

Abstract
Introduction and importance:
Dandy–Walker syndrome (DWS) is a rare congenital posterior fossa malformation, affecting 1 in 25 000–30 000 live births. Prenatal diagnosis is possible via ultrasound and MRI. This case highlights a rare Dandy–Walker variant with bilateral optic atrophy and status epilepticus, emphasizing the need for awareness of progressive neurological and visual impairment in Dandy–Walker spectrum disorders. The case adds to the evolving knowledge on the diverse phenotypic presentations of the Dandy–Walker variant, particularly in resource-limited settings where diagnostic and therapeutic interventions may be constrained.
Case presentation:
A 27-month-old female with recurrent seizures since infancy presented with generalized tonic–clonic movements, ocular deviation, and frothing, lasting 2 h. Examination revealed fever, tachycardia, tachypnea, and hypertonia. MRI at 5 months confirmed Dandy–Walker spectrum disorder with cerebellar vermis hypoplasia, a posterior fossa cyst, and fourth ventricle malformation. Electroencephalogram at 14 months showed diffuse encephalopathy with multifocal seizures, and fundoscopy revealed bilateral optic atrophy. TORCH (toxoplasmosis, rubella cytomegalovirus, herpes simplex, and HIV) screening was unremarkable. Despite antiseizure therapy with levetiracetam and phenytoin, persistent seizures required pediatric intensive care unit admission. Valproate (15 mg/kg/day, titrated to 20 mg/kg/day) achieved seizure control.
Clinical discussion:
This case underscores the complexity of Dandy–Walker spectrum disorder with status epilepticus in a 27-month-old female. No infectious source was identified, and lumbar puncture was declined. Seizure control required escalation of antiseizure therapy. Long-term neurodevelopmental, genetic, and neurometabolic evaluation is crucial for comprehensive management.
Conclusion:
This case highlights the need for long-term neurodevelopmental, genetic, and neurometabolic evaluation for comprehensive management.
Keywords: case report, Dandy–Walker spectrum, optic atrophy, seizure disorder
Introduction
Dandy–Walker (DW) syndrome (DWS) is a congenital brain malformation affecting the posterior fossa, classified by Barkovich into Complex A [classic Dandy–Walker malformation (DWM) and its variant] and Complex B (“mega cisterna magna”)1. However, this classification is debated, with evolving terminology in contemporary literature. The incidence is estimated at 1 in 25 000–30 000 live births2. DWS results from the persistence of Blake’s pouch, which normally regresses by the fourth month of gestation. Key features of DWS include cerebellar vermis agenesis or hypoplasia, cystic dilation of the fourth ventricle, and posterior fossa enlargement, while the variant presents with vermian hypoplasia and fourth ventricular dilation without posterior fossa enlargement2. Prenatal diagnosis is possible through ultrasound and MRI3.
HIGHLIGHTS
Dandy–Walker syndrome (DWS) is a rare congenital posterior fossa malformation resulting from failure of normal closure of the fourth ventricle with persistence of Blake’s pouch.
DWS is characterized by (1) agenesis or hypoplasia of the vermis, (2) cystic dilation of the fourth ventricle with communication to a large cystic dilated posterior fossa, (3) upward displacement of tentorium and torcula (torcular-lambdoid inversion), and (4) posterior fossa enlargement.
Seizures occur, often in association with increased intracranial pressure, attenuated cortical inhibition leading to lower seizure threshold, and supratentorial malformations.
It presents a multifaceted clinical challenge, particularly when comorbid with non-neurological conditions, and remains an area of ongoing research.
Herein, we present a case of 27-month-old female with bilateral optic atrophy with Dandy–Walker variant (DWV) with status epilepticus.
This case report was drafted in accordance with the CARE Case Report Guidelines 20134.
Case presentation
A 27-month-old South Asian female child presented to the emergency department with complaints of abnormal body movements characterized by jerky movements of all four limbs, accompanied by up-rolling of the eyes and frothing from the mouth. These symptoms persisted for approximately 2 h prior to admission. There was a history of fever, blinking movements, and loss of consciousness.
On examination, her temperature was 101.6°F, heart rate was 160 beats per minute, respiratory rate was 78 breaths per minute, and oxygen saturation was 99% while receiving oxygen via a face mask. General examination revealed no pallor, icterus, lymphadenopathy, clubbing, cyanosis, edema, or dehydration. Respiratory system examination showed bilateral equal air entry with diffuse bilateral wheezing. Cardiovascular examination revealed a regular sinus rhythm with normal first and second heart sounds. Abdominal examination indicated a soft, non-tender, non-distended abdomen with normal bowel sounds. Neurological examination showed increased muscle tone, normal cry, and normal activity, with a Glasgow Coma Scale score of 15/15. A random blood glucose measurement was 359 mg/dL. Upon subsequent measurement, the child’s blood glucose levels were found to be within normal range, and there was no clinical or biochemical evidence suggestive of underlying diabetes mellitus or metabolic disorder.
The child was born at term via emergency lower-segment caesarean section for a previous lower-segment caesarean section with oligohydramnios. Antenatal fetal ultrasound scanning was reported to be normal. The child had her first seizure episode at 4 months of age and was initiated on oral levetiracetam (100 mg per 1 mL) at a dose of 12.5 mg/kg/day once daily. A second seizure episode occurred at 5 months of age, followed by a third episode at 7 months of age, prompting an increase in her antiseizure medication dosage to levetiracetam (1/100) at a dose of 18.75 mg/kg/day oral once daily and phenytoin 3.125 mg/kg/day per oral once daily.
The patient is the second-born child in the family. The first child, a 5-year-old female, exhibits normal growth and neurodevelopment with no history of seizures or systemic illness.The mother’s prior pregnancy and perinatal course were uneventful, and no antenatal complications were reported. There is no known family history of seizure disorders, congenital brain malformations, neurodevelopmental delay, or other genetic syndromes. Both paternal and maternal family histories were unremarkable.
An MRI performed at 5 months of age demonstrated cerebellar vermis hypoplasia with a posterior fossa cystic structure communicating with a poorly formed fourth ventricle, along with hypoplasia of both cerebellar hemispheres, confirming a diagnosis of DW spectrum disorder (Fig. 1).
Figure 1.
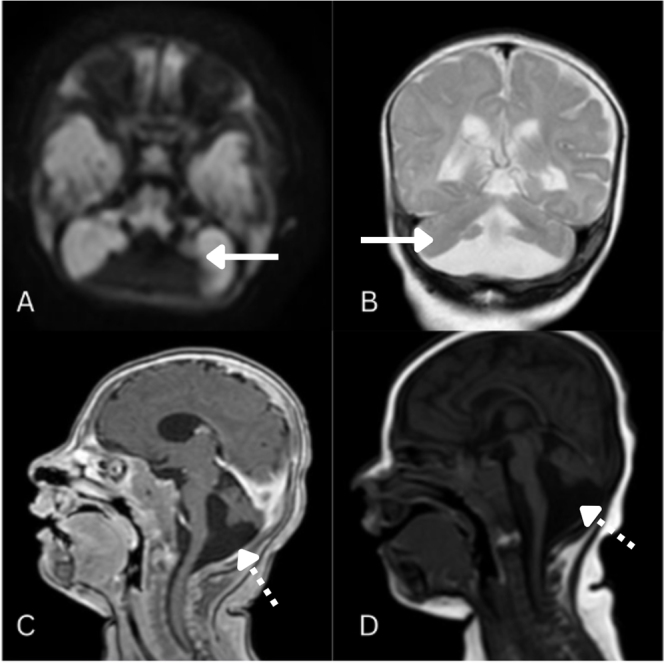
(A) FLAIR axial, (B) T2 coronal, (C) T1 post-contrast sagittal, and (D) T1 sagittal MRI images showing cerebellar vermis hypoplasia with a posterior fossa cystic structure communication with a poorly formed fourth ventricle, along with hypoplasia of both cerebellar hemispheres. Solid arrow: cerebellar vermis hypoplasia. Dotted arrow: posterior fossa cystic structure communicating with a poorly formed fourth ventricle.
A 20-min electroencephalogram (EEG) conducted at 14 months of age revealed diffusely slow, monotonous, undifferentiated, and disorganized background cerebral activity, with high-voltage slow waves in both hemispheres. Frequent spikes and sharp waves were observed in the right and left frontal and temporal regions, suggestive of moderate to severe diffuse encephalopathy with multifocal seizure activity. Fundoscopic evaluation demonstrated bilateral optic atrophy. Serological testing for toxoplasmosis (IgG and IgM), rubella (IgG and IgM), cytomegalovirus (IgG and IgM), and herpes simplex virus 1 and 2 (IgG and IgM) showed titers within normal limits.
The patient was admitted to the pediatric intensive care unit (PICU) for further evaluation of recurrent seizures. A lumbar puncture was recommended to assess cerebrospinal fluid for potential central nervous system infections, but the legal guardians declined the procedure. The patient was initiated on levetiracetam at 40 mg/kg/day and phenytoin at 3.125 mg/kg/day once daily. However, on the second day of admission, she developed myoclonic seizures, prompting the initiation of valproate at 15 mg/kg/day, which was gradually increased to 20 mg/kg/day over 2 days. Following this adjustment, the seizures were successfully controlled.
Discussion
The most acceptable classification of DWS is given by Barkovich, who classified the spectrum/complex into DW complex A, which includes classic DWM and the DWV, and DW complex B, which includes DW “mega cisterna magna”[5,6]. However, the validity of this classification is now subject to debate, and terms such as “variant” or “complex” are increasingly discouraged in contemporary literature. Incidence of DWM and related variants has been reported to be 1 in 25 000–30 000 of live births7.
The Blake’s pouch is a transient, normal embryological structure that usually regresses starting from the ninth week of development when it fenestrates and develops the foramen of Magendie and usually completes by the fourth month of gestation8. Failure of normal closure of the fourth ventricle with persistence of Blake’s pouch leads to a complex yet rare posterior fossa anomaly, commonly referred to as DWM. DWS is characterized by (1) agenesis or hypoplasia of the vermis, (2) cystic dilation of the fourth ventricle with communication to a large cystic dilated posterior fossa, (3) upward displacement of tentorium and torcula (torcular-lambdoid inversion), and (4) posterior fossa enlargement[3,9,10]. DWV is characterized by cerebellar vermian hypoplasia, cystic fourth ventricular dilatation, and normal posterior fossa volume11.
Ultrasonographic assessment by measuring the size of ventricles or measurements of the brainstem-vermis angle enables prenatal diagnosis after the eighteenth week of gestation, corresponding to the complete development of the cerebellar vermis[12,13]. Magnetic resonance imaging provides an adjunctive diagnostic modality for confirmation14. In patients with the DWV, discrepancies between prenatal and postnatal imaging findings may occur due to the variability in cerebellar vermian development15.
DWS is associated with a range of neurodevelopmental abnormalities, primarily due to cerebellar involvement, which is integral to motor coordination as well as certain cognitive and behavioral functions16. Clinical manifestations typically become evident during early infancy, with macrocephaly, signs of increased intracranial pressure, and spastic paraparesis being the most frequently observed features17. Additional early findings may include hypotonia, delayed motor development, and intellectual impairment18. Less commonly, patients may exhibit focal neurological deficits such as strabismus, nystagmus, cranial nerve palsies, truncal ataxia, and speech disturbances19. Furthermore, seizures may occur in some cases, often in association with increased intracranial pressure, attenuated cortical inhibition leading to lower seizure threshold, and supratentorial malformations20. Optic nerve atrophy had been noted in 11.1% of the cases as reported by Stambolliu et al17.
DWS presents a multifaceted clinical challenge, particularly when comorbid with non-neurological conditions, and remains an area of ongoing research. It has been frequently associated with severe psychiatric disorders, including schizophrenia, bipolar disorder, major depressive disorder, and impulse control disorders21. Additionally, a significant proportion of individuals with DWS exhibit craniofacial dysmorphisms and structural anomalies of the central nervous system, such as corpus callosum agenesis22, rachischisis, and ectopic cerebral or cerebellar tissue17. Congenital abnormalities affecting the cardiovascular23, urogenital24, and gastrointestinal systems25 have also been widely reported. Furthermore, neoplastic processes and neurocutaneous disorders occur with variable prevalence26. DWS is often linked to genetic syndromes and chromosomal anomalies, including 3q syndrome – where the ZIC1 and ZIC4 genes associated with DWS are located – as well as trisomy 18, further contributing to the complexity of its pathophysiology, clinical course, and management strategies[2,27].
In patients with DWM, the presence of aqueductal obstruction, as demonstrated on preoperative neuroimaging, is a critical determinant in treatment planning, necessitating concurrent drainage of both supra- and infratentorial compartments to mitigate the risk of a transtentorial pressure gradient28. With the development of neuroendoscopic techniques, endoscopic third ventriculostomy (ETV) is considered a reasonable initial therapeutic approach, particularly in patients with a patent aqueduct, with ventriculoperitoneal (VP) or cystoperitoneal shunting reserved for cases where ETV fails29. In less common instances involving aqueductal stenosis, restoration of communication between compartments through aqueductoplasty or stent placement, in conjunction with third ventriculostomy, is required to address the obstruction effectively5.
The prognosis for individuals with DWM is highly variable and depends on multiple clinical factors, including the extent of cerebellar and posterior fossa abnormalities, the presence of hydrocephalus, and the timing of medical intervention30. Early diagnosis and appropriate neurosurgical and supportive management can improve outcomes, particularly in cases with minimal cerebellar dysfunction and low intracranial pressure. In such cases, individuals may experience relatively preserved neurological function and achieve near-normal developmental milestones31.
Hydrocephalus in DWM is a critical determinant of long-term outcomes. However, in cases with severe cerebellar hypoplasia, enlarged posterior fossa cysts, and significant hydrocephalus, the risk of neurodevelopmental impairment is considerably higher. These patients are more likely to present with intellectual disabilities, motor dysfunction, gait disturbances, and coordination deficits due to cerebellar involvement. The presence of hydrocephalus further complicates prognosis, as elevated intracranial pressure can lead to progressive neurocognitive decline and optic atrophy if left untreated32. Neurosurgical interventions such as VP shunting can mitigate these risks by controlling cerebrospinal fluid accumulation and preventing further neurological deterioration29.
Prenatal imaging, particularly obstetric ultrasound, plays a crucial role in prognosis. Fetal lateral ventricle measurements can serve as an important prognostic indicator; studies suggest that ventriculomegaly between 11 and 15 mm is associated with a 21% risk of developmental delay, while measurements exceeding 15 mm increase this risk to over 50%33.
Additionally, individuals with DWM are at an increased risk of epilepsy, with studies indicating an overall seizure prevalence of approximately 30% in patients with non-tumoral hydrocephalus19.
In a retrospective study of 28 children with DWM, Di Nora et al reported frequent associated anomalies. Hydrocephalus was found in 48% (13/28), with 10% (3/28) also having corpus callosum anomalies. Corpus callosum, cardiac, and genitourinary anomalies were seen in 7% (2/28), 10% (3/28), and 10% (3/28), respectively. Developmental delay (67%, 19/28) and epilepsy (32%, 9/28) were the most common clinical features. MRI was the initial diagnostic tool in 61% (17/28), followed by ultrasound and CT19.
In a study done by Agrawal et al, a 3-day-old full-term neonate was diagnosed with DEM characterized by cystic dilatation of the fourth ventricle, hypoplasia of the cerebellar vermis, and an enlarged posterior fossa. Although hydrocephalus commonly develops in about 90% of cases, this neonate did not show hydrocephalus initially, and thus no shunting procedure was performed at diagnosis34.
In a study done by Pradhan et al, a male neonate delivered via cesarean section at 41 weeks of gestation presented with multiple petechiae and peripheral cyanosis. Neuroimaging revealed mild hydrocephalus with lateral ventricle dilatation and a cystic lesion in the posterior fossa consistent with a DWV characterized by hypoplastic cerebellar vermis. Cerebrospinal fluid analysis showed decreased glucose and elevated protein levels, confirming meningitis. The neonate developed generalized tonic–clonic seizures, which were managed successfully with levetiracetam. Neurosurgical intervention with VP shunting was planned but postponed due to meningitis35.
Given the complexity and heterogeneity of DWM, a multidisciplinary approach involving neurology, neurosurgery, developmental pediatrics, and rehabilitation specialists is essential to optimizing patient outcomes. Such collaboration ensures comprehensive evaluation and management of the diverse clinical manifestations associated with DWM. Early intervention and coordinated care can significantly improve the patient’s neurological development and quality of life36.
Functional outcome is subject to several factors, which include other structural brain abnormalities, extra-CNS manifestations, epilepsy, motor, visual, or hearing impairment, and other congenital abnormalities11.
Conclusion
The current study shows a 27-month-old female child with a diagnosed DWS disorder and a history of recurrent seizures presented with a prolonged seizure episode characterized by generalized jerky movements, up-rolling of the eyes, and frothing from the mouth. Initial stabilization included oxygen support and antiseizure medications. MRI findings confirmed DWS with the findings of cerebellar vermis hypoplasia with a posterior fossa cyst, while EEG demonstrated multifocal epileptiform discharges with moderate to severe diffuse encephalopathy. Fundoscopic examination further revealed bilateral optic atrophy, suggesting potential progressive neurodegeneration. Despite an elevated temperature, no clear infectious source was identified. Persistent convulsive seizures can themselves cause hyperthermia due to increased metabolic activity and muscle contractions. Therefore, in the absence of evidence for infection, the hyperthermia observed in this case is likely secondary to prolonged seizure activity; cerebrospinal fluid analysis could not be performed due to the legal guardian’s refusal of lumbar puncture. Seizure activity was initially persistent, necessitating an escalation of antiseizure therapy, including levetiracetam, phenytoin, and valproate, leading to eventual seizure control. Given her underlying structural brain anomaly, multifocal epilepsy, and optic atrophy, she requires continued close follow-up for comprehensive neurodevelopmental assessment, metabolic and genetic evaluation, and long-term seizure management.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Published online 22 July 2025
Contributor Information
Bipesh Kumar Shah, Email: shahbipeshkumar@gmail.com.
Karun Bhattarai, Email: anelitekarun99@gmail.com.
Ethical approval
Ethical approval is not required for a case report.
Consent
Written informed consent was obtained from the patient’s guardian for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
Sources of funding
Not applicable.
Author contributions
B.K.S.: analyzed and interpreted the data, conceptualization, investigations, visualization, supervision, and writing – review and editing. K.B.: data curation, writing – original draft, and writing – review and editing. All authors read and approved the final manuscript.
Conflicts of interest disclosure
None.
Research registration unique identifying number (UIN)
Not applicable.
Guarantor
Bipesh Kumar Shah and Karun Bhattarai.
Provenance and peer review
Not commissioned, externally peer-reviewed.
Data availability statement
Not applicable.
References
- 1.Barkovich AJ, Millen KJ, Dobyns WB. A developmental classification of malformations of the brainstem. Ann Neurol 2007;62:625–39. [DOI] [PubMed] [Google Scholar]
- 2.Sun Y, Wang T, Zhang N, et al. Clinical features and genetic analysis of Dandy-Walker syndrome. BMC Pregnancy Childbirth 2023;23:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Correa GG, Amaral LF, Vedolin LM. Neuroimaging of Dandy-Walker malformation: new concepts. Top Magn Reson Imaging 2011;22:303–12. [DOI] [PubMed] [Google Scholar]
- 4.Riley DS, Barber MS, Kienle GS, et al. CARE guidelines for case reports: explanation and elaboration document. J Clin Epidemiol 2017;89:218–35. [DOI] [PubMed] [Google Scholar]
- 5.Spennato P, Mirone G, Nastro A, et al. Hydrocephalus in Dandy-Walker malformation. Childs Nerv Syst 2011;27:1665–81. [DOI] [PubMed] [Google Scholar]
- 6.Bosemani T, Orman G, Boltshauser E, et al. Congenital abnormalities of the posterior fossa. Radiographics 2015;35:200–20. [DOI] [PubMed] [Google Scholar]
- 7.Sun Y, Zhang N, Tian H, et al. Prenatal diagnosis of the Dandy-Walker malformation associated with partial trisomy 12p and distal 15q deletion. J Genet. 2021;100:40. [PubMed] [Google Scholar]
- 8.Ramaswamy S, Rangasami R, Suresh S, et al. Spontaneous resolution of Blake’s pouch cyst. Radiol Case Rep 2013;8:877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Treviño Alanís MG, González Cantú N, Montes Cruz JV, et al. [Dandy Walker malformation]. Arch Argent Pediatr 2014;112:103–04. [DOI] [PubMed] [Google Scholar]
- 10.Jurcă MC, Kozma K, Petcheşi CD, et al. Anatomic variants in Dandy-Walker complex. Rom J Morphol Embryol 2017;58:1051–55. [PubMed] [Google Scholar]
- 11.Spennato P, Cascone D, Di Martino G, et al. Dandy–Walker Malformations/Variants. In: Di Rocco C, Pang D, Rutka JT, eds. Textbook of Pediatric Neurosurgery. Springer International Publishing; 2020. 831–56. [Google Scholar]
- 12.Kline-Fath BM, Calvo-Garcia MA. Prenatal imaging of congenital malformations of the brain. Semin Ultrasound CT MR 2011;32:167–88. [DOI] [PubMed] [Google Scholar]
- 13.Prieto LJ, Ruiz Y, Pérez L.et al. The brainstem-vermis and brainstem-tentorium angles in the fetus: a study of their reproducibility by fetal magnetic resonance imaging and their evolution along the gestation. Front Med (Lausanne) 2022;9:878906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu F, Fu L, Xu C, et al. Prenatal magnetic resonance imaging helps discover cerebellar dysplasia or malformations in foetuses. Curr Med Imaging 2023;2020:e15734056256514. doi: 10.2174/0115734056256514231020103822. [DOI] [PubMed] [Google Scholar]
- 15.Harper T, Fordham LA, Wolfe HM. The fetal Dandy Walker complex: associated anomalies, perinatal outcome and postnatal imaging. Fetal Diagn Ther 2007;22:277–81. [DOI] [PubMed] [Google Scholar]
- 16.Basson MA, Wingate RJ. Congenital hypoplasia of the cerebellum: developmental causes and behavioral consequences. Front Neuroanat 2013;7:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stambolliu E, Ioakeim-Ioannidou M, Kontokostas K, et al. The most common comorbidities in Dandy-Walker syndrome patients: a systematic review of case reports. J Child Neurol 2017;32:886–902. [DOI] [PubMed] [Google Scholar]
- 18.Monteagudo A. Dandy-Walker malformation. Am J Obstet Gynecol 2020;223:B38–B41. [DOI] [PubMed] [Google Scholar]
- 19.Di Nora A, Costanza G, Pizzo F, et al. Dandy-Walker malformation and variants: clinical features and associated anomalies in 28 affected children-a single retrospective study and a review of the literature. Acta Neurol Belg 2023;123:903–09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Y, Zhu J, Zhang D, et al. Refractory psychiatric symptoms and seizure associated with Dandy-Walker syndrome: a case report and literature review. Medicine (Baltimore) 2022;101:e31421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.El Tahir M, Ahmed M, Salman S, et al. Dandy-Walker malformation and intermittent explosive disorder: a case report. SAGE Open Med Case Rep 2022;10:2050313x221103355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paudel S, Poudel SK, Shah R, et al. Dandy Walker variant with agenesis of corpus callosum diagnosed late prenatally by foetal ultrasound: a case report. Ann Med Surg 2024;86:2301–04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Donahue ML, Ryan RM. Interstitial deletion of 8q21–>22 associated with minor anomalies,congenital heart defect, and Dandy-Walker variant. Am J Med Genet 1995;56:97–100. [DOI] [PubMed] [Google Scholar]
- 24.Zaki MS, Masri A, Gregor A, et al. Dandy-Walker malformation, genitourinary abnormalities, and intellectual disability in two families. Am J Med Genet A 2015;167a:2503–07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sasaki-Adams D, Elbabaa SK, Jewells V, et al. The Dandy-Walker variant: a case series of 24 pediatric patients and evaluation of associated anomalies, incidence of hydrocephalus, and developmental outcomes. J Neurosurg Pediatr 2008;2:194–99. [DOI] [PubMed] [Google Scholar]
- 26.Swar MO, Mahgoub SM, Yassin RO, et al. Dandy-Walker malformation and neurocutaneous melanosis in a three-month-old infant. Sudan J Paediatr 2013;13:61–65. [PMC free article] [PubMed] [Google Scholar]
- 27.Ferraris A, Bernardini L, Sabolic Avramovska V, et al. Dandy-Walker malformation and Wisconsin syndrome: novel cases add further insight into the genotype-phenotype correlations of 3q23q25 deletions. Orphanet J Rare Dis 2013;8:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dastoli P, Nicacio J, Da Costa MD, et al. Hydrocephalus and Dandy- Walker malformation: a review. Arch Pediatr Neurosurg 2020;2:e442020. [Google Scholar]
- 29.Mohanty A, Biswas A, Satish S, et al. Treatment options for Dandy-Walker malformation. J Neurosurg 2006;105:348–56. [DOI] [PubMed] [Google Scholar]
- 30.Alexiou GA, Prodromou N. Dandy-Walker Malformations. In: Manto M, Gruol D, Schmahmann J, Koibuchi N, Sillitoe R, eds. Handbook of the Cerebellum and Cerebellar Disorders. Springer International Publishing; 2020. 1–8. [Google Scholar]
- 31.Kumar R, Jain M, Chhabra D. Dandy-Walker syndrome: different modalities of treatment and outcome in 42 cases. Child’s Nerv Syst 2001;17:348–52. [DOI] [PubMed] [Google Scholar]
- 32.Ulrich J, Caird J, Crimmins D. Predicting outcomes in Dandy-Walker malformation: a retrospective cohort study. J Neurosurg Pediatr 2021;28:710–15. [DOI] [PubMed] [Google Scholar]
- 33.Yashon D, Jane JA, Sugar O. The course of severe untreated infantile hydrocephalus. Prognostic significance of the cerebral mantle. J Neurosurg 1965;23:509–16. [DOI] [PubMed] [Google Scholar]
- 34.Agrawal S, Thakur P. Dandy–Walker malformation. BMJ Case Rep 2009;2009:bcr0720080559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pradhan B, Sharma B, Acharya P, et al. Dandy-Walker malformation with neonatal meningitis: a case report. JNMA J Nepal Med Assoc 2024;62:142–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zamora EA, Das JM, Ahmad T. Dandy-Walker Malformation. In: StatPearls. StatPearls Publishing LLC; 2025. https://www.ncbi.nlm.nih.gov/books/NBK538197/. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.