 2004. A model based background adjustment for oligonucleotide expression arrays. [Google Scholar]
- Dobbin KK, Beer DG, Meyerson M, Yeatman TJ, Gerald WL, Jacobson JW, Conley B, Buetow KH, Heiskanen M, Simon RM, Minna JD, Girard L, Misek DE, Taylor JM, Hanash S, Naoki K, Hayes DN, Ladd-Acosta C, Enkemann SA, Viale A, Giordano TJ. Interlaboratory comparability study of cancer gene expression analysis using oligonucleotide microarrays. Clin Cancer Res. 2005;11:565–572. [PubMed] [Google Scholar]
- Cui X, Hwang JT, Qiu J, Blades NJ, Churchill GA. Improved statistical tests for differential gene expression by shrinking variance components estimates. Biostatistics. 2005;6:59–75. doi: 10.1093/biostatistics/kxh018. [DOI] [PubMed] [Google Scholar]
- Allison DB, Gadbury GL, Heo M, Fernandez JR, Lee C, Prolla TA, Weindruch R. A mixture model approach for the analysis of microarray gene expression data. Comput Stati Data Anal. 2002;39:1–20. doi: 10.1016/S0167-9473(01)00046-9. [DOI] [Google Scholar]
- Woo Y, Affourtit J, Daigle S, Viale A, Johnson K, Naggert J, Churchill G. A comparison of cDNA, oligonucleotide, and Affymetrix GeneChip gene expression microarray platforms. J Biomol Tech. 2004;15:276–284. [PMC free article] [PubMed] [Google Scholar]
- Wang H, He X, Band M, Wilson C, Liu L. A study of inter-lab and inter-platform agreement of DNA microarray data. BMC Genomics. 2005;6:71. doi: 10.1186/1471-2164-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shedden K, Chen W, Kuick R, Ghosh D, Macdonald J, Cho KR, Giordano TJ, Gruber SB, Fearon ER, Taylor JM, Hanash S. Comparison of seven methods for producing Affymetrix expression scores based on False Discovery Rates in disease profiling data. BMC Bioinformatics. 2005;6:26. doi: 10.1186/1471-2105-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller SC, Gregg JP, Hagerman P, Rocke DM. Transformation and normalization of oligonucleotide microarray data. Bioinformatics. 2003;19:1817–1823. doi: 10.1093/bioinformatics/btg245. [DOI] [PubMed] [Google Scholar]
- Thygesen HH, Zwinderman AH. Comparing transformation methods for DNA microarray data. BMC Bioinformatics. 2004;5:77. doi: 10.1186/1471-2105-5-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin LX, Kerr KF, Contributing Members of the Toxicogenomics Research Consortium Empirical evaluation of data transformations and ranking statistics for microarray analysis. Nucleic Acids Res. 2004;32:5471–5479. doi: 10.1093/nar/gkh866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pounds S, Cheng C. Statistical development and evaluation of microarray gene expression data filters. J Comput Biol. 2005;12:482–495. doi: 10.1089/cmb.2005.12.482. [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:2. doi: 10.1093/bioinformatics/19.2.185. http://bioinformatics.oxfordjournals.org/cgi/reprint/19/2/185 [DOI] [PubMed] [Google Scholar]
- Toxicogenomics Research Consortium Standardizing global gene expression analysis between laboratories and across platforms. Nat Methods. 2005;2:351–356. doi: 10.1038/nmeth754. [DOI] [PubMed] [Google Scholar]
- Larkin JE, Frank BC, Gavras H, Sultana R, Quackenbush J. Independence and reproducibility across microarray platforms. Nat Methods. 2005;2:337–344. doi: 10.1038/nmeth757. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Warren D, Spencer F, Kim IF, Biswal S, Frank BC, Gabrielson E, Garcia JG, Geoghegan J, Germino G, Griffin C, Hilmer SC, Hoffman E, Jedlicka AE, Kawasaki E, Martinez-Murillo F, Morsberger L, Lee H, Petersen D, Quackenbush J, Scott A, Wilson M, Yang Y, Ye SQ, Yu W. Multiple-laboratory comparison of microarray platforms. Nat Methods. 2005;2:345–350. doi: 10.1038/nmeth756. [DOI] [PubMed] [Google Scholar]
- Ihaka R, Gentleman R. A language for data analysis and graphics. J Comp Graph Stat. 1996;5:299–314. [Google Scholar]