

Abstract
Alzheimer's disease (AD) is an insidious, progressive, and irreversible neurodegenerative disease characterized by the deposition of extracellular amyloid β-protein (Aβ) to form senile plaques and abnormal phosphorylation of intracellular tau protein to form neuronal fiber tangles. The pathogenesis of AD is complex, and there are several hypotheses, primarily including the Aβ cascade hypothesis, the neurofibrillary tangle hypothesis, the inflammatory hypothesis, and the cholinergic hypothesis. It has been suggested that the dysregulation of multiple energy metabolic pathways, especially mitochondria metabolism, may be related to the severity of AD pathology and disease symptoms in the brain. The modification of histone (lysine) methylation, an actively regulated and reversible process, is closely related to energy metabolism and plays a crucial role in AD development. In summary, histone methylation, energy metabolism, and AD restricted and regulated each other. Here, we review the advances in the correlation between histone methylation, energy metabolism, and AD. This can provide further insights into the mechanisms underlying AD pathogenesis and its control.
Keywords: Histone methylation, Energy metabolism, Alzheimer's disease
1. Introduction
Alzheimer's disease (AD) is an insidious, progressive, irreversible, and incurable brain disorder. According to statistics, there were approximately 57 million adults over 40 years of age with AD worldwide in 2019, and this number is expected to grow to 153 million by 2050 [1]. The risk factors of AD mainly include age, genetics, and environment [2, 3]. The pathological features of AD mainly include abnormal deposition of amyloid β-protein (Aβ) and neurofibrillary tangles (NFTs) caused by hyperphosphorylated tau protein. The pathogenesis of AD is complex, primarily including the Aβ cascade hypothesis, the neurofibrillary tangles hypothesis, the inflammation hypothesis, and the cholinergic neuron hypothesis. Patients with AD can be classified into familial and sporadic types. AD is one of the aging-related neurodegenerative diseases. According to the age at diagnosis, AD is classified as early-onset and late-onset AD. Early-onset AD is closely associated with the mutation of Aβ-related genes, such as amyloid precursor protein (APP) and presenilin-1 (PS1). Apolipoprotein E4 (APOE4) is the major genetic risk factor for late-onset AD. With aging, energy metabolism in the body is impaired and the activity of histone methylation-modifying enzymes is altered. López-Otin et al. [4] identified nine hallmarks of aging process, including genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, mitochondrial dysfunction, cellular senescence, deregulated nutrient sensing, stem cell exhaustion, and altered intercellular communication. Numerous findings have revealed that these hallmarks are all closely related to AD. Mitochondria is the center of energy metabolism in the body, and its dysfunction leads to severely impaired energy metabolism. Epigenetics modifications include DNA methylation, histone modifications, chromatin remodeling, and RNA modifications. Histone modifications play important roles in epigenetic modifications, among which histone methylation is a more complex, enduring, and stable modification. There is growing evidence that the dysregulation of multiple energy metabolic pathways may be associated with the pathological changes in AD brain and the severity of disease symptoms. In addition, histone methylation is a prevalent epigenetic modification closely related to energy metabolism and has emerged as an essential player in the onset and development of AD. Besides, a large number of studies have shown that energy metabolism and histone methylation are mutually regulated, but neither has been fully elucidated in relation to AD. Therefore, understanding the interconnection between histone methylation, energy metabolism, and AD will provide further insight into the mechanisms of AD pathogenesis and its control.
2. Histone methylation and AD
2.1. Histone methylation
The nucleosome is the basic structural unit of chromatin and consists of histone octamer and double-stranded DNA. Histone octamer is composed of four different histone proteins: H2A, H2B, H3, and H4. Various covalent modifications, including methylation, phosphorylation, acetylation, and ubiquitination, occur at the ends of histones. These modifications can regulate gene expression by directly changing the interaction between histones and DNA, or by recruiting proteins or protein complexes with specific functions. These post-translational modifications of histones are also called the “histone code” because the histone modification sites and their interactions with other regulatory proteins form a network that regulates gene expression. It is worth noting that histone modification sites do not function independently but interact with and regulate each other. Histone post-translational modifications play an essential role in epigenetic regulation and are closely related to the regulation of gene expression. Different modifications exhibit different characteristics. For example, histone methylation modifications are relatively stable, whereas acetylation modifications are highly dynamic, and phosphorylation, ubiquitination, and other modifications are unstable. Histone methylation can occur in the lysine (Lys or K) and arginine residues. Owing to the diversity of modification sites and the fact that the same modification site can be methylated to different degrees, methylation is a more complex modification than other post-translational modifications of histones. Lysine can be mono-methylated (me1), di-methylated (me2), or tri-methylated (me3), while arginine can be mono-methylated and symmetrically or asymmetrically di-methylated. Depending on the methylation site, lysine methylation of H3 and H4 can regulate transcriptional activation and repression. Methylation of histone 3 lysine 4 (H3K4), H3K36, and H3K79 is primarily involved in gene activation, and methylation of H3K9, H3K27, and H4K20 is mainly associated with transcriptional repression. Arginine methylation has been found to promote transcriptional activation.
Histone methylation is an actively regulated and reversible process, among which histone (lysine) methylation modification has been most commonly studied. It involves histone lysine methyltransferases (KMTs)-mediated methylation and histone lysine demethylases (KDMs)-mediated demethylation (Fig. 1). Based on their catalytic domain sequence, KMTs are classified into SET-containing and non-SET-containing domains, of which only KMT4 (DOT1L) (Table 1) is a non-SET-containing methyltransferase. KDMs include lysine-specific demethylases (LSD) and the Jumonji C domain-containing (JMJD) protein family (Table 2). The process of KMTs-mediated histone methylation is intricate, as KMTs become activated only after forming a complex with the corresponding protein and subsequently recognize specific histone modification sites for methylation, thereby regulating gene expression. For instance, pre-existing H3K9me2 or H3K9me3 is specifically recognized by the chromodomain of heterochromatin protein 1, which subsequently recruits the catalytic KMT1A (SUV39H1) to tri-methylate neighboring H3K9 sites, thereby establishing a positive feedback loop [5]. H3K27 methylation represses gene expression by recruiting poly-comb repressive complexes in which the poly-comb is a structural domain that recognizes the H3K27 site [6]. Unlike KDMs, KMTs do not require the involvement of other proteins and function directly.
Figure 1.
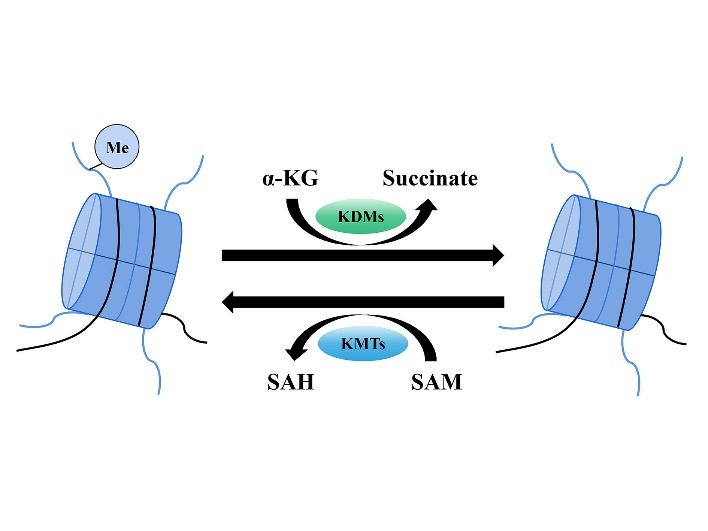
Histone methylation and demethylation process. Note: Histone methylation refers to the transfer of methyl groups to the lysine residues of histones catalyzed by KMTs, with SAM as the methyl donor, whereas SAM itself becomes SAH. Histone demethylation refers to the change of histone modification with α-KG as the substrate and the removal of methyl groups from the lysine residues catalyzed by KDMs, while α-KG becomes succinate. SAM: s-adenosyl-L-methionine; SAH: s-adenosyl-L-homocysteine; α-KG: α-ketoglutarate; Me: methyl.
Table 1.
Classification and modification Loci of KMTs.
| Nomenclature | Official Symbol/Alias | Loci | References |
|---|---|---|---|
| KMT1A | SUV39H1 | H3K9 | [194, 195] |
| KMT1B | SUV39H2 | H3K9 | [196] |
| KMT1C | G9a/EHMT2 | H3K9 | [197] |
| KMT1D | GLP/EHMT1 | H3K9 | [7] |
| KMT1E | SETDB1/ESET | H3K9 | [198] |
| KMT1F | SETDB2 | H3K4 | [199] |
| KMT2A | MLL1 | H3K4 | [200] |
| KMT2B | MLL2 | H3K4 | [201, 202] |
| KMT2C | MLL3 | H3K4 | [203] |
| KMT2D | MLL4 | H3K4 | [204] |
| KMT2E | MLL5 | H3K4 | [205] |
| KMT2F | SET1A | H3K4 | [206] |
| KMT2G | SET1B | H3K4 | [207] |
| KMT2H | ASH1L | H3K4, H3K9, H3K36, H4K20 | [208, 209] |
| KMT3A | SETD2 | H3K36 | [210] |
| KMT3B | NSD1 | H3K36, H4K20 | [211] |
| KMT3C | SMYD2 | H3K4, H3K36 | [212, 213] |
| KMT3D | SMYD3 | H3K4 | [214] |
| KMT3E | WHSC1L1/NSD3 | H3K4, H3K27, H3K36 | [215, 216] |
| KMT3F | WHSC1/NSD2 | H3K4, H3K27, H3K36, H4K20 | [215, 217, 218] |
| KMT4 | DOT1L | H3K79 | [219] |
| KMT5A | SET8/PR-SET7/SETD8 | H4K20 | [220, 221] |
| KMT5B | SUV20H1 | H4K20 | [222] |
| KMT5C | SUV20H2 | H4K20 | [222] |
| KMT6 | EZH2 | H3K27 | [223] |
| KMT7 | SET7/SET9/SETD7 | H3K4 | [214, 221] |
| KMT8A | PRDM2/RIZ1 | H3K9 | [224] |
| KMT8B | PRDM9 | H3K4 | [225] |
| KMT8C | PRDM6 | H4K20 | [226] |
| KMT8D | PRDM8 | H3K9 | [227] |
| KMT8E | PRDM3 | H3K9 | [228] |
| KMT8F | PRDM16 | H3K9 | [228] |
Table 2.
Classification and modification Loci of KDMs.
| Nomenclature | Official Symbol/Alias | Loci | References |
|---|---|---|---|
| KDM1A | LSD1 | H3K4, H3K9 | [229, 230] |
| KDM1B | LSD2/AOF1 | H3K4 | [231] |
| KDM2A | FBXL11/JHDM1A | H3K36 | [232] |
| KDM2B | FBXL10/JHDM1B | H3K36 | [233] |
| KDM3A | JMJD1A/JHDM2A | H3K9 | [234] |
| KDM3B | JMJD1B/JHDM2B | H3K9 | [235] |
| KDM3C | JMJD1C/JHDM2C | H3K9 | [236] |
| KDM4A | JMJD2A/JHDM3A | H3K9, H3K36 | [237] |
| KDM4B | JMJD2B/JHDM3B | H3K9, H3K36 | [238] |
| KDM4C | JMJD2C/JHDM3C/GASC1 | H3K9, H3K36 | [239, 240] |
| KDM4D | JMJD2D/JHDM3D | H3K9, H3K36 | [240] |
| KDM5A | JARID1A/RBP2 | H3K4 | [241] |
| KDM5B | JARID1B/PLU1 | H3K4 | [242] |
| KDM5C | JARID1C/SMCX | H3K4 | [243] |
| KDM5D | JARID1D/SMCY | H3K4 | [243] |
| KDM6A | UTX | H3K27 | [244, 245] |
| KDM6B | JMJD3 | H3K27 | [244, 245] |
| KDM7A | JHDM1D/KIAA1718 | H3K9, H3K27 | [246] |
| KDM7B | JHDM1F/PHF8 | H3K9, H3K27, H4K20 | [247] |
| KDM7C | PHF2 | H3K9, H3K4 | [248] |
| KDM8 | JMJD5 | H3K36 | [249] |
2.2. KMTs and AD
Numerous studies have shown that KMTs are involved in AD pathogenesis by affecting the expression of related proteins and genes.
2.2.1. G9a, GLP, and EZH1/2
Synaptic plasticity is recognized as the neurobiological basis of learning and memory and is involved in the development of AD. Brain-derived neurotrophic factor (BDNF), predominantly synthesized in the brain and abundant in the cerebral cortex and hippocampus, regulates long-term potentiation (LTP) which is one of the most widely cited cellular models underlying synaptic plasticity. Glutamate receptors are also involved in synaptic plasticity. They are classified as ionotropic and metabotropic types, which include the N-methyl-D-aspartic receptor (NMDAR) and the α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor. G9a and GLP are histone methyltransferases that mono- and di-methylate H3K9. They share approximately 80% sequence identity in the SET-containing domain to form a heterodimer [7]. Reportedly, BDNF levels are reduced in patients with AD [8, 9]. Sharma et al. [10] demonstrated that the inhibitors of G9a and GLP, such as BIX01294 and UNC0642, upregulated BDNF expression in the CA1 region of acute hippocampal slices from five- to seven-week-old male Wistar rats treated with Aβ1-42 oligomers and further maintained late-LTP. In late-stage five familial AD mutations mice, it has been reported that the inhibitors of G9a and GLP rescued cognitive deficits by relieving H3K9me2-mediated repression of glutamate receptors transcription [11]. In addition, the knockdown of G9a was found to reverse the reduction of hypoxia-induced neprilysin (NEP) in mouse primary cortical and hippocampal neurons, facilitating Aβ clearance [12]. In the prefrontal cortex of the P301S Tau mouse model, Wang et al. [13] observed that UNC0642 had an effect on improving spatial and recognition memory in five- to seven-month-old mice (late stage of cognitive impairment associated with NFT deposits) by inhibiting tau protein hyperphosphorylation. However, the mechanism of the effect of the inhibitor on tau protein phosphorylation is still unclear. The cytoskeleton plays vital roles in coordinating neuronal transport and maintaining synaptic function [14]. This study also found that UNC0642 restored approximately 22% of the down-regulated genes (enriched in the regulatory cytoskeleton) in mice, which facilitated the restoration of neuronal excitability and synaptic transmission in the prefrontal cortex and improved cognition [13]. Furthermore, the decrease in autophagy is associated with an increase in the proportion of damaged mitochondria, and consequently leads to increased accumulation of reactive oxygen species (ROS) and Aβ. Autophagosome formation requires the participation of microtubule-associated protein 1 light chain 3 beta, diabetes- and obesity-regulated protein, and WD repeat domain phosphoinositide-interacting protein 1 [15-17]. Studies have shown that G9a inhibits the expression of these proteins, thereby controlling autophagy [18], which may be detrimental to the expression of various aging phenotypes.
Postsynaptic membrane density (PSD) is the structural basis of postsynaptic signal transduction and integration. PSD95, the major scaffolding protein in the PSD, integrates NMDAR signals and regulates synaptic plasticity and learning memory. Griñán-Ferré et al. [19] found that UNC0642 treatment increased PSD95 protein levels. EZH2, a negative regulator of dendritic morphogenesis in mammalian neurons, has been documented to interact with the transcriptional repressor chromodomain Y-like transcription co-repressor to upregulate H3K27me3 levels in the BDNF promoter region, thereby limiting dendritic development [20]. EZH1 and EZH2 play antagonistic roles in the regulation of PSD95 transcription [21], but none have been observed in AD.
2.2.2. KMT2 family, SMYD3, and SETD7
KMT2A modulates memory by influencing the expression of genes involved in synaptic plasticity [22]. Cao et al. [23] found that administration of the SET1/MLL inhibitor WRD5-0103 inhibited the increase in H3K4me3 levels and the down-regulation of glutamatergic synaptic receptor expression, and restored synaptic plasticity in the prefrontal cortex, thus alleviating cognitive deficits in P301S Tau mice and five familial AD mutations mice. Among the up-regulated genes reversed by WRD5-0103 treatment in the prefrontal cortex of AD mice, many showed increased H3K4me3 enrichment at their promoters, including serum- and glucocorticoid-regulated kinase 1 (SGK1) and early growth response 1 (EGR1). Evidence showed that SGK1, a member of the serine/threonine protein kinase family, was significantly elevated in the prefrontal cortex of patients with AD. The administration of specific SGK1 inhibitor GSK650394 in AD mice reduced tau protein hyperphosphorylation [23]. Overexpression of SGK1 in the hippocampus was reported to increase Aβ degradation mediated by insulin-degrading enzyme (IDE) and non-amyloid formation of APP mediated by a disintegrin and metalloprotease 10 (ADAM10) (a type of α-secretase), thereby affecting the reduction of Aβ production and deposition. However, the amyloid formation of APP, mediated by β-secretase 1 (BACE1), was not affected [24]. EGR1 is a member of the immediate early gene family. It has been reported that EGR1 is associated with Aβ production, tau protein phosphorylation [25, 26], and synaptic plasticity [27, 28], but whether EGR1 is a deleterious or protective factor in the AD is still controversial and needs further explored. SETDB1 (ESET) was found to upregulate the level of H3K9me3 in the striatum, which consequently decreased the expression of EGR1, ultimately causing impaired neurological function in the striatum in an R6/2 mouse model of Huntington's disease, resulting in various behavioral deficits [29]. A recent study has discovered that SETDB1 (ESET) inhibitors decreased H3K9me3 levels, and improved motor function and neuropathological symptoms in Huntington's chorea transgenic mice [30]. Given that EGR1 has been implicated in the pathological changes in AD and can be modulated by SET1/MLL and SETDB1 (ESET) through the methylation pathway, we hypothesize that the two histone methyltransferases may also influence the pathological progression of AD by affecting EGR1.
In addition, a recent study has demonstrated that the histone methyltransferase SMYD3, which primarily catalyzes H3K4me2/3, was significantly elevated in the prefrontal cortex of AD patients and P301S Tau mice. BCI-121, the inhibitor of SMYD3, has been found to rescue NMDAR and cognitive deficits in P301S Tau mice [31]. In a study conducted by Bichmann et al. [32] showed that knock down and inhibitor studies supported by proteomics data led to the identification of SETD7 as a novel lysine methyltransferase for tau. The above study also revealed that SETD7 specifically methylated tau at K132, an event that facilitated subsequent methylation at K130.
2.2.3. Others
In the hippocampus of aged mice, ETP69 (an inhibitor of SUV39H1) was demonstrated to decrease the H3K9me3 level of the BDNF promoter [33]. In addition, PRDM5, which is associated with astrocyte proliferation and neuronal apoptosis, may be involved in inflammatory processes in the central nervous system [34]; however, these have not been observed in AD.
Apart from the KMTs inhibitors mentioned above, a large number of specific inhibitors of different KMTs have been developed over the past few years. Among the KMTs inhibitors, the inhibitors of EZH2 and DOT1L have entered the stage of clinical trials. For example, tazemetostat (EPZ6438) is an oral EZH2 inhibitor that can be used to treat sarcoma [35]. Valemetostat is the first dual EZH1/2 inhibitor approved for the treatment of adult T-cell leukemia/lymphoma [36]. Besides, pinometostat (EPZ5676), the inhibitor of DOT1L, is being studied and tested for the treatment of diseases such as MLL and AML leukemia [37]. Current studies mainly focus on the role of these inhibitors in the treatment of some cancers and many blood malignancies, but whether they have effects on the development of AD has not yet been reported.
2.3. KDMs and AD
There are two main types of KDMs, including flavin adenine dinucleotide (FAD)-dependent lysine-specific demethylases (KDM1A/1B or LSD1/2) and JmjC domain-containing lysine demethylases. The latter has diverse catalytic substrates that require both α-KG and divalent iron ions (Fe2+) and has the potential to remove trimethyl groups from the histone lysine residues.
2.3.1. LSD
LSD, the first histone demethylase discovered, belongs to the superfamily of FAD-dependent amine oxidases. It specifically removes mono- and di-methylated modifications of H3K4 in vitro and mono- and di-methylated modifications of H3K9 in vivo with the participation of FAD, but fails to demethylate H3K4me3. It has been demonstrated that increased LSD1-mediated histone demethylation attenuated tau protein-mediated neurodegeneration in PS19 Tau mice that mimic tau protein aggregation [38]. Longaretti et al. [39] indicated that LSD1/neuroLSD1 (a neuron-specific splice variant) levels in the hippocampus decreased significantly with aging in individuals aged 20-94 years, regardless of sex. Additionally, LSD1 was found to influence synaptic plasticity-related proteins in neuroLSD1KO mice under environmental stress conditions. However, the impact of LSD1 on AD remains unclear and warrants further exploration.
The KDMs inhibitors, which are similar to KMTs inhibitors, are still in the research phases. Pharmacological inhibition of LSD1 has shown promising therapeutic benefits of AD. ORY-2001, one of the LSD1 inhibitors, has completed the phase Ⅱa clinical trial in patients diagnosed with mild to moderate AD and undergone the phase Ⅱa clinical trial in patients diagnosed with moderate to severe AD [40, 41]. Besides, ORY-2001 has been reported to rescue cognitive function and reduce the inflammation signature in the SAMP8 (senescence accelerated mouse prone 8) mice, which is used to study AD [42].
2.3.2. The Jumonji C domain-containing (JMJD) family histone demethylases
The JMJD family of demethylases consists primarily of the KDM1~KDM7 family. The KDM2 family, including KDM2A/JHDM1A and KDM2B/JHDM1B, participates in the demethylation of H3K4me3 and H3K36me1/2. It is highly expressed in the forebrain, cerebellum, occipital lobe, and postcentral gyrus during early mammalian embryonic development, and changes with developmental stage and age [43]. Moreover, mutations and abnormal expression of these genes can alter the transcriptional levels of target genes by affecting their demethylation, thereby regulating the expression of downstream signaling pathways and affecting neurogenesis, apoptosis, oxidative stress, and energy metabolism [44-47]. KDM2B affects the formation of neuroinflammatory amyloid plaques in the prodromal phase of AD and is directly involved in AD pathogenesis [48]. The susceptibility genes rs28604990 and rs7955747, which are located within a haplotype block that includes intron 6 and intron 12 of KDM2B, respectively, are most substantially associated with AD in APOE4 non-carriers [49].
According to bioinformatic analysis, the expression levels of JHDM1A, JHDM2A/2B, and JHDM3A/3B were significantly higher in the postmortem brain tissue of AD patients than in non-demented controls, whereas JHDM1B level was downregulated. However, the results need to be further verified by more experiments. In tauR406W-induced transgenic Drosophila model, the knockdown of KDM4A (homologous to human JHDM3A) was found to ameliorate tauR406W-induced locomotion defects by restoring heterochromatin [50]. Griñán-Ferré et al. [51] discovered that JARID1A (KDM5A) was associated with Aβ in five familial AD mutations mice model. The KDM6 family of H3K27me2/3 demethylases includes JMJD3 and UTX. High expression of JMJD3 has been documented to enhance H3K27me3 demethylation of disstal-less homeobox2, mixed lineage leukemia 1, mammalian achaete-scute homologue-1, and other genes related to neuronal development [52]; however, whether it has this effect on AD is unknown. Evidence showed that medium and high doses of curcumin inhibited Aβ aggregation and attenuated mitochondrial stress response by modulating the JMJD3-H3K27me3-BDNF pathway in APP/PS1 mice and SH-SY5Y cells [53]. In addition, a study in caenorhabditis elegans discovered that elevated UTX activated the insulin/IGF-1 signaling pathway (IIS) and prevented the transcription factor DAF-16 from entering the nucleus to activate anti-aging genes. Similar aging alterations have also been observed in brain samples from rhesus macaques [54]. However, whether UTX plays the same role in AD development remains to be confirmed.
Chen et al. [55] found that the mTOR signaling pathway was overactive in the hippocampus of PHF8 knockdown mice, resulting in LTP attenuation and cognitive deficits. However, whether PHF8 affects AD in the way mentioned above remains to be explored.
Besides, researchers observed changes in other histone methylation marks in AD patients compared with controls, with mainly increased H4K20me2 and H3K79me1 levels and decreased H3K79me2, H3K36me2, H4K20me3, H3K27me1, and H3K56me1 marks [56].
Taken together, KMTs and KDMs have effects on proteins and genes involved in AD-related pathologies, such as synaptic plasticity, Aβ formation and clearance, and tau protein hyperphosphorylation by modifying lysines at different sites on histones (Table 3). For the drugs addressed before, such as EZH2, DOT1L, and LSD1 inhibitors, they are safe and tolerable and most of them have their best clinical response after weeks to months of continuous therapy. However, some problems such as selectivity as well as resistance of single-target drugs and potential off-target effects limit their clinical application. Besides, the inhibitors show remarkable promise in preclinical studies, but their clinical efficacy has been more modest. Currently, the forthcoming focus in the field of drug development is anticipated to be on the development of inhibitors which is founded on a multitarget approach. In addition, different combination strategies are being pursued in clinical trials, including the combination of epigenetic therapies with targeted therapies and immunotherapy. Given the fundamental roles of dysregulated KMTs and KDMs in AD development, these inhibitors may hold the key to regulating AD-related pathologic changes by inhibiting the main molecular pathways and emerge as an effective and promising strategy for AD therapy.
Table 3.
KMTs/KDMs and AD.
| KMTs/KDMs | Category | Affected pathways/genes/proteins | Pathologies/phenotypes in AD | Experimental subjects | References |
|---|---|---|---|---|---|
| KMTs | G9a/GLP (KMT1C/1D) | BIX01294, UNC0642 (G9a, GLP inhibitors) →BDNF, Glutamate receptors (NMDAR), PSD95↑; Hypoxia→H3K9me2 level in the NEP promoter↑→NEP↓ (reversed by G9a knockdown) | Synaptic plasticity↑; Aβ clearance↑; Tau protein hyperphosphorylation↓; | Rats, mice; Cells | [10-13, 19] |
| KMTs | MLL1 (KMT2A) | Genes related to AD and LTP | Synaptic plasticity | Mice | [22] |
| KMTs | SET1/MLL family (SET1A/1B (SETD1A/1B)) | WRD5-0103 (SET1/MLL inhibitor) →Synaptic plasticity in prefrontal cortex↑; WRD5-0103→EGR1, SGK1↓; GSK650394 (SGK1 inhibitor) →Hyperphosphorylation of tau protein↓; SGK1↑→IDE↑/ADAM10↑→Aβ degradation↑/Aβ production↓ (BACE1-mediated APP amyloid formation process is not affected) | Synaptic plasticity↑; Aβ degradation↑, Aβ production↓; Tau protein hyperphosphorylation↓ | Mice, cells | [23-28] |
| KMTs | SMYD3 | BCI-121 (SMYD3 inhibitor) →NMDAR↑ | Cognitive function↑ | Human tissues, mice, and cells | [31] |
| KDMs | LSD1 (KDM1A) | None | Tau protein phosphorylation↓; Cognitive function↓; Inflammation↑ | Mice | [38, 42] |
| KDMs | JHDM1B (KDM2B) | None | Aβ formation↑ | Human brain tissues | [49] |
| KDMs | KDM4A (homologous to human JHDM3A) | None | TauR406W-induced locomotion defects | Drosophila | [50] |
| KDMs | JARID1A (KDM5A) | ①BACE1↑, sAPPβ↑; ②p-tau↑; ①SOD1↑, 4-HNE↑, Aldh2↓; ④TNF-α↑, IL-6↑; ⑤SYP↓ | ①Aβ↑; ②Tau protein phosphorylation↑; ①Oxidative stress↑; ④Neuroinflammation↑; ⑤Synaptic plasticity↓ | Mice | [51] |
| KDMs | JMJD3(KDM6B) | CUR→JMJD3-H3K27me3-BDNF→Aβ aggregation, mitochondrial stress response↓ | Aβ aggregation, mitochondrial stress response↓ | Mice, cells | [53] |
Note: IL-6: interleukin 6; TNF-α: tumor necrosis factor alpha; sAPPβ: soluble amyloid precursor protein β; SOD: superoxidedismutase; 4-HNE: 4-Hydroxynonenal; Aldh2: aldehyde dehydrogenase-2; SYP: synaptophysin; CUR: curcumin (“↑” denotes increased levels; “↓” denotes reduced levels).
3. Energy metabolism and AD
Energy metabolism is the process of releasing, transferring, storing, and utilizing energy as part of the material metabolism process, in which the mitochondria play an important role. Dysfunctional energy metabolism in the brain is considered a precipitating factor and a central link in many neurodegenerative diseases such as AD. It has been shown that disorders of energy metabolism occur in the central nervous system and periphery early in AD concurrent with the development of AD pathology. These disruptions include mitochondrial dysfunction and glucose, lipid, and amino acid metabolism disorders. The tricarboxylic acid cycle (TCA cycle), also known as the Krebs cycle, is the final metabolic pathway of the three major nutrients and the pivot of the three major metabolic processes (Fig. 2). Conversely, pathological changes in AD can affect energy metabolism homeostasis. Oxidative stress induced by Aβ42 oligomers, for example, may impair glucose metabolism [57]. Consequently, there is a complex interplay between energy metabolism and AD.
Figure 2.

Glucose, fatty acid, and amino acid metabolism and TCA cycle. Note: ACO2: aconitase 2; IDH3A: isocitrate dehydrogenase 3A; α-KG: α-ketoglutaric acid; OGDH: α-ketoglutarate dehydrogenase; SUCLG2: succinyl- CoA synthetase; SDH: succinate dehydrogenase; FH: fumarate hydratase; MDH2: malate dehydrogenase 2; OAA: oxaloacetic acid; CS: citrate synthase; Glu: glutamate; Gln: glutamine; GLUD: glutamate dehydrogenase; GLS/GLS2: glutaminase; GSH: glutathione; GAD: glutamic acid decarboxylase; GABA-T: γ-aminobutyric acid transaminase; GABA: γ-aminobutyric acid; SSA: succinic semialdehyde; SSADH: succinate semialdehyde dehydrogenase
3.1. Glucose metabolism and AD
Glucose is the primary source of energy in the brain. Because it cannot be synthesized or stored in the central nervous system, glucose must enter the brain from the periphery through the blood-brain barrier (BBB) with the help of sodium-dependent glucose transporter 1/2 and glucose transporters (GLUTs) [58, 59].
3.1.1. Decreased glucose intake
Several positron emission tomography studies have shown that the entorhinal cortex and parietal lobes have a deficit in glucose uptake in mild cognitive impairment, suggesting that impaired glucose metabolism plays a key role in the early pathogenesis of AD [60]. Glucose deprivation has been shown to increase the expression of the unfolded protein response, which facilitated tau protein phosphorylation in vivo and in vitro models [61]. Besides, the experiment using the postmortem brain samples from AD patients and age-matched non-AD patients demonstrated that impaired glucose metabolism inhibited nonamyloidogenic α-secretase processing of APP via the peroxisome proliferator activated receptor gamma coactivator 1 alpha/forkhead box protein O3a (PGC-1α/FoxO3a) pathway, thus promoting Aβ generation [62]. The increase in glucose uptake was found to boost memory by changing the distinctive chromatin acetylation status of the promoter regions of BDNF and fibroblast growth factor 1 in neuronal cells, astrocytes, and microglial cells of the murine hippocampus [63]. Moreover, a study conducted in both cell culture and animals showed that high glucose levels led to neuronal mitochondrial dysfunction and resultant insulin resistance in an adenosine monophosphate-activated protein kinase-dependent manner [64]. However, these regulatory mechanisms have not been observed in AD. In the brain, GLUT1 is expressed mainly in glia and endothelial cells [65]. Reduced GLUT1 expression in BBB worsens cerebrovascular degeneration, neuropathology, and cognitive function in AD [66]. In addition, glycogen synthase kinase-3β (GSK-3β) is a highly conserved multifunctional serine/threonine protein kinase whose activity is negatively regulated by protein kinase B (PKB/AKT). The advanced glycation end products (AGEs) play a key role in AD [67]. They were discovered to cause spatial memory deficit and tau protein hyperphosphorylation through receptor for advanced glycation end products (RAGE)-mediated AKT/GSK-3β pathway in the human neuroblastoma SK-N-SH cells, hippocampal neuron, and the rat brain [68].
3.1.2. Insulin resistance
Phosphatidylinositol 3-kinase (PI3K), an intracellular phosphatidylinositol kinase with serine/threonine kinase and phosphatidylinositol kinase activities, is involved in the regulation of various cellular functions such as glucose transport. It has been shown that insulin binds to its receptor and is essential for the metabolism of Aβ and tau protein via the PI3K/AKT signaling pathway [69]. Insulin was also found to promote the expression of NEP and IDE in astrocytes through the activation of extracellular signal-regulated kinase (ERK)-mediated pathway [70]. However, Steen et al. [71] reported that impaired insulin signaling caused by aberrant fluctuations in blood glucose levels resulted in dephosphorylation of GSK-3, which in turn encouraged the Aβ deposition and tau protein phosphorylation in the postmortem brain tissues of AD and control cases. Additionally, a study conducted by Ho et al. [72] in AD mouse model demonstrated that insulin resistance affected the production and clearance of Aβ by taking effect on γ-secretase and IDE.
3.1.3. O-GlcNAcylation
Phosphorylated and O-GlcNAcylated tau proteins in the brain interact to promote the formation of NFTs. Impaired cerebral glucose metabolism due to the absence of GLUT1/3 can decrease tau protein glycosylation, while increasing tau protein phosphorylation [73]. O-GlcNAcase insufficiency can elevate the level of O-GlcNAcylation, which inhibits the activation of necroptosis factors and the excessive activation of astrocytes and microglia, ultimately ameliorating neuronal death and neuroinflammation in AD. Furthermore, increased O-GlcNAc level is associated with restored mitochondrial function, normal mitochondrial morphology, and reduced Aβ deposition [74], but the detailed mechanisms are still unclear.
3.1.4. Ketone body metabolism
Ketone bodies, a generic term for acetoacetic acid, β-hydroxybutyric acid (βOHB), and acetone, are metabolized more rapidly than glucose and can bypass glycolysis to enter directly into the TCA cycle. A large number of studies have shown that ketone bodies can reduce AD pathologies including extracellular Aβ plagues, NFTs, and neuronal loss through various pathways [75]. Ketone bodies can promote Aβ1-40 transendothelial transport via enhancing the function of lipoprotein receptor-related protein 1 (LRP1) and P-glycoprotein transporter and phosphatidylinositol-binding clathrin assembly protein (PICALM), which are responsible for Aβ clearance [76]. It has been found that βOHB increased the level of NEP via G protein-coupled receptor 109A in the five familial AD mutations mice and HT22 cells [77] and prevented the increase in the expression of BACE1 caused by GLUT inhibitor in SH-SY5Y cells [78]. βOHB was also discovered to reduce Aβ plague formation and microgliosis in the five familial AD mutations mice by inhibiting nod-like receptor pyrin domain containing 3 (NLRP3) inflammasome activation [79]. In addition, a recent study has demonstrated that βOHB ameliorated the downregulation of tyrosine kinase receptor A (TrkA) expression in Aβ-treated SH-SY5Y cells by inhibiting histone deacetylase 1/3 (HDAC1/3) [80]. In APP/PS1/Tau transgenic mice, circulating βOHB concentrations increased by alternative-day-fasting have been shown to improve the expression of PSD95 and synapsin and reduce Aβ oligomers as well as tau hyperphosphorylation [81].
3.2. Lipid metabolism and AD
Most solid components of the brain consist of lipids, including triglycerides, fatty acids, phospholipids, sterol lipids, and sphingolipids. These lipids are important for maintaining BBB function and are involved in oxidative stress and energy balance. Studies have shown that increases in sphingolipids and triglycerides are associated with Aβ production. The increase of ceramides, a kind of sphingolipids, can promote lipid peroxidation, oxidative stress, mitochondrial dysfunction, and neuronal death. Additionally, the decreased levels of specialized pro-resolution mediators from eicosapentaenoic acid and docosahexaenoic acid and the increased production of prostaglandin E from arachidonic acid can promote inflammation, and thus increasing the risk of AD [82].
Genome-wide association studies have identified some AD-related genes involved in lipid trafficking or metabolism, such as APOE, clusterin/apolipoprotein J, triggering receptor expressed on myeloid cells 2 (TREM2), PICALM, sterol regulatory-element binding protein 2 (SREBP2), and the ATP-binding cassette subfamily A (ABCA) [83].
3.2.1. APOE4
APOE is a polymorphic protein that regulates lipoproteins conversion and metabolism. APOE4, an isoform of APOE, is considered the most significant risk factor for late-onset AD. Numerous studies have shown that APOE4 exerts important roles in Aβ metabolism, tau protein phosphorylation, neuroinflammation, synaptic plasticity, and the functions of BBB and mitochondria. APOE4 was shown to enhance APP transcription and Aβ synthesis by activating the non-canonical mitogen-activated protein (MAP)-kinase cascade involving dual leucine-zipper kinase (DLK), mitogen-activated protein kinase kinase 7 (MKK7), and ERK1/2 in vivo and in vitro [84]. LRP1, a key receptor for Aβ clearance at the BBB, can regulate the Aβ clearance by binding to APOE and PICALM [85, 86]. Conversely, Wan et al. [87] discovered that Aβ stimulated lipolysis in a manner that appeared partially dependent on the PKA/ERK signaling pathway and induced leptin and IL-6 secretion in cultured human adipose tissue. This process leads to the release of fatty acids, triggering lipid deposition and insulin resistance, consequently affecting lipid degradation.
APOE4 was also found to induce the phosphorylation of tau proteins at certain sites through the activation of calpain-dependent kinase 5 (CDK5) signaling pathway in AD mouse model [88]. The present study has provided compelling evidence that the APOE epsilon4 affected the immune system through down-regulation of the IL-7/IL-7R signaling pathway, contributing to neuroinflammation in AD patients [89]. In terms of morphology and function of synapses, induced pluripotent stem cells-derived cerebral organoids from AD patients carrying APOE epsilon4/epsilon4 have been shown a decrease in synaptic integrity [90]. APOE4 also impairs hippocampal LTP through reducing the phosphorylation of cAMP response element binding protein (CREB) as well as Ca2+/calmodulin-dependent protein kinase IIα (CaMKIIα) [91]. Another study using in vivo models discovered that APOE4 led to fewer dendritic spine numbers by elevating the activity of calcium/calmodulin-dependent protein phosphatase 2B (PP2B) [92]. However, the exact mechanisms are unclear. Besides, Yin et al. [93] found that the PGC-1α-sirtuin3 signaling pathway mediated synaptic damage by APOE4, ultimately resulting in cognitive impairment in vivo and in vitro models. It has been demonstrated that humanized APOE4 mice showed a leaky BBB, increased matrix metallopeptidase 9, impaired tight junctions, and reduced astrocyte end-foot coverage of blood vessels [94]. APOE epsilon4 carriers were found to have lower expression levels of mitochondrial fusion- and division-related proteins compared to non-carriers [95].
3.2.2. TREM2, SREBP2, and ABCA family
TREM2, located on the surface of microglia, can affect the metabolism of cholesterol, myelin, and phospholipids. TREM2 can increase the degradation of amyloid plaques by promoting microglial phagocytosis [96], and act as a ligand for phospholipids, lipoproteins, and apolipoproteins [97]. Conversely, a study using AD mice found that TREM2 knockdown reduced microglia activation and aggregation around Aβ plagues, which increased Aβ plague pathology [98]. Deficiency of TREM2 was demonstrated to impair the microglial response to tau accumulation in the brain of hTau and PS19 mice [99]. Besides, Zhong et al. [100] reported that TREM2 suppressed the activation of classical complement cascade and complement-mediated synaptic loss in AD mice models by directly binding to C1q, the initiator of the classical complement pathway.
SREBP2, a sterol regulatory transcription factor, has an essential role in regulating cholesterol and fatty acid homeostasis. Overexpression of SREBP2 accelerates oxidative damage, Aβ accumulation, neuroinflammation, and cognitive decline [101]. Specific knockdown of SREBP2 in astrocytes was discovered to significantly decrease Aβ and tau protein levels [102]. On the contrary, oligomeric Aβ42 inhibited SREBP2 activation through AKT inhibition [103].
The ABCA transporters family plays a central role in the maintenance of cholesterol-homeostasis in the brain and has a prominent role in AD development. These transporters have been reported to target the primary hallmark of AD pathogenesis, namely, the Aβ hypothesis. Among the ABCA transporters family, ABCA1, 2, 5, and 7 are all associated with Aβ metabolism. It has been found that the increased ABCA1 led to an increase in lipidation of APOE. The lipidated APOE was discovered to facilitate the proteolytic degradation of Aβ by NEP and IDE and promote the clearance of Aβ in the hippocampus of male Wistar rats [104]. It has been reported that ABCA2 was associated with β/γ-secretase, which hydrolyzed APP in the N2a mouse neuroblastoma cells [105]. Another study showed that the overexpression of ABCA2 increased low-density lipoprotein receptor density, which decreased Aβ deposition in vivo [106]. ABCA5 can reduce Aβ production but does not promote Aβ clearance [107]. In ABCA7 haplodeficient microglia, Aikawa et al. [108] reported that proper endosomal-lysosomal trafficking might be disturbed, resulting in abnormal accumulation of Aβ. In addition, deletion of ABCA7 facilitated the processing of APP to Aβ through increasing the levels of BACE1 and SREBP2 in primary neurons and mouse brains. This study also demonstrated that knockdown of ABCA7 expression in neurons caused endoplasmic reticulum stress highlighted by increased levels of protein kinase R-like endoplasmic reticulum kinase (PERK) and increased phosphorylation of eukaryotic initiation factor 2α (eIF2α), resulting in cognitive decline [109]. Apart from targeting the Aβ hypothesis, lacking ABCA1 was found to result in an impaired hippocampal neurite morphology in APP/PS1 mice [110]. In summary, lipid substances and related regulators of lipid metabolism may be involved in the pathogenesis of AD by interacting with crucial mechanisms involved in the etiology of AD.
3.3. Amino acid metabolism and AD
As neurotransmitters in the central nervous system, amino acids are involved in the synthesis and metabolism of various active substances in the body and play important roles in learning, memory neurotransmission, and energy metabolism. Disorders of amino acid metabolism have also been reported in patients with AD [111].
3.3.1. Glutamate, γ-aminobutyric acid, glutamine, and branched-chain amino acids
Excitatory amino acids such as glutamate (Glu) have excitatory effects on neurons. Their release can damage neuronal cells and lead to other pathologies, including learning and memory impairment. Inhibitory amino acids, like γ-aminobutyric acid (GABA) have postsynaptic inhibitory effects and can protect neurons from damage.
Glutamine (Gln) is widely distributed in the brain and is the most abundant amino acid in cerebrospinal fluid. It plays a significant role in lipid and protein synthesis, nucleic acid synthesis, and autophagy [112]. The level of Gln in AD is decreased and is proven to have protective effects on AD [113]. For instance, Gln was found to protect against oxidative stress-induced injury in vitro and in vivo through activating the Wnt3a/β-catenin signaling pathway [114]. Besides, Gln is an important precursor of Glu and GABA. Following its entry into the cell through the transporter protein, Gln generates Glu under GLS or GLS2, followed by α-KG and GABA under the catalytic action of GLUD and GAD respectively. Under normal physiological conditions, Glu and GABA maintain the balance and normal function of the nervous system, but both are significantly decreased in AD [115, 116]. GABA reacts with α-KG to generate SSA and, ultimately, succinate, which constitutes the GABA shunt. The GABA shunt, a bypass of the TCA cycle, provides approximately 20% of the energy to the brain, thereby altering AD pathology to some extent. It has been documented that Glu promoted the polarization of M2 macrophage and reduced the inflammatory response through the α-KG-JMJD3 pathway [117]. Succinate is generated from Gln via GABA bypass. It has been demonstrated to stabilize hypoxia-inducible factor-1α (HIF-1α) by inhibiting prolyl hydroxylase or enhancing ROS enzyme activity, thereby promoting the release of inflammatory factors including IL-1β [118, 119]. However, these regulatory mechanisms have not yet been reported in AD. Considering that HIF-1α is associated with multiple pathological features of AD and microglia is an important macrophage in the brain, it is worth to investigate whether Gln in microglia can also influence the release of pro- and anti-inflammatory factors via the α-KG-JMJD3 or HIF-1α pathway to delay the development of AD.
Branched-chain amino acids (BCAAs) include leucine, isoleucine, and valine. The level of BCAAs is elevated in AD. A recent study has found that the supplementation of BCAAs increased phosphorylated tau in HT22 hippocampal neurons. The restriction of BCAAs effectively lowered amyloid and tau pathology in APP/PS1 mice. This study also reported that the application of BT2, a BCAAs-lowering compound, reduced Aβ42 and enhanced cortical and hippocampal neurotransmitter levels in five familial AD mutations mice [120]. However, the specific mechanisms by which BCAAs affect AD pathology need to be further studied. Additionally, BCAAs, especially leucine, play critical roles in the brain as key nitrogen donors for Glu synthesis [121], and Glu is a precursor of GABA. Consequently, disturbances in BCAAs levels may lead to a serious imbalance in these key neurotransmitters.
3.3.2. Serine and tryptophan
L-serine is a non-essential amino acid and is produced mainly in glial cells, especially astrocytes. L-serine is the obligatory precursor of D-serine, a co-agonist of synaptic NMDARs required for synaptic plasticity in the hippocampus. L-serine is approved by FDA for supplemental use, while D-serine is not. It has been found that impaired synaptic plasticity and memory in triple transgenic AD mice were rescued by the dietary supplement of L-serine [122]. A recent study reported that D-serine levels were elevated in the serum and cerebrospinal fluid of AD patients compared with controls [123] and Aβ oligomers might lead to elevated D-serine levels by upregulating serine racemase in cultured hippocampal neurons as well as APP/PS1 transgenic mice [124]. Reducing D-serine in the brain was shown to partially attenuate neuronal loss and reactive astrogliosis in AD mouse model [125]. However, Juliette et al. [122] discovered that the level of D-serine in the hippocampus was lower in AD mice than in control mice and D-serine supplement was found to rescue the spatial memory deficits. Another study showed that the concentrations of D-serine in the serum and cerebrospinal fluid were unaltered in AD patients [126]. It can be seen that the expression of D-serine in AD is controversial, and further studies are needed in the future to clarify the regulatory mechanisms of D-serine in AD progression.
Tryptophan is an essential amino acid whose metabolism is carried majorly through the kynurenine pathway. The kynurenine pathway is intricately linked to AD pathogenesis owing to the influence of kynurenine metabolites on amyloid aggregation, modulation of neuroinflammation, oxidative stress, and so on [127-129]. Recently, bioinformatics analysis identified five tryptophan metabolism-related genes associated with AD, including propionyl-CoA carboxylase subunit beta (PCCB), TEA domain transcription factor 1 (TEAD1), phenylalanyl-tRNA synthetase subunit beta (FARSB), neurofascin (NFASC), and ezrin (EZR). The expressions of PCCB and NFASC were down-regulated in the hippocampus of APP/PS1 mice [130], but the exact mechanisms have not yet been clarified.
In summary, glucose, lipids, and proteins, the three primary energy-providing components, play critical roles in the pathogenesis of AD (Table 4).
Table 4.
Energy metabolism and AD.
| Energy metabolism | Direction (→/←) | Affected pathways/genes/proteins | Pathologies/phenotypes in AD | Experimental subjects | References |
|---|---|---|---|---|---|
| Glucose metabolism | → | Glucose deprivation →Unfolded protein response↑ | Tau protein phosphorylation↑ | Hamsters, cells | [61] |
| → | Impaired glucose metabolism→PGC-1α↓→FoxO3a↑→APP processing (α- secretase) ↓ | Aβ generation↑ | Human brain tissues, cells | [62] | |
| → | AGEs→ RAGE- mediated AKT/GSK-3β pathway | Spatial memory deficit↑; Tau protein hyperphosphorylation↑ | Mice, cells | [68] | |
| → | Impaired insulin signaling→ GSK3 dephosphorylation | Aβ deposition and tau protein phosphorylation↑ | Human brain tissues | [71] | |
| → | Insulin →activation of ERK- mediated pathway →NEP, IDE↑; Insulin resistance →γ-secretase↑, IDE↓ | Aβ production↑ and Aβ clearance↑ | Cells, mice | [70, 72] | |
| → | Lack of GLUT1/3→Tau protein glycosylation↓→Tau protein phosphorylation↑ | NFT↑ | Human brain tissues, mice, and cells | [73] | |
| → | O-GlcNAcase insufficiency→O-GlcNAcylation↑→Necrotrophic apoptotic factor↓ and excessive activation of astrocytes and microglia | Neuronal death and neuroinflammation in the brain↓; Mitochondrial function↑, normal mitochondrial morphology↑, and Aβ deposition↓ | Human brain tissues, mice, and cells | [74] | |
| → | Ketone bodies→LRP1, P-glycoprotein transporter and PICALM↑→Aβ1-40 transendothelial transport↑ | Aβ clearance↑ | Cells | [76] | |
| → | ①βOHB→G protein-coupled receptor 109A→NEP↑; ②βOHB→BACE1↓; ①βOHB→NLRP3 inflammasome activation↓; ④βOHB→HDAC1/3↓→TrkA↑; ⑤βOHB↑ (increased by alternative-day-fasting) →PSD95↑, synapsin↑, Aβ oligomers↓, tau hyperphosphorylation↓ | ①Aβ degradation↑; ②Aβ generation↓; ①Aβ plague formation, microgliosis↓; ④Aβ-induced neurotoxicity↓; ⑤Synaptic plasticity↑, Aβ oligomers↓, tau hyperphosphorylation↓ | ①Cells; ②Mice; Cells; ①Human samples, mice, and cells; ④Cells; ⑤Mice | [77-81] | |
| ← | Aβ42→Oxidative stress → Impaired glucose metabolism → Synaptic dysfunction and neuronal death | Aβ42↑; Synaptic dysfunction and neuronal death↑ | Human brain tissues | [57] | |
| Lipid metabolism | → | APOE4→Non-canonical DLK→MKK7→ERK1/2 MAP-kinase cascade →APP↑ | Aβ synthesis↑ | Mice, cells | [84] |
| → | LRP1+APOE; LRP1+PICALM | Aβ clearance↑ | Human brain tissues, mice, and cells | [85, 86] | |
| → | APOE4→Activation of CDK5 signaling pathway | Tau protein phosphorylation↑ | Mice | [88] | |
| → | APOE4→IL-7/IL-7R signaling pathway↓ | Neuroinflammation↑ | Patients | [89] | |
| → | ①APOE4→ Phosphorylation of CREB and CaMKⅡα↓→Hippocampal LTP↓; ②APOE4→PP2B↑→Dendritic spine numbers↓; ①APOE4→PGC-1α-Sirtuin3 signaling pathway→ Synaptic damage↑ | Synaptic function↓, cognitive dysfunction↑ | ①Mice; ②Mice; ①Mice, cells | [93, 95, 250, 251] | |
| → | ①TREM2→Microglial phagocytosis↑→Aβ degradation↑; ②TREM2 knockdown→ Microglia activation and aggregation around Aβ plagues↓; ①TREM2 deficiency→ The microglial response to tau accumulation↓; ④TREM2→ The activation of classical complement cascade and complement-mediated synaptic loss↓ | ①Aβ degradation↑; ②Aβ plague↑; ①Tau accumulation↓; ④Synaptic function↓ | ①Mice, cells; ②Mice, cells; ①Mice; ④Human brain tissues, mice | [96, 98-100] | |
| → | Overexpression of SREBP2; Knockdown of SREBP2 | Oxidative damage, Aβ accumulation, neuroinflammation, and cognitive decline↑; Aβ and tau protein↓ | Mice, cells | [101, 102] | |
| ← | Oligomeric Aβ42→AKT inhibition→SREBP2 activation↓ | Oligomeric Aβ42 | Mice, cells | [103] | |
| → | ①ABCA1→Lipidation of APOE→ Proteolytic degradation of Aβ by NEP, IDE; ②ABCA2→β/γ-secretase; ①ABCA2 overexpression →Low-density lipoprotein receptor density↑; ④ABCA5 →Aβ production↓; ⑤ABCA7 knockout→Endosomal-lysosomal trafficking; ⑥ABCA7 deletion→SREBP2, BACE1↑; ABCA7 knockdown→ Endoplasmic reticulum stress→PERK-eIF2α pathway | Aβ clearance↑; Aβ production↓; Aβ deposition↑; Aβ accumulation↑; Cognitive function | ①Rats; ②Cells; ①Mice; ④Human brain tissues, mice, cells; ⑤Mice, cells; ⑥Mice, cells; Mice, cells | [104-109] | |
| → | ABCA1↓→impaired hippocampal neurite morphology | Cognitive function↓ | Mice | [110] | |
| ← | Aβ→PKA/ERK signaling pathway →Lipolysis; Aβ→Leptin and IL-6 secretion | Aβ↑ | Cells | [87] | |
| Amino acid metabolism | → | Gln→Wnt3a/β-catenin signaling pathway↑→SOD↑, glutathione peroxidase↑, malondialdehyde↓ | Oxidative stress-induced injury↓ | Mice, cells | [114] |
| → | BCAAs↓ →Amyloid and tau pathology↓, cortical and hippocampal neurotransmitter levels↑ | Aβ↓; Tau phosphorylation↓; Cognitive function↑ | Mice, cells | [120] | |
| → | L-serine↑→Stimulates the NMDAR | Synaptic plasticity and memory↑ | Human brain tissues, mice | [122] | |
| → | D-serine↓ →Neuronal loss and reactive astrogliosis↓; D-serine supplement →Spatial memory deficits↓ | Neuronal loss and reactive astrogliosis↓; Spatial memory deficits↓ | Mice; Human brain tissues, mice | [122, 125] | |
| → | Tryptophan →Kynurenine pathway (kynurenine metabolites) | Aβ aggregation; Modulation of neuroinflammation; Oxidative stress | Elderly individuals; Human brain tissues, cells; Rats | [127-129] | |
| → | Tryptophan metabolism-related genes (PCCB, TEAD1, FARSB, NFASC, EZR) | None | Mice | [130] | |
| ← | Aβ oligomers→ serine racemase↑→D-serine↑ | Aβ oligomers↑ | Mice, cells | [124] |
Note: “→” denotes the role of energy metabolism on AD; “←” denotes the role of AD on energy metabolism; “↑” denotes increased levels; “↓” denotes reduced levels.
3.4. Mitochondrial dysfunction
Mitochondria are the major organelles involved in energy metabolism, where metabolic activities, such as glucolipid metabolism, amino acid metabolism, and the TCA cycle interact with each other. Mitochondrial biosynthesis, dynamics, autophagy, and ROS production mediated by apoptosis affect the maintenance of mitochondrial homeostasis. In addition, the mitochondrial cascade hypothesis proposes that mitochondrial dysfunction triggers AD pathology and may lead to neurodegenerative lesions, such as Aβ deposition, NFTs, synaptic loss, and axonal transport defects.
Mitochondrial homeostasis is achieved through continuous division and fusion. In AD, inhibition of dynamin-related protein 1 is associated with loss of neuronal synapse and dendritic spine, defection of axonal transport, Aβ deposition, and tau protein hyperphosphorylation [131, 132]. Overexpression of mitochondrial fusion proteins including optic atrophy 1 was demonstrated to alleviate the overproduction of ROS in the mitochondria of AD brains [133]. On the contrary, Aβ deposition and tau hyperphosphorylation affect mitochondria, leading to mitochondrial fragmentation and interfering with energy metabolism [134, 135]. In addition to kinetic abnormalities, mitochondrial biosynthesis and mitophagy were impaired in human AD brain samples, AD-induced pluripotent stem cell-derived neurons, and transgenic animal models of AD. Defects in mitophagy induce the accumulation of dysfunctional mitochondria, thereby promoting AD pathology [136].
Essential enzymes and intermediate metabolites of the TCA cycle are closely associated with AD development. The activities of key enzymes, such as IDH, SDH, and pyruvate dehydrogenase, are significantly decreased in AD, affecting ATP production and energy metabolism [137]. Moreover, the antioxidant function of the organism gradually decreases with age, resulting in excessive production of ROS, which attacks ACO2 during isocitric acid synthesis, thus affecting mitochondrial function [138]. Intermediate metabolites enter the cytoplasm via carriers on the inner mitochondrial membrane. These metabolites participate in mitochondria-to-nucleus retrograde signaling, which regulates AD by influencing mitochondrial homeostasis.
4. Energy metabolism and histone methylation
Of note, multiple studies have reported crosstalk between energy metabolism and histone methylation.
4.1. Energy metabolism affects histone methylation
In energy metabolism, particularly in the TCA cycle, FAD acts as a coenzyme for the conversion of succinate to fumarate. LSD1 is a FAD-dependent demethylase that catalyzes the demethylation of H3K4me1/2 and H3K9me1/2 [139]. The catalytic activity of LSD1 may be directly related to the cellular metabolic state via fluctuations in the FAD/FADH2 (the reduced form of FAD) ratio, depending on FAD oxidation processes such as the TCA cycle [140]. Moreover, as a crucial intermediate in the TCA cycle, α-KG not only plays a role in DNA demethylation but also serves as a co-factor in histone demethylases of the JMJD family. This intermediate can be supplied directly or indirectly by mitochondria and affects the epigenetic regulation of the nuclear genome. When mitochondrial metabolism is disturbed, α-KG decreases histone methylation, whereas succinate and fumarate may exert opposite effects [141]. Obesity, caused by lipid metabolism disorders, is one of the main causes of AD pathogenesis. Adipose tissues include white adipose tissue, brown adipose tissue, and beige adipose tissue. Promoting the thermogenic function of brown adipose tissue and browning of beige adipose tissue in white adipose tissue can reduce obesity. Peroxisome proliferator-activated receptor γ (PPARγ) belongs to one of the intranuclear hormone receptor superfamily members. PPARγ and other receptors from this family such as PPARα, play important roles in adipocyte production and energy metabolisms. There is evidence that uncoupling protein 1 (UCP1) is the center of brown adipose tissue thermogenesis and systemic energy homeostasis. It was found that 1% α-KG dissolved in drinking water promoted the formation of beige fat and prevented obesity in middle-aged mice [142]. In addition, IDH1 is an essential metabolic enzyme that catalyzes the conversion of isocitric acid to α-KG. Chromatin immunoprecipitation analysis revealed that IDH1-mediated increase of α-KG reduced the level of H3K4me3 in the promoters of brown adipogenic genes, including PPARγ and UCP1, which was accompanied by their decreased expression in vivo and in vitro, respectively [143]. However, whether α-KG may affect AD-like pathologies by regulating the expression of genes related to brown adipogenesis, such as PPARγ and UCP1, needs to be further explored.
In addition to being influenced by related enzyme activities and intermediate metabolites, histone methylation is affected by changes in SAM levels. SAM is produced through the coupling of folate and methionine cycles in the cytoplasm, enters the nucleus as a methyl source for histones, and participates in histone methylation. Thus, SAM levels are regulated by methionine. When methionine levels are deficient, SAM levels decrease, which subsequently affects the transcription of genes related to Aβ processing, nerve growth factor, and epigenetic modifications [144]. Additionally, mitochondrial bifunctional enzymes can use methylenetetrahydrofolate to synthesize formyltetra-hydrofolate, while consuming one-carbon groups during purine biosynthesis. When mitochondrial bifunctional enzymes are not expressed during purine biosynthesis, the consumption of one-carbon groups is decreased. Accordingly, the synthesis of serine increases, whereas excess serine in the cytoplasm generates cysteine, which reacts with ATP to synthesize SAM, ultimately increasing histone methylation levels [145].
Studies have found that βOHB induced a decrease in the H3K27me3 as well as an increase in the H3K4me3 occupied in BDNF promoters and both were dependent on the activation of cAMP/PKA signaling pathways in the primary hippocampal neurons and the murine hippocampal neuronal cell line HT22 [146, 147]. PR/SET domain containing protein 9 (PRDM9) is a histone methyltransferase that can tri-methylate H3K4 and H3K36 at surrounding nucleosomes. Gln was demonstrated to promote transcriptional induction of brown adipogenic and thermogenic genes, such as PPARγ and UCP1, through C/EBPβ-PRDM9-mediated H3K4me3 in vivo and in vitro models [148].
4.2. Histone methylation affects energy metabolism
SETD2 is a methyltransferase that regulates H3K36me3 levels. A recent study reported that SETD2 deficiency inhibited the expression of cholesterol efflux genes such as ABCA1, resulting in lipid accumulation in vivo and in vitro [144]. It has been shown that PPARγ can be inhibited by G9a and EZH2 and activated by MLL3/4 [149-151]. Another study has shown that JHDM2A played an essential role in directly regulating PPARα and UCP1 in vivo and in vitro experiments, two of the important genes involved in controlling energy balance [152]. LSD1 mediates demethylation of H3K9me1 and H3K9me2. Sambeat et al. [153] observed that LSD1 was recruited to the UCP1 promoter by directly interacting with zinc finger protein 516 for transcriptional activation by H3K9 demethylation at the UCP1 promoter region, which promoted the function of thermogenic adipose tissue. The knockdown of PHF2 was found to result in a reduction in the expression of major genes related to fat cell differentiation, including PPARγ and CCAAT/enhancer binding protein α (C/EBPα) in vitro [154]. PR domain-containing 16 (PRDM16) is a dominant activator of the biogenesis of beige adipocytes and an attractive target for improving metabolic health. Wang et al. [155] found that EHMT1 stabilized PRDM16 protein by competing with amyloid protein-binding protein 2 for direct binding to PRDM16 and blocking PRDM16 polyubiquitination, thereby promoting beige adipocyte biogenesis.
The administrations of PPARγ and PPARα agonists can decrease amyloid levels, reduce inflammation, and ameliorate synaptic function [156-159]. It has been found that the deletion of UCP1 in Tg2576 mice increased body temperature, which elevated Aβ and tau levels and decreased synaptic protein levels [160]. However, whether PPARα, PPARγ, and UCP1 mediate the regulation of AD-related pathologies by G9a, EZH2, MLL3/4, JHDM2A, LSD1, PHF2, and EHMT1 need to be further explored.
SREBP1 and SREBP2 are responsible for regulating fatty acid and cholesterol synthesis, respectively [161]. In vivo and in vitro models, inhibition of DOT1L was found to lead to suppressed SREBP1/2 lipid synthetic pathways that consequently enhanced macrophage inflammatory activation [162]. A recent study has demonstrated that SETD8 modulated SREBP1 by mono-methylation of H4K20 to promote the lipogenesis of renal cell carcinoma [163]. Tang et al. [164] observed that UTX promoted prostate tumorigenesis and lipid metabolism by binding to the promoter of SREBP1c, one of the isoforms of SREBP1, to increase SREBP1c transcription in vivo and in vitro experiments. Besides, ligand-activated liver X receptors (LXRs) are transcription factors that play an essential role in promoting lipogenesis. The activation of LXRα, one of the subtypes of LXRs, was discovered to increase JMJD2B recruitment to LXR response elements and reduce H3K9me2/3 enrichment in the promoter of LXRα-dependent lipogenic genes such as SREBP1 [165]. The relationship between SREBP2 and AD has been explained previously, and the knockdown of SREBP1 has been reported to reduce the palmitate-induced increase in BACE1 expression and subsequent Aβ genesis in the mouse hippocampus and mouse Neuro-2a neuroblastoma cells [166]. However, it remains unknown whether DOT1L, SETD8, UTX, and JMJD2B affect AD-related pathologies via SREBP1 or SREBP2.
In addition, pancreatic duodenal homeobox 1, an essential transcription factor for pancreatic differentiation and development, can promote the expression of insulin, glucokinase, and other important genes in pancreatic β-cells. Studies have shown that pancreatic duodenal homeobox 1 recruited SET7/9, which methylated H3K4 in the promoter region of insulin secretion-related genes, such as β-cell-specific transcription factor MafA, the glucose transporter Glut2 (Slc2a2), and preproinsulin (Ins1/2) [167, 168]. Insulin can cross the BBB, bind to the insulin receptor (IR) which are mainly located in the olfactory bulbs, hypothalamus, cerebral cortex, and hippocampus [169], and lead to IR autophosphorylation. The phosphorylated IR then phosphorylates the insulin receptor substrate. The activated insulin receptor substrate not only regulates glucose metabolism, but also inhibits GSK-3α and GSK-3β to regulate APP expression, Aβ clearance, and tau protein phosphorylation levels through the PI3K/AKT pathway [170-172]. AMP-activated protein kinase is a vital energy sensor, and its activation comes with metabolic benefits. Sirtuin 3 plays an important role in maintaining mitochondrial function and energy metabolism. A previous study has found that the overexpression of KDM5B triggered mitochondrial glucose and lipid metabolism disorder and oxidative stress via transcriptional inhibition of sirtuin 3 expression by demethylating H3K4me3 or inactivation of AMP-activated protein kinase/sirtuin 3 pathways in the models of diabetic peripheral neuropathy model [173]. Diabetic peripheral neuropathy is one of the most common diabetes-related complications and diabetes is proven to be a risk factor for the development of AD, but whether this regulatory mechanism exists in AD needs further explored.
In conclusion, a complex connection exists between energy metabolism and histone methylation (Table 5), both of which are critical for the development of AD. However, the underlying mechanisms remain unknown. Therefore, understanding the relationship between energy metabolism and histone methylation is important for AD.
Table 5.
Energy metabolism and histone methylation.
| Energy metabolism | Direction (→/←) | KMTs/KDMs | Experimental subjects | References |
|---|---|---|---|---|
| FAD | → | LSD1 (FAD-dependent demethylase) demethylation of H3K4me1/2 and H3K9me1/2 | None | [139] |
| α-KG | → | JMJD family (α-KG-dependent demethylase) | None | [141] |
| IDH1 | → | H3K4me3 level in the promoters of PPARγ and UCP1↓ | Mice, cells | [143] |
| SAM | → | Methyl donor for HMTs | None | [145] |
| βOHB | → | cAMP/PKA signaling pathways→BDNF promoters (H3K27me3↓, H3K4me3↑) | Mice, cells | [146, 147] |
| Gln | → | C/EBPβ/PRDM9 pathways→Brown adipogenic and thermogenic genes↑ | Human tissues, mice, and cells | [148] |
| ABCA1↓→lipid accumulation | ← | SETD2 deficiency | Mice, cells | [144] |
| PPARγ | ← | Inhibited by EHMT2 (G9a) and EZH2; Activated by MLL3/4 | Cells; Mice, cells; Mice | [149-151] |
| PPARα and UCP1↑ | ← | JHDM2A | Mice, cells | [152] |
| UCP1 promoter (H3K9 demethylation) | ← | LSD1+ zinc finger protein 516 | Mice, cells | [153] |
| PPARγ, C/EBPα↓ | ← | PHF2 knockdown | Cells | [154] |
| Beige adipocyte biogenesis↑ | ← | EHMT1 (stabilizes PRDM16) | Mice, cells | [155] |
| ①SREBP1/2 lipid synthetic pathways↓; ②SREBP1 (H4K20me); ①SREBP1c | ← | ①DOT1L; ②SETD8; ①UTX | ①Mice, cells; ②Human tissues, cells; ①Mice, cells | [162-164] |
| SREBP1 (H3K9me2/3) | ← | JMJD2B (recruited by LXRα) | Mice, cells | [165] |
| MafA, Slc2a2, and Ins1/2↑ | ← | SET7/9 (recruited by pancreatic duodenal homeobox 1) | Mice, cells | [167, 168] |
| Mitochondrial glucose and lipid metabolism disorder and oxidative stress (Sirtuin 3 expression↓ (H3K4me3) or AMP-activated protein kinase/Sirtuin 3 pathways↓) | ← | KDM5B | Mice, cells | [173] |
Note: “→” denotes the role of energy metabolism on AD; “←” denotes the role of AD on energy metabolism; “↑” denotes increased levels; “↓” denotes reduced levels.
5. Discussion
AD is a chronic neurodegenerative disorder, and its complexity makes its cure an unsolved problem for the time being. Currently, as for drugs for AD treatment, anticholinesterase inhibitors, N-methyl-D-aspartate receptor antagonists [174], and antibodies against Aβ such as aducanumab and lecanemab [175], have been approved by FDA. However, there are many limitations in the application of these drugs. Therefore, it is urgent to develop new treatments that can prevent or slow AD progression, for example, the inhibitors of KMTs and KDMs. In addition to medicine, environmental factors such as diet, physical activities, and stress, are all closely related to the development of AD. This review mainly focuses on the correlation between histone methylation, energy metabolism, and AD, so the search for dietary and lifestyle interventions that affect histone methylation or energy metabolism can be beneficial in advancing the translation of research findings into the clinical arena. For instance, curcumin was demonstrated to upregulate JMJD3 and reduce the level of H3K27me3 in the BDNF promoter region, improving mitochondrial function and reducing the level of Aβ [53]. Ascorbic acid serves as a potential coenzyme for specific histone demethylases of the JMJD family [176]. It was reported to have therapeutic benefits in addressing age-related diseases, such as AD [177, 178]. Sadli et al. [179] found that docosahexaenoic acid decreased concentrations of di-methylation of H3K4, H3K9, H3K27, H3K36, and H3K79 in M17 human neuronal cells. However, further research is necessary to determine whether ascorbic acid and docosahexaenoic acid have therapeutic effects on AD through histone methylation. Additionally, a recent study has discovered that oral supplementation with a multi-strain probiotic formulation reduced Aβ aggregates and brain damage by affecting lipid metabolism in a triple transgenic AD mouse model [180]. Reducing folate and vitamin B12 in neuroblastoma cell lines might also lead to a decrease in SAM levels, thereby increasing PS1 and BACE levels and Aβ production [181]. Apart from folate and vitamin B12, some micronutrients such as choline, methionine, and betaine, are essential for the generation of SAM, and they are proven to have important roles in ameliorating AD-like pathologies [182-184]. Therefore, increasing the amount of the above nutrients in the diet can be effective in preventing AD to some extent. Histone methylation can be dynamically regulated by stressors, including physical and psychological stress. A study using SAMP8 animals showed that chronic mild stress treatment increased H3K9 methylation [185]. Chronic social defeat stress in adult mice was found to significantly increase the levels of the H3K27me3 at the promoter of BDNF which is associated with synaptic plasticity and learning [186]. In terms of physical activities, exercise is associated with a lower risk for AD mortality. A previous study using APP/PS1 mice demonstrated that regular exercise alleviated cognitive impairment via increasing brain regional GLUT1- and GLUT3-mediated glucose metabolism [187]. Running exercise was reported to inhibit TREM2 shedding and maintain TREM2 protein levels, which were accompanied by the promotion of brain glucose metabolism, microglial glucose metabolism, and morphological plasticity in the hippocampus of AD mice [188]. Besides, Ying et al. [189] observed that amino acid metabolism was affected in the cortex and hypothalamus after high-intensity interval training in APP/PS1 mice. However, the relationship between lifestyle changes and histone methylation has not yet been clearly reported and needs to be further explored. Therefore, keeping a good diet, having regular exercises, and reducing stress can help to prevent and treat patients with AD.
6. Conclusion, limitations, and future directions
In this review, we have integrated recent studies on histone methylation, energy metabolism, and AD to better understand the relationship between the three of them (Fig. 3). Histone methylation, energy metabolism, and AD are closely associated with each other. Abnormal energy metabolism, especially in mitochondria, affects histone methylation by regulating the activities of relevant enzymes and the metabolism of intermediates. These enzymes and their intermediates promote or inhibit the expression of related genes, resulting in AD phenotypes. Various factors contributing to the pathological changes in AD, such as hypoxia, high ROS levels, calcium overload, and the pathological characteristics of AD itself, have been found to affect energy metabolism. Meanwhile, histone methylation regulates energy metabolism in the body. The interplay between histone methylation, energy metabolism, and AD is intricate and bidirectional. Further studies on their interactions may provide new insights into elucidating the pathogenesis of AD and controlling its progression.
Figure 3.

The relationship and regulatory mechanisms of histone methylation, energy metabolism, and AD. Note: The content not reported in AD is marked in red.
However, there are some limitations remaining in this review. First, this review mainly focuses on the relationship between histone methylation and AD, but does not consider the effects of other types of histone modifications and the interactions of different types of histone modifications on pathological changes in AD. Second, in terms of AD treatment, this review concentrates on medication, diet, and lifestyle interventions targeting histone methylation and energy metabolism, however, the potential therapeutic targets of other epigenetic modification types have not been taken into consideration. Moreover, the studies included in the review are mainly based on in vivo and in vitro experiments with a low quality of evidence, so the specific roles of related indicators of histone methylation or energy metabolism in the brains of AD patients remain further explored.
Therefore, based on this review, there is still a lot of work to be done in the search for biomarkers, the innovation of mechanisms as well as experimental techniques and interventions, and the translation of research results in AD. Firstly, more in-depth studies on the effects of interactions between different epigenetic modifications, such as DNA methylation, histone modifications, and non-coding RNAs, on AD-related genes and signaling pathways. Secondly, the integration of multi-genomic approaches, such as genomics, transcriptomics, and proteomics with epigenetics, helps advance our understanding of AD. In recent years, with the advance of genetics, biochemistry, imaging and other disciplines, the combination of multiple disciplines can effectively improve the accuracy of early diagnosis of AD. For example, a research team used emerging technologies, such as artificial intelligence, to analyze CSF proteomics, and then identified novel biomarkers of important clinical value, which improves diagnostic accuracy and provides the possibility of early and precise diagnosis of AD [190]. Researchers have also found that the combination of participants’ electroencephalogram biomarkers, demographic information, CSF biomarkers, and APOE phenotype are effective in assessing individual cognitive function and disease progression [191]. In terms of treatment, advanced scientific methods, such as central targeting technology, nano drug delivery system, and deep brain stimulation, are used to overcome the difficulty of drugs penetrating and reaching the brain tissue and to control drug-resistant symptoms, so as to improve the therapeutic effect of drugs. Combining different types of therapies may provide a synergistic effect. For example, the integration of histone methylation or energy metabolism with conventional therapies, such as anti-amyloid treatments, and the combined therapies between epigenetic modifications and energy metabolism. It has also been suggested that a tight interlink between nutritional counseling, supervised physical training, oral health, and cognitive-oriented communication training would be beneficial in maintaining quality of life in AD patients [192]. Besides, attention has been given to the potential therapeutic targeting of other types of epigenetic modifications. More importantly, because the human organism exhibits higher complexity in answering to external factors, it is not easy to translate the findings from animal models into human interventions. Therefore, some of the findings mentioned in this review need to be confirmed by further clinical trials to reduce bias and enhance the reliability and credibility of the studies.
In conclusion, AD is the most common type of dementia. The pathogenesis of AD is influenced by multiple risk factors and is the result of the combined effects of multiple genes, multiple epigenetic modifications mechanisms, multiple energy metabolism pathways, and different brain regions. According to the latest report, effective invention on 14 risk factors, such as obesity, diabetes, air pollution, and lack of physical activities, may prevent or delay the occurrence of dementia by 45% [193]. Consequently, strengthening the communication and cooperation among various disciplines, exploring the mechanisms deeply by which risk factors affect AD, and adopting corresponding interventions to delay and prevent the occurrence of AD will be of great potential benefit to the aging population.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant number: 82173518).
Funding Statement
This work was supported by the National Natural Science Foundation of China (Grant number: 82173518).
Author contributions
Jiaqi Fu: conceptualization, writing - original draft; Li An: conceptualization, writing - review & editing, supervision.
Declaration of Interest Statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Collaborators. GDF (2022). Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health, 7:e105-e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rosario ER, Chang L, Stanczyk FZ, Pike CJ (2004). Age-related testosterone depletion and the development of Alzheimer disease. Jama, 292:1431-1432. [DOI] [PubMed] [Google Scholar]
- [3].Migliore L, Coppedè F (2022). Gene-environment interactions in Alzheimer disease: the emerging role of epigenetics. Nat Rev Neurol, 18:643-660. [DOI] [PubMed] [Google Scholar]
- [4].López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G (2023). Hallmarks of aging: An expanding universe. Cell, 186:243-278. [DOI] [PubMed] [Google Scholar]
- [5].Millán-Zambrano G, Burton A, Bannister AJ, Schneider R (2022). Histone post-translational modifications - cause and consequence of genome function. Nat Rev Genet, 23:563-580. [DOI] [PubMed] [Google Scholar]
- [6].Yuan W, Wu T, Fu H, Dai C, Wu H, Liu N, et al. (2012). Dense chromatin activates Polycomb repressive complex 2 to regulate H3 lysine 27 methylation. Science, 337:971-975. [DOI] [PubMed] [Google Scholar]
- [7].Tachibana M, Ueda J, Fukuda M, Takeda N, Ohta T, Iwanari H, et al. (2005). Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev, 19:815-826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ng TKS, Ho CSH, Tam WWS, Kua EH, Ho RC (2019). Decreased Serum Brain-Derived Neurotrophic Factor (BDNF) Levels in Patients with Alzheimer's Disease (AD): A Systematic Review and Meta-Analysis. Int J Mol Sci, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Piancatelli D, Aureli A, Sebastiani P, Colanardi A, Del Beato T, Del Cane L, et al. (2022). Gene- and Gender-Related Decrease in Serum BDNF Levels in Alzheimer's Disease. Int J Mol Sci, 23:14599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sharma M, Dierkes T, Sajikumar S (2017). Epigenetic regulation by G9a/GLP complex ameliorates amyloid-beta 1-42 induced deficits in long-term plasticity and synaptic tagging/capture in hippocampal pyramidal neurons. Aging Cell, 16:1062-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zheng Y, Liu A, Wang ZJ, Cao Q, Wang W, Lin L, et al. (2019). Inhibition of EHMT1/2 rescues synaptic and cognitive functions for Alzheimer's disease. Brain, 142:787-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wang Z, Yang D, Zhang X, Li T, Li J, Tang Y, et al. (2011). Hypoxia-induced down-regulation of neprilysin by histone modification in mouse primary cortical and hippocampal neurons. PLoS One, 6:e19229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang W, Cao Q, Tan T, Yang F, Williams JB, Yan Z (2021). Epigenetic treatment of behavioral and physiological deficits in a tauopathy mouse model. Aging Cell, 20:e13456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Guedes-Dias P, Holzbaur ELF (2019). Axonal transport: Driving synaptic function. Science, 366:eaaw9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mauvezin C, Orpinell M, Francis VA, Mansilla F, Duran J, Ribas V, et al. (2010). The nuclear cofactor DOR regulates autophagy in mammalian and Drosophila cells. EMBO Rep, 11:37-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mauthe M, Jacob A, Freiberger S, Hentschel K, Stierhof YD, Codogno P, et al. (2011). Resveratrol-mediated autophagy requires WIPI-1-regulated LC3 lipidation in the absence of induced phagophore formation. Autophagy, 7:1448-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Proikas-Cezanne T, Robenek H (2011). Freeze-fracture replica immunolabelling reveals human WIPI-1 and WIPI-2 as membrane proteins of autophagosomes. J Cell Mol Med, 15:2007-2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Artal-Martinez de Narvajas A, Gomez TS, Zhang JS, Mann AO, Taoda Y, Gorman JA, et al. (2013). Epigenetic regulation of autophagy by the methyltransferase G9a. Mol Cell Biol, 33:3983-3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Griñán-Ferré C, Marsal-García L, Bellver-Sanchis A, Kondengaden SM, Turga RC, Vázquez S, et al. (2019). Pharmacological inhibition of G9a/GLP restores cognition and reduces oxidative stress, neuroinflammation and β-Amyloid plaques in an early-onset Alzheimer's disease mouse model. Aging (Albany NY), 11:11591-11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Qi C, Liu S, Qin R, Zhang Y, Wang G, Shang Y, et al. (2014). Coordinated regulation of dendrite arborization by epigenetic factors CDYL and EZH2. J Neurosci, 34:4494-4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Henriquez B, Bustos FJ, Aguilar R, Becerra A, Simon F, Montecino M, et al. (2013). Ezh1 and Ezh2 differentially regulate PSD-95 gene transcription in developing hippocampal neurons. Mol Cell Neurosci, 57:130-143. [DOI] [PubMed] [Google Scholar]
- [22].Kerimoglu C, Sakib MS, Jain G, Benito E, Burkhardt S, Capece V, et al. (2017). KMT2A and KMT2B Mediate Memory Function by Affecting Distinct Genomic Regions. Cell Rep, 20:538-548. [DOI] [PubMed] [Google Scholar]
- [23].Cao Q, Wang W, Williams JB, Yang F, Wang ZJ, Yan Z (2020). Targeting histone K4 trimethylation for treatment of cognitive and synaptic deficits in mouse models of Alzheimer's disease. Sci Adv, 6:eabc8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lian B, Liu M, Lan Z, Sun T, Meng Z, Chang Q, et al. (2020). Hippocampal overexpression of SGK1 ameliorates spatial memory, rescues Abeta pathology and actin cytoskeleton polymerization in middle-aged APP/PS1 mice. Behav Brain Res, 383:112503. [DOI] [PubMed] [Google Scholar]
- [25].Qin X, Wang Y, Paudel HK (2017). Inhibition of Early Growth Response 1 in the Hippocampus Alleviates Neuropathology and Improves Cognition in an Alzheimer Model with Plaques and Tangles. Am J Pathol, 187:1828-1847. [DOI] [PubMed] [Google Scholar]
- [26].Zhao X, Wang X, Su G, Sun Q, Fu J, Zhang H, et al. (2019). The effect of early growth response 1 on levels of Amyloid-beta 40 peptide in U87MG cells. J Cell Biochem, 120:3514-3519. [DOI] [PubMed] [Google Scholar]
- [27].Koldamova R, Schug J, Lefterova M, Cronican AA, Fitz NF, Davenport FA, et al. (2014). Genome-wide approaches reveal EGR1-controlled regulatory networks associated with neurodegeneration. Neurobiol Dis, 63:107-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cho C, MacDonald R, Shang J, Cho MJ, Chalifour LE, Paudel HK (2017). Early growth response-1-mediated down-regulation of drebrin correlates with loss of dendritic spines. J Neurochem, 142:56-73. [DOI] [PubMed] [Google Scholar]
- [29].Lee J, Hwang YJ, Kim Y, Lee MY, Hyeon SJ, Lee S, et al. (2017). Remodeling of heterochromatin structure slows neuropathological progression and prolongs survival in an animal model of Huntington's disease. Acta Neuropathol, 134:729-748. [DOI] [PubMed] [Google Scholar]
- [30].Hwang YJ, Hyeon SJ, Kim Y, Lim S, Lee MY, Kim J, et al. (2021). Modulation of SETDB1 activity by APQ ameliorates heterochromatin condensation, motor function, and neuropathology in a Huntington's disease mouse model. J Enzyme Inhib Med Chem, 36:856-868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Williams JB, Cao Q, Wang W, Lee YH, Qin L, Zhong P, et al. (2023). Inhibition of histone methyltransferase Smyd3 rescues NMDAR and cognitive deficits in a tauopathy mouse model. Nat Commun, 14:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bichmann M, Prat Oriol N, Ercan-Herbst E, Schöndorf DC, Gomez Ramos B, Schwärzler V, et al. (2021). SETD7-mediated monomethylation is enriched on soluble Tau in Alzheimer's disease. Mol Neurodegener, 16:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Snigdha S, Prieto GA, Petrosyan A, Loertscher BM, Dieskau AP, Overman LE, et al. (2016). H3K9me3 Inhibition Improves Memory, Promotes Spine Formation, and Increases BDNF Levels in the Aged Hippocampus. J Neurosci, 36:3611-3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang Y, Liu X, Xue H, Liu X, Dai A, Song Y, et al. (2016). Upregulation of PRDM5 Is Associated with Astrocyte Proliferation and Neuronal Apoptosis Caused by Lipopolysaccharide. J Mol Neurosci, 59:146-157. [DOI] [PubMed] [Google Scholar]
- [35].Weiss MC, Agulnik M (2020). Tazemetostat as a treatment for epithelioid sarcoma. Expert Opinion on Orphan Drugs, 8:311-315. [Google Scholar]
- [36].Yamagishi M, Kuze Y, Kobayashi S, Nakashima M, Morishima S, Kawamata T, et al. (2024). Mechanisms of action and resistance in histone methylation-targeted therapy. Nature, 627:221-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Stein EM, Garcia-Manero G, Rizzieri DA, Tibes R, Berdeja JG, Savona MR, et al. (2018). The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood, 131:2661-2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Engstrom AK, Walker AC, Moudgal RA, Myrick DA, Kyle SM, Bai Y, et al. (2020). The inhibition of LSD1 via sequestration contributes to tau-mediated neurodegeneration. Proc Natl Acad Sci U S A, 117:29133-29143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Longaretti A, Forastieri C, Toffolo E, Caffino L, Locarno A, Miseviciute I, et al. (2020). LSD1 is an environmental stress-sensitive negative modulator of the glutamatergic synapse. Neurobiol Stress, 13:100280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mehndiratta S, Liou JP (2020). Histone lysine specific demethylase 1 inhibitors. RSC Med Chem, 11:969-981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Shen L, Wang B, Wang SP, Ji SK, Fu MJ, Wang SW, et al. (2024). Combination Therapy and Dual-Target Inhibitors Based on LSD1: New Emerging Tools in Cancer Therapy. J Med Chem, 67:922-951. [DOI] [PubMed] [Google Scholar]
- [42].Maes T, Mascaró C, Rotllant D, Lufino MMP, Estiarte A, Guibourt N, et al. (2020). Modulation of KDM1A with vafidemstat rescues memory deficit and behavioral alterations. PLoS One, 15:e0233468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Labonne JD, Lee KH, Iwase S, Kong IK, Diamond MP, Layman LC, et al. (2016). An atypical 12q24.31 microdeletion implicates six genes including a histone demethylase KDM2B and a histone methyltransferase SETD1B in syndromic intellectual disability. Hum Genet, 135:757-771. [DOI] [PubMed] [Google Scholar]
- [44].Kawakami E, Tokunaga A, Ozawa M, Sakamoto R, Yoshida N (2015). The histone demethylase Fbxl11/Kdm2a plays an essential role in embryonic development by repressing cell-cycle regulators. Mech Dev, 135:31-42. [DOI] [PubMed] [Google Scholar]
- [45].Hong X, Xu Y, Qiu X, Zhu Y, Feng X, Ding Z, et al. (2016). MiR-448 promotes glycolytic metabolism of gastric cancer by downregulating KDM2B. Oncotarget, 7:22092-22102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kurt IC, Sur I, Kaya E, Cingoz A, Kazancioglu S, Kahya Z, et al. (2017). KDM2B, an H3K36-specific demethylase, regulates apoptotic response of GBM cells to TRAIL. Cell Death Dis, 8:e2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chavdoula E, Anastas V, La Ferlita A, Aldana J, Carota G, Spampinato M, et al. (2024). Transcriptional regulation of amino acid metabolism by KDM2B, in the context of ncPRC1.1 and in concert with MYC and ATF4. Metabolism, 150:155719. [DOI] [PubMed] [Google Scholar]
- [48].De Jager PL, Srivastava G, Lunnon K, Burgess J, Schalkwyk LC, Yu L, et al. (2014). Alzheimer's disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci, 17:1156-1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Jiang S, Yang W, Qiu Y, Chen HZ (2015). Identification of novel quantitative traits-associated susceptibility loci for APOE ε 4 non-carriers of Alzheimer's disease. Curr Alzheimer Res, 12:218-227. [DOI] [PubMed] [Google Scholar]
- [50].Park SY, Seo J, Chun YS (2019). Targeted Downregulation of kdm4a Ameliorates Tau-engendered Defects in Drosophila melanogaster. J Korean Med Sci, 34:e225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Griñán-Ferré C, Sarroca S, Ivanova A, Puigoriol-Illamola D, Aguado F, Camins A, et al. (2016). Epigenetic mechanisms underlying cognitive impairment and Alzheimer disease hallmarks in 5XFAD mice. Aging (Albany NY), 8:664-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Park DH, Hong SJ, Salinas RD, Liu SJ, Sun SW, Sgualdino J, et al. (2014). Activation of neuronal gene expression by the JMJD3 demethylase is required for postnatal and adult brain neurogenesis. Cell Rep, 8:1290-1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Li J, Wang S, Zhang S, Cheng D, Yang X, Wang Y, et al. (2021). Curcumin slows the progression of Alzheimer's disease by modulating mitochondrial stress responses via JMJD3-H3K27me3-BDNF axis. Am J Transl Res, 13:13380-13393. [PMC free article] [PubMed] [Google Scholar]
- [54].Jin C, Li J, Green CD, Yu X, Tang X, Han D, et al. (2011). Histone demethylase UTX-1 regulates C. elegans life span by targeting the insulin/IGF-1 signaling pathway. Cell Metab, 14:161-172. [DOI] [PubMed] [Google Scholar]
- [55].Chen X, Wang S, Zhou Y, Han Y, Li S, Xu Q, et al. (2018). Phf8 histone demethylase deficiency causes cognitive impairments through the mTOR pathway. Nat Commun, 9:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Nativio R, Lan Y, Donahue G, Sidoli S, Berson A, Srinivasan AR, et al. (2020). An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer's disease. Nat Genet, 52:1024-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Butterfield DA, Halliwell B (2019). Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci, 20:148-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Nishizaki T, Kammesheidt A, Sumikawa K, Asada T, Okada Y (1995). A sodium- and energy-dependent glucose transporter with similarities to SGLT1-2 is expressed in bovine cortical vessels. Neurosci Res, 22:13-22. [DOI] [PubMed] [Google Scholar]
- [59].Kyrtata N, Emsley HCA, Sparasci O, Parkes LM, Dickie BR (2021). A Systematic Review of Glucose Transport Alterations in Alzheimer's Disease. Front Neurosci, 15:626636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Cunnane SC, Trushina E, Morland C, Prigione A, Casadesus G, Andrews ZB, et al. (2020). Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat Rev Drug Discov, 19:609-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].van der Harg JM, Nolle A, Zwart R, Boerema AS, van Haastert ES, Strijkstra AM, et al. (2014). The unfolded protein response mediates reversible tau phosphorylation induced by metabolic stress. Cell Death Dis, 5:e1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Qin W, Haroutunian V, Katsel P, Cardozo CP, Ho L, Buxbaum JD, et al. (2009). PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch Neurol, 66:352-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Hossain MS, Oomura Y, Katafuchi T (2018). Glucose Can Epigenetically Alter the Gene Expression of Neurotrophic Factors in the Murine Brain Cells. Mol Neurobiol, 55:3408-3425. [DOI] [PubMed] [Google Scholar]
- [64].Peng Y, Liu J, Shi L, Tang Y, Gao D, Long J, et al. (2016). Mitochondrial dysfunction precedes depression of AMPK/AKT signaling in insulin resistance induced by high glucose in primary cortical neurons. J Neurochem, 137:701-713. [DOI] [PubMed] [Google Scholar]
- [65].Shah K, Desilva S, Abbruscato T (2012). The role of glucose transporters in brain disease: diabetes and Alzheimer's Disease. Int J Mol Sci, 13:12629-12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Winkler EA, Nishida Y, Sagare AP, Rege SV, Bell RD, Perlmutter D, et al. (2015). GLUT1 reductions exacerbate Alzheimer's disease vasculo-neuronal dysfunction and degeneration. Nat Neurosci, 18:521-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Srikanth V, Maczurek A, Phan T, Steele M, Westcott B, Juskiw D, et al. (2011). Advanced glycation endproducts and their receptor RAGE in Alzheimer's disease. Neurobiol Aging, 32:763-777. [DOI] [PubMed] [Google Scholar]
- [68].Li XH, Lv BL, Xie JZ, Liu J, Zhou XW, Wang JZ (2012). AGEs induce Alzheimer-like tau pathology and memory deficit via RAGE-mediated GSK-3 activation. Neurobiol Aging, 33:1400-1410. [DOI] [PubMed] [Google Scholar]
- [69].Gabbouj S, Ryhänen S, Marttinen M, Wittrahm R, Takalo M, Kemppainen S, et al. (2019). Altered Insulin Signaling in Alzheimer's Disease Brain - Special Emphasis on PI3K-Akt Pathway. Front Neurosci, 13:629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Yamamoto N, Ishikuro R, Tanida M, Suzuki K, Ikeda-Matsuo Y, Sobue K (2018). Insulin-signaling Pathway Regulates the Degradation of Amyloid beta-protein via Astrocytes. Neuroscience, 385:227-236. [DOI] [PubMed] [Google Scholar]
- [71].Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, et al. (2005). Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease - is this type 3 diabetes? Journal of Alzheimers Disease, 7:63-80. [DOI] [PubMed] [Google Scholar]
- [72].Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, et al. (2004). Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer's disease. Faseb j, 18:902-904. [DOI] [PubMed] [Google Scholar]
- [73].Liu F, Shi J, Tanimukai H, Gu J, Gu J, Grundke-Iqbal I, et al. (2009). Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer's disease. Brain, 132:1820-1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Park J, Ha HJ, Chung ES, Baek SH, Cho Y, Kim HK, et al. (2021). O-GlcNAcylation ameliorates the pathological manifestations of Alzheimer's disease by inhibiting necroptosis. Sci Adv, 7:eabd3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Ramezani M, Fernando M, Eslick S, Asih PR, Shadfar S, Bandara EMS, et al. (2023). Ketone bodies mediate alterations in brain energy metabolism and biomarkers of Alzheimer's disease. Front Neurosci, 17:1297984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Versele R, Corsi M, Fuso A, Sevin E, Businaro R, Gosselet F, et al. (2020). Ketone Bodies Promote Amyloid-β(1-40) Clearance in a Human in Vitro Blood-Brain Barrier Model. Int J Mol Sci, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Wu Y, Gong Y, Luan Y, Li Y, Liu J, Yue Z, et al. (2020). BHBA treatment improves cognitive function by targeting pleiotropic mechanisms in transgenic mouse model of Alzheimer's disease. Faseb j, 34:1412-1429. [DOI] [PubMed] [Google Scholar]
- [78].Chandan G, Ganguly U, Pal S, Singh S, Saini RV, Chakrabarti SS, et al. (2023). GLUT inhibitor WZB117 induces cytotoxicity with increased production of amyloid-beta peptide in SH-SY5Y cells preventable by beta-hydroxybutyrate: implications in Alzheimer's disease. Pharmacol Rep, 75:482-489. [DOI] [PubMed] [Google Scholar]
- [79].Shippy DC, Wilhelm C, Viharkumar PA, Raife TJ, Ulland TK (2020). β-Hydroxybutyrate inhibits inflammasome activation to attenuate Alzheimer's disease pathology. J Neuroinflammation, 17:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Li X, Zhan Z, Zhang J, Zhou F, An L (2020). β-Hydroxybutyrate Ameliorates Aβ-Induced Downregulation of TrkA Expression by Inhibiting HDAC1/3 in SH-SY5Y Cells. Am J Alzheimers Dis Other Demen, 35:1533317519883496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Ye Y, Fu C, Li Y, Sun J, Li X, Chai S, et al. (2024). Alternate-day fasting improves cognitive and brain energy deficits by promoting ketone metabolism in the 3xTg mouse model of Alzheimer's disease. Exp Neurol, 381:114920. [DOI] [PubMed] [Google Scholar]
- [82].Kao YC, Ho PC, Tu YK, Jou IM, Tsai KJ (2020). Lipids and Alzheimer's Disease. Int J Mol Sci, 21:1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Yin F (2023). Lipid metabolism and Alzheimer's disease: clinical evidence, mechanistic link and therapeutic promise. FEBS J, 290:1420-1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Huang YA, Zhou B, Wernig M, Sudhof TC (2017). ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Abeta Secretion. Cell, 168:427-441 e421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Zhao Z, Sagare AP, Ma Q, Halliday MR, Kong P, Kisler K, et al. (2015). Central role for PICALM in amyloid-beta blood-brain barrier transcytosis and clearance. Nat Neurosci, 18:978-987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Ma Q, Zhao Z, Sagare AP, Wu Y, Wang M, Owens NC, et al. (2018). Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-beta42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol Neurodegener, 13:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wan Z, Mah D, Simtchouk S, Kluftinger A, Little JP (2015). Role of amyloid beta in the induction of lipolysis and secretion of adipokines from human adipose tissue. Adipocyte, 4:212-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Zhou M, Huang T, Collins N, Zhang J, Shen H, Dai X, et al. (2016). APOE4 Induces Site-Specific Tau Phosphorylation Through Calpain-CDK5 Signaling Pathway in EFAD-Tg Mice. Curr Alzheimer Res, 13:1048-1055. [DOI] [PubMed] [Google Scholar]
- [89].Zhang YJ, Cheng Y, Tang HL, Yue Q, Cai XY, Lu ZJ, et al. (2024). APOE ε4-associated downregulation of the IL-7/IL-7R pathway in effector memory T cells: Implications for Alzheimer's disease. Alzheimers Dement. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Zhao J, Fu Y, Yamazaki Y, Ren Y, Davis MD, Liu CC, et al. (2020). APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer's disease patient iPSC-derived cerebral organoids. Nat Commun, 11:5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Qiao F, Gao XP, Yuan L, Cai HY, Qi JS (2014). Apolipoprotein E4 impairs in vivo hippocampal long-term synaptic plasticity by reducing the phosphorylation of CaMKIIα and CREB. J Alzheimers Dis, 41:1165-1176. [DOI] [PubMed] [Google Scholar]
- [92].Li Y, Xia B, Li R, Yin D, Wang Y, Liang W (2017). Expression of brain-derived neurotrophic factors, neurotrophin-3, and neurotrophin-4 in the nucleus accumbens during heroin dependency and withdrawal. Neuroreport, 28:654-660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Yin J, Nielsen M, Carcione T, Li S, Shi J (2019). Apolipoprotein E regulates mitochondrial function through the PGC-1α-sirtuin 3 pathway. Aging (Albany NY), 11:11148-11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Jackson RJ, Meltzer JC, Nguyen H, Commins C, Bennett RE, Hudry E, et al. (2022). APOE4 derived from astrocytes leads to blood-brain barrier impairment. Brain, 145:3582-3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Simonovitch S, Schmukler E, Masliah E, Pinkas-Kramarski R, Michaelson DM (2019). The Effects of APOE4 on Mitochondrial Dynamics and Proteins in vivo. J Alzheimers Dis, 70:861-875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell, 160:1061-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M (2016). TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron, 91:328-340. [DOI] [PubMed] [Google Scholar]
- [98].Griciuc A, Patel S, Federico AN, Choi SH, Innes BJ, Oram MK, et al. (2019). TREM2 Acts Downstream of CD33 in Modulating Microglial Pathology in Alzheimer's Disease. Neuron, 103:820-835.e827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Leyns CEG, Ulrich JD, Finn MB, Stewart FR, Koscal LJ, Remolina Serrano J, et al. (2017). TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc Natl Acad Sci U S A, 114:11524-11529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Zhong L, Sheng X, Wang W, Li Y, Zhuo R, Wang K, et al. (2023). TREM2 receptor protects against complement-mediated synaptic loss by binding to complement C1q during neurodegeneration. Immunity, 56:1794-1808.e1798. [DOI] [PubMed] [Google Scholar]
- [101].Barbero-Camps E, Fernandez A, Martinez L, Fernandez-Checa JC, Colell A (2013). APP/PS1 mice overexpressing SREBP-2 exhibit combined Abeta accumulation and tau pathology underlying Alzheimer's disease. Hum Mol Genet, 22:3460-3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Wang H, Kulas JA, Wang C, Holtzman DM, Ferris HA, Hansen SB (2021). Regulation of beta-amyloid production in neurons by astrocyte-derived cholesterol. Proc Natl Acad Sci U S A, 118:e2102191118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Mohamed A, Viveiros A, Williams K, Posse de Chaves E (2018). Aβ inhibits SREBP-2 activation through Akt inhibition. J Lipid Res, 59:1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Sarlak Z, Moazzami M, Attarzadeh Hosseini M, Gharakhanlou R (2019). The effects of aerobic training before and after the induction of Alzheimer's disease on ABCA1 and APOE mRNA expression and the level of soluble Aβ1-42 in the hippocampus of male Wistar rats. Iran J Basic Med Sci, 22:399-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Davis W Jr. (2010). The ATP-binding cassette transporter-2 (ABCA2) increases endogenous amyloid precursor protein expression and Aβ fragment generation. Curr Alzheimer Res, 7:566-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Katsouri L, Georgopoulos S (2011). Lack of LDL receptor enhances amyloid deposition and decreases glial response in an Alzheimer's disease mouse model. PLoS One, 6:e21880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Fu Y, Hsiao JH, Paxinos G, Halliday GM, Kim WS (2015). ABCA5 regulates amyloid-beta peptide production and is associated with Alzheimer's disease neuropathology. J Alzheimers Dis, 43:857-869. [DOI] [PubMed] [Google Scholar]
- [108].Aikawa T, Ren Y, Yamazaki Y, Tachibana M, Johnson MR, Anderson CT, et al. (2019). ABCA7 haplodeficiency disturbs microglial immune responses in the mouse brain. Proc Natl Acad Sci U S A, 116:23790-23796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Sakae N, Liu CC, Shinohara M, Frisch-Daiello J, Ma L, Yamazaki Y, et al. (2016). ABCA7 Deficiency Accelerates Amyloid-beta Generation and Alzheimer's Neuronal Pathology. J Neurosci, 36:3848-3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Fitz NF, Carter AY, Tapias V, Castranio EL, Kodali R, Lefterov I, et al. (2017). ABCA1 Deficiency Affects Basal Cognitive Deficits and Dendritic Density in Mice. J Alzheimers Dis, 56:1075-1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Martinez M, Frank A, Diez-Tejedor E, Hernanz A (1993). Amino acid concentrations in cerebrospinal fluid and serum in Alzheimer's disease and vascular dementia. J Neural Transm Park Dis Dement Sect, 6:1-9. [DOI] [PubMed] [Google Scholar]
- [112].Altman BJ, Stine ZE, Dang CV (2016). From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer, 16:619-634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Sun L, Guo D, Jia Y, Shi M, Yang P, Wang Y, et al. (2022). Association between Human Blood Metabolome and the Risk of Alzheimer's Disease. Ann Neurol, 92:756-767. [DOI] [PubMed] [Google Scholar]
- [114].Wang Y, Wang Q, Li J, Lu G, Liu Z (2019). Glutamine Improves Oxidative Stress through the Wnt3a/beta-Catenin Signaling Pathway in Alzheimer's Disease In Vitro and In Vivo. Biomed Res Int, 2019:4690280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Rupsingh R, Borrie M, Smith M, Wells JL, Bartha R (2011). Reduced hippocampal glutamate in Alzheimer disease. Neurobiol Aging, 32:802-810. [DOI] [PubMed] [Google Scholar]
- [116].Bai X, Edden RA, Gao F, Wang G, Wu L, Zhao B, et al. (2015). Decreased γ-aminobutyric acid levels in the parietal region of patients with Alzheimer's disease. J Magn Reson Imaging, 41:1326-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, et al. (2017). alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol, 18:985-994. [DOI] [PubMed] [Google Scholar]
- [118].Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. (2013). Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature, 496:238-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. (2016). Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell, 167:457-470 e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Siddik MAB, Mullins CA, Kramer A, Shah H, Gannaban RB, Zabet-Moghaddam M, et al. (2022). Branched-Chain Amino Acids Are Linked with Alzheimer's Disease-Related Pathology and Cognitive Deficits. Cells, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Yudkoff M, Daikhin Y, Lin ZP, Nissim I, Stern J, Pleasure D, et al. (1994). Interrelationships of leucine and glutamate metabolism in cultured astrocytes. J Neurochem, 62:1192-1202. [DOI] [PubMed] [Google Scholar]
- [122].Le Douce J, Maugard M, Veran J, Matos M, Jégo P, Vigneron PA, et al. (2020). Impairment of Glycolysis-Derived l-Serine Production in Astrocytes Contributes to Cognitive Deficits in Alzheimer's Disease. Cell Metab, 31:503-517.e508. [DOI] [PubMed] [Google Scholar]
- [123].Chang CH, Kuo HL, Ma WF, Tsai HC (2020). Cerebrospinal Fluid and Serum d-Serine Levels in Patients with Alzheimer's Disease: A Systematic Review and Meta-Analysis. J Clin Med, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Madeira C, Lourenco MV, Vargas-Lopes C, Suemoto CK, Brandão CO, Reis T, et al. (2015). d-serine levels in Alzheimer's disease: implications for novel biomarker development. Transl Psychiatry, 5:e561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Ni X, Inoue R, Wu Y, Yoshida T, Yaku K, Nakagawa T, et al. (2023). Regional contributions of D-serine to Alzheimer's disease pathology in male App(NL-G-F/NL-G-F) mice. Front Aging Neurosci, 15:1211067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Nuzzo T, Miroballo M, Casamassa A, Mancini A, Gaetani L, Nisticò R, et al. (2020). Cerebrospinal fluid and serum d-serine concentrations are unaltered across the whole clinical spectrum of Alzheimer's disease. Biochim Biophys Acta Proteins Proteom, 1868:140537. [DOI] [PubMed] [Google Scholar]
- [127].Santamaría A, Galván-Arzate S, Lisý V, Ali SF, Duhart HM, Osorio-Rico L, et al. (2001). Quinolinic acid induces oxidative stress in rat brain synaptosomes. Neuroreport, 12:871-874. [DOI] [PubMed] [Google Scholar]
- [128].Rahman A, Ting K, Cullen KM, Braidy N, Brew BJ, Guillemin GJ (2009). The excitotoxin quinolinic acid induces tau phosphorylation in human neurons. PLoS One, 4:e6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Chatterjee P, Zetterberg H, Goozee K, Lim CK, Jacobs KR, Ashton NJ, et al. (2019). Plasma neurofilament light chain and amyloid-β are associated with the kynurenine pathway metabolites in preclinical Alzheimer's disease. J Neuroinflammation, 16:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Song Z, Wu Z, Luo R, He C, Li Z, Yang M, et al. (2023). Identification of tryptophan metabolism-related genes in immunity and immunotherapy in Alzheimer's disease. Aging (Albany NY), 15:13077-13099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Kandimalla R, Manczak M, Fry D, Suneetha Y, Sesaki H, Reddy PH (2016). Reduced dynamin-related protein 1 protects against phosphorylated Tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer's disease. Hum Mol Genet, 25:4881-4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Baek SH, Park SJ, Jeong JI, Kim SH, Han J, Kyung JW, et al. (2017). Inhibition of Drp1 Ameliorates Synaptic Depression, Abeta Deposition, and Cognitive Impairment in an Alzheimer's Disease Model. J Neurosci, 37:5099-5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, et al. (2009). Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci, 29:9090-9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Manczak M, Reddy PH (2012). Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer's disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum Mol Genet, 21:2538-2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Zysk M, Beretta C, Naia L, Dakhel A, Pavenius L, Brismar H, et al. (2023). Amyloid-beta accumulation in human astrocytes induces mitochondrial disruption and changed energy metabolism. J Neuroinflammation, 20:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, et al. (2019). Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer's disease. Nat Neurosci, 22:401-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE (2005). Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol, 57:695-703. [DOI] [PubMed] [Google Scholar]
- [138].Cho YH, Kim GH, Park JJ (2021). Mitochondrial aconitase 1 regulates age-related memory impairment via autophagy/mitophagy-mediated neural plasticity in middle-aged flies. Aging Cell, 20:e13520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Noce B, Di Bello E, Fioravanti R, Mai A (2023). LSD1 inhibitors for cancer treatment: Focus on multi-target agents and compounds in clinical trials. Front Pharmacol, 14:1120911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Hino S, Sakamoto A, Nagaoka K, Anan K, Wang Y, Mimasu S, et al. (2012). FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nat Commun, 3:758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Etchegaray JP, Mostoslavsky R (2016). Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol Cell, 62:695-711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Tian Q, Zhao J, Yang Q, Wang B, Deavila JM, Zhu MJ, et al. (2020). Dietary alpha-ketoglutarate promotes beige adipogenesis and prevents obesity in middle-aged mice. Aging Cell, 19:e13059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Kang HS, Lee JH, Oh KJ, Lee EW, Han BS, Park KY, et al. (2020). IDH1-dependent alpha-KG regulates brown fat differentiation and function by modulating histone methylation. Metabolism, 105:154173. [DOI] [PubMed] [Google Scholar]
- [144].Li XJ, Li QL, Ju LG, Zhao C, Zhao LS, Du JW, et al. (2021). Deficiency of Histone Methyltransferase SET Domain-Containing 2 in Liver Leads to Abnormal Lipid Metabolism and HCC. Hepatology, 73:1797-1815. [DOI] [PubMed] [Google Scholar]
- [145].Naviaux RK (2008). Mitochondrial control of epigenetics. Cancer Biol Ther, 7:1191-1193. [DOI] [PubMed] [Google Scholar]
- [146].Hu E, Du H, Zhu X, Wang L, Shang S, Wu X, et al. (2018). Beta-hydroxybutyrate Promotes the Expression of BDNF in Hippocampal Neurons under Adequate Glucose Supply. Neuroscience, 386:315-325. [DOI] [PubMed] [Google Scholar]
- [147].Hu E, Du H, Shang S, Zhang Y, Lu X (2020). Beta-Hydroxybutyrate Enhances BDNF Expression by Increasing H3K4me3 and Decreasing H2AK119ub in Hippocampal Neurons. Front Neurosci, 14:591177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Pan X, Ye L, Guo X, Wang W, Zhang Z, Wang Q, et al. (2023). Glutamine Production by Glul Promotes Thermogenic Adipocyte Differentiation Through Prdm9-Mediated H3K4me3 and Transcriptional Reprogramming. Diabetes, 72:1574-1596. [DOI] [PubMed] [Google Scholar]
- [149].Lee J, Saha PK, Yang QH, Lee S, Park JY, Suh Y, et al. (2008). Targeted inactivation of MLL3 histone H3-Lys-4 methyltransferase activity in the mouse reveals vital roles for MLL3 in adipogenesis. Proc Natl Acad Sci U S A, 105:19229-19234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Wang L, Xu S, Lee JE, Baldridge A, Grullon S, Peng W, et al. (2013). Histone H3K9 methyltransferase G9a represses PPARgamma expression and adipogenesis. EMBO J, 32:45-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Lan T, Li P, Zhang SJ, Liu SY, Zeng XX, Chai F, et al. (2024). Paeoniflorin promotes PPARgamma expression to suppress HSCs activation by inhibiting EZH2-mediated histone H3K27 trimethylation. Phytomedicine, 128:155477. [DOI] [PubMed] [Google Scholar]
- [152].Tateishi K, Okada Y, Kallin EM, Zhang Y (2009). Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature, 458:757-761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Sambeat A, Gulyaeva O, Dempersmier J, Tharp KM, Stahl A, Paul SM, et al. (2016). LSD1 Interacts with Zfp516 to Promote UCP1 Transcription and Brown Fat Program. Cell Rep, 15:2536-2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Lee KH, Ju UI, Song JY, Chun YS (2014). The histone demethylase PHF2 promotes fat cell differentiation as an epigenetic activator of both C/EBPalpha and C/EBPdelta. Mol Cells, 37:734-741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Wang Q, Li H, Tajima K, Verkerke ARP, Taxin ZH, Hou Z, et al. (2022). Post-translational control of beige fat biogenesis by PRDM16 stabilization. Nature, 609:151-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Searcy JL, Phelps JT, Pancani T, Kadish I, Popovic J, Anderson KL, et al. (2012). Long-term pioglitazone treatment improves learning and attenuates pathological markers in a mouse model of Alzheimer's disease. J Alzheimers Dis, 30:943-961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Domingues C, da Cruz ESOAB, Henriques AG (2017). Impact of Cytokines and Chemokines on Alzheimer's Disease Neuropathological Hallmarks. Curr Alzheimer Res, 14:870-882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Chandra S, Roy A, Jana M, Pahan K (2019). Cinnamic acid activates PPARα to stimulate Lysosomal biogenesis and lower Amyloid plaque pathology in an Alzheimer's disease mouse model. Neurobiol Dis, 124:379-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Saez-Orellana F, Leroy T, Ribeiro F, Kreis A, Leroy K, Lalloyer F, et al. (2021). Regulation of PPARalpha by APP in Alzheimer disease affects the pharmacological modulation of synaptic activity. JCI Insight, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Jung CG, Yamashita H, Kato R, Zhou C, Matsushita H, Takeuchi T, et al. (2023). Deletion of UCP1 in Tg2576 Mice Increases Body Temperature and Exacerbates Alzheimer's Disease-Related Pathologies. Int J Mol Sci, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].DeBose-Boyd RA, Ye J (2018). SREBPs in Lipid Metabolism, Insulin Signaling, and Beyond. Trends Biochem Sci, 43:358-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Willemsen L, Prange KHM, Neele AE, van Roomen C, Gijbels M, Griffith GR, et al. (2022). DOT1L regulates lipid biosynthesis and inflammatory responses in macrophages and promotes atherosclerotic plaque stability. Cell Rep, 41:111703. [DOI] [PubMed] [Google Scholar]
- [163].Li X, Liu Z, Xia C, Yan K, Fang Z, Fan Y (2022). SETD8 stabilized by USP17 epigenetically activates SREBP1 pathway to drive lipogenesis and oncogenesis of ccRCC. Cancer Lett, 527:150-163. [DOI] [PubMed] [Google Scholar]
- [164].Tang D, Dai Y, Xu S (2021). Targeting KDM6A Suppresses SREBP1c-Dependent Lipid Metabolism and Prostate Tumorigenesis. Cancer Res. [DOI] [PubMed] [Google Scholar]
- [165].Kim JH, Jung DY, Kim HR, Jung MH (2020). Histone H3K9 Demethylase JMJD2B Plays a Role in LXRα-Dependent Lipogenesis. Int J Mol Sci, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Marwarha G, Claycombe-Larson K, Lund J, Ghribi O (2019). Palmitate-Induced SREBP1 Expression and Activation Underlies the Increased BACE 1 Activity and Amyloid Beta Genesis. Mol Neurobiol, 56:5256-5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Deering TG, Ogihara T, Trace AP, Maier B, Mirmira RG (2009). Methyltransferase Set7/9 maintains transcription and euchromatin structure at islet-enriched genes. Diabetes, 58:185-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Maganti AV, Maier B, Tersey SA, Sampley ML, Mosley AL, Ozcan S, et al. (2015). Transcriptional activity of the islet beta cell factor Pdx1 is augmented by lysine methylation catalyzed by the methyltransferase Set7/9. J Biol Chem, 290:9812-9822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Unger J, McNeill TH, Moxley RT 3rd, White M, Moss A, Livingston JN (1989). Distribution of insulin receptor-like immunoreactivity in the rat forebrain. Neuroscience, 31:143-157. [DOI] [PubMed] [Google Scholar]
- [170].Deng Y, Li B, Liu Y, Iqbal K, Grundke-Iqbal I, Gong CX (2009). Dysregulation of insulin signaling, glucose transporters, O-GlcNAcylation, and phosphorylation of tau and neurofilaments in the brain: Implication for Alzheimer's disease. Am J Pathol, 175:2089-2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Plaschke K, Kopitz J, Siegelin M, Schliebs R, Salkovic-Petrisic M, Riederer P, et al. (2010). Insulin-resistant brain state after intracerebroventricular streptozotocin injection exacerbates Alzheimer-like changes in Tg2576 AbetaPP-overexpressing mice. J Alzheimers Dis, 19:691-704. [DOI] [PubMed] [Google Scholar]
- [172].Chen Y, Deng Y, Zhang B, Gong CX (2014). Deregulation of brain insulin signaling in Alzheimer's disease. Neurosci Bull, 30:282-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Jiao Y, Li YZ, Zhang YH, Cui W, Li Q, Xie KL, et al. (2023). Lysine demethylase KDM5B down-regulates SIRT3-mediated mitochondrial glucose and lipid metabolism in diabetic neuropathy. Diabet Med, 40:e14964. [DOI] [PubMed] [Google Scholar]
- [174].Nunes D, Loureiro JA, Pereira MC (2022). Drug Delivery Systems as a Strategy to Improve the Efficacy of FDA-Approved Alzheimer's Drugs. Pharmaceutics, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Söderberg L, Johannesson M, Nygren P, Laudon H, Eriksson F, Osswald G, et al. (2023). Lecanemab, Aducanumab, and Gantenerumab - Binding Profiles to Different Forms of Amyloid-Beta Might Explain Efficacy and Side Effects in Clinical Trials for Alzheimer's Disease. Neurotherapeutics, 20:195-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Maity J, Majumder S, Pal R, Saha B, Mukhopadhyay PK (2023). Ascorbic acid modulates immune responses through Jumonji-C domain containing histone demethylases and Ten eleven translocation (TET) methylcytosine dioxygenase. Bioessays, 45:e2300035. [DOI] [PubMed] [Google Scholar]
- [177].Hamid M, Mansoor S, Amber S, Zahid S (2022). A quantitative meta-analysis of vitamin C in the pathophysiology of Alzheimer's disease. Front Aging Neurosci, 14:970263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Sampaio I, Quatroni FD, Pincela Lins PM, Nascimento AS, Zucolotto V (2022). Modulation of beta-amyloid aggregation using ascorbic acid. Biochimie, 200:36-43. [DOI] [PubMed] [Google Scholar]
- [179].Sadli N, Ackland ML, De Mel D, Sinclair AJ, Suphioglu C (2012). Effects of zinc and DHA on the epigenetic regulation of human neuronal cells. Cell Physiol Biochem, 29:87-98. [DOI] [PubMed] [Google Scholar]
- [180].Bonfili L, Cuccioloni M, Gong C, Cecarini V, Spina M, Zheng Y, et al. (2022). Gut microbiota modulation in Alzheimer's disease: Focus on lipid metabolism. Clin Nutr, 41:698-708. [DOI] [PubMed] [Google Scholar]
- [181].Fuso A, Cavallaro RA, Zampelli A, D'Anselmi F, Piscopo P, Confaloni A, et al. (2007). gamma-Secretase is differentially modulated by alterations of homocysteine cycle in neuroblastoma and glioblastoma cells. J Alzheimers Dis, 11:275-290. [DOI] [PubMed] [Google Scholar]
- [182].Judd JM, Jasbi P, Winslow W, Serrano GE, Beach TG, Klein-Seetharaman J, et al. (2023). Inflammation and the pathological progression of Alzheimer's disease are associated with low circulating choline levels. Acta Neuropathol, 146:565-583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Zhang Y, Jia J (2023). Betaine Mitigates Amyloid-β-Associated Neuroinflammation by Suppressing the NLRP3 and NF-κB Signaling Pathways in Microglial Cells. J Alzheimers Dis, 94:S9-s19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Wang C, Hei Y, Liu Y, Bajpai AK, Li Y, Guan Y, et al. (2024). Systems genetics identifies methionine as a high risk factor for Alzheimer's disease. Front Neurosci, 18:1381889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Puigoriol-Illamola D, Martínez-Damas M, Griñán-Ferré C, Pallàs M (2020). Chronic Mild Stress Modified Epigenetic Mechanisms Leading to Accelerated Senescence and Impaired Cognitive Performance in Mice. Int J Mol Sci, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ (2006). Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci, 9:519-525. [DOI] [PubMed] [Google Scholar]
- [187].Pang R, Wang X, Pei F, Zhang W, Shen J, Gao X, et al. (2019). Regular Exercise Enhances Cognitive Function and Intracephalic GLUT Expression in Alzheimer's Disease Model Mice. J Alzheimers Dis, 72:83-96. [DOI] [PubMed] [Google Scholar]
- [188].Zhang SS, Zhu L, Peng Y, Zhang L, Chao FL, Jiang L, et al. (2022). Long-term running exercise improves cognitive function and promotes microglial glucose metabolism and morphological plasticity in the hippocampus of APP/PS1 mice. J Neuroinflammation, 19:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Ying N, Luo H, Li B, Gong K, Shu Q, Liang F, et al. (2023). Exercise Alleviates Behavioral Disorders but Shapes Brain Metabolism of APP/PS1 Mice in a Region- and Exercise-Specific Manner. J Proteome Res, 22:1649-1659. [DOI] [PubMed] [Google Scholar]
- [190].Guo Y, Chen SD, You J, Huang SY, Chen YL, Zhang Y, et al. (2024). Multiplex cerebrospinal fluid proteomics identifies biomarkers for diagnosis and prediction of Alzheimer's disease. Nat Hum Behav. [DOI] [PubMed] [Google Scholar]
- [191].Jiao B, Li R, Zhou H, Qing K, Liu H, Pan H, et al. (2023). Neural biomarker diagnosis and prediction to mild cognitive impairment and Alzheimer's disease using EEG technology. Alzheimers Res Ther, 15:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Ablinger I, Dressel K, Rott T, Lauer AA, Tiemann M, Batista JP, et al. (2022). Interdisciplinary Approaches to Deal with Alzheimer's Disease-From Bench to Bedside: What Feasible Options Do Already Exist Today? Biomedicines, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Livingston G, Huntley J, Liu KY, Costafreda SG, Selbæk G, Alladi S, et al. (2024). Dementia prevention, intervention, and care: 2024 report of the Lancet standing Commission. Lancet, 404:572-628. [DOI] [PubMed] [Google Scholar]
- [194].Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, et al. (2000). Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature, 406:593-599. [DOI] [PubMed] [Google Scholar]
- [195].Wang T, Xu C, Liu Y, Fan K, Li Z, Sun X, et al. (2012). Crystal structure of the human SUV39H1 chromodomain and its recognition of histone H3K9me2/3. PLoS One, 7:e52977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].O'Carroll D, Scherthan H, Peters AH, Opravil S, Haynes AR, Laible G, et al. (2000). Isolation and characterization of Suv39h2, a second histone H3 methyltransferase gene that displays testis-specific expression. Mol Cell Biol, 20:9423-9433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, et al. (2002). G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev, 16:1779-1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ 3rd (2002). SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev, 16:919-932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Falandry C, Fourel G, Galy V, Ristriani T, Horard B, Bensimon E, et al. (2010). CLLD8/KMT1F is a lysine methyltransferase that is important for chromosome segregation. J Biol Chem, 285:20234-20241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, et al. (2006). Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol, 13:713-719. [DOI] [PubMed] [Google Scholar]
- [201].Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, et al. (2004). Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell, 13:587-597. [DOI] [PubMed] [Google Scholar]
- [202].Andreu-Vieyra CV, Chen R, Agno JE, Glaser S, Anastassiadis K, Stewart AF, et al. (2010). MLL2 is required in oocytes for bulk histone 3 lysine 4 trimethylation and transcriptional silencing. PLoS Biol, 8:e1000453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Goo YH, Sohn YC, Kim DH, Kim SW, Kang MJ, Jung DJ, et al. (2003). Activating signal cointegrator 2 belongs to a novel steady-state complex that contains a subset of trithorax group proteins. Mol Cell Biol, 23:140-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Nightingale KP, Gendreizig S, White DA, Bradbury C, Hollfelder F, Turner BM (2007). Cross-talk between histone modifications in response to histone deacetylase inhibitors: MLL4 links histone H3 acetylation and histone H3K4 methylation. J Biol Chem, 282:4408-4416. [DOI] [PubMed] [Google Scholar]
- [205].Emerling BM, Bonifas J, Kratz CP, Donovan S, Taylor BR, Green ED, et al. (2002). MLL5, a homolog of Drosophila trithorax located within a segment of chromosome band 7q22 implicated in myeloid leukemia. Oncogene, 21:4849-4854. [DOI] [PubMed] [Google Scholar]
- [206].Wysocka J, Myers MP, Laherty CD, Eisenman RN, Herr W (2003). Human Sin3 deacetylase and trithorax-related Set1/Ash2 histone H3-K4 methyltransferase are tethered together selectively by the cell-proliferation factor HCF-1. Genes Dev, 17:896-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Lee JH, Tate CM, You JS, Skalnik DG (2007). Identification and characterization of the human Set1B histone H3-Lys4 methyltransferase complex. J Biol Chem, 282:13419-13428. [DOI] [PubMed] [Google Scholar]
- [208].Gregory GD, Vakoc CR, Rozovskaia T, Zheng X, Patel S, Nakamura T, et al. (2007). Mammalian ASH1L is a histone methyltransferase that occupies the transcribed region of active genes. Mol Cell Biol, 27:8466-8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].An S, Yeo KJ, Jeon YH, Song JJ (2011). Crystal structure of the human histone methyltransferase ASH1L catalytic domain and its implications for the regulatory mechanism. J Biol Chem, 286:8369-8374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Sun XJ, Wei J, Wu XY, Hu M, Wang L, Wang HH, et al. (2005). Identification and characterization of a novel human histone H3 lysine 36-specific methyltransferase. J Biol Chem, 280:35261-35271. [DOI] [PubMed] [Google Scholar]
- [211].Wang GG, Cai L, Pasillas MP, Kamps MP (2007). NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat Cell Biol, 9:804-812. [DOI] [PubMed] [Google Scholar]
- [212].Brown MA, Sims RJ 3rd, Gottlieb PD, Tucker PW (2006). Identification and characterization of Smyd2: a split SET/MYND domain-containing histone H3 lysine 36-specific methyltransferase that interacts with the Sin3 histone deacetylase complex. Mol Cancer, 5:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Abu-Farha M, Lambert JP, Al-Madhoun AS, Elisma F, Skerjanc IS, Figeys D (2008). The tale of two domains: proteomics and genomics analysis of SMYD2, a new histone methyltransferase. Mol Cell Proteomics, 7:560-572. [DOI] [PubMed] [Google Scholar]
- [214].Hamamoto R, Furukawa Y, Morita M, Iimura Y, Silva FP, Li M, et al. (2004). SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat Cell Biol, 6:731-740. [DOI] [PubMed] [Google Scholar]
- [215].Kim SM, Kee HJ, Eom GH, Choe NW, Kim JY, Kim YS, et al. (2006). Characterization of a novel WHSC1-associated SET domain protein with H3K4 and H3K27 methyltransferase activity. Biochem Biophys Res Commun, 345:318-323. [DOI] [PubMed] [Google Scholar]
- [216].He C, Li F, Zhang J, Wu J, Shi Y (2013). The methyltransferase NSD3 has chromatin-binding motifs, PHD5-C5HCH, that are distinct from other NSD (nuclear receptor SET domain) family members in their histone H3 recognition. J Biol Chem, 288:4692-4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Kuo AJ, Cheung P, Chen K, Zee BM, Kioi M, Lauring J, et al. (2011). NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol Cell, 44:609-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Pei H, Zhang L, Luo K, Qin Y, Chesi M, Fei F, et al. (2011). MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature, 470:124-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Feng Q, Wang H, Ng HH, Erdjument-Bromage H, Tempst P, Struhl K, et al. (2002). Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol, 12:1052-1058. [DOI] [PubMed] [Google Scholar]
- [220].Fang J, Feng Q, Ketel CS, Wang H, Cao R, Xia L, et al. (2002). Purification and functional characterization of SET8, a nucleosomal histone H4-lysine 20-specific methyltransferase. Curr Biol, 12:1086-1099. [DOI] [PubMed] [Google Scholar]
- [221].Nishioka K, Rice JC, Sarma K, Erdjument-Bromage H, Werner J, Wang Y, et al. (2002). PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol Cell, 9:1201-1213. [DOI] [PubMed] [Google Scholar]
- [222].Yang H, Pesavento JJ, Starnes TW, Cryderman DE, Wallrath LL, Kelleher NL, et al. (2008). Preferential dimethylation of histone H4 lysine 20 by Suv4-20. J Biol Chem, 283:12085-12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. (2002). Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science, 298:1039-1043. [DOI] [PubMed] [Google Scholar]
- [224].Kim KC, Geng L, Huang S (2003). Inactivation of a histone methyltransferase by mutations in human cancers. Cancer Res, 63:7619-7623. [PubMed] [Google Scholar]
- [225].Hayashi K, Yoshida K, Matsui Y (2005). A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature, 438:374-378. [DOI] [PubMed] [Google Scholar]
- [226].Wu Y, Ferguson JE 3rd, Wang H, Kelley R, Ren R, McDonough H, et al. (2008). PRDM6 is enriched in vascular precursors during development and inhibits endothelial cell proliferation, survival, and differentiation. J Mol Cell Cardiol, 44:47-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Eom GH, Kim K, Kim SM, Kee HJ, Kim JY, Jin HM, et al. (2009). Histone methyltransferase PRDM8 regulates mouse testis steroidogenesis. Biochem Biophys Res Commun, 388:131-136. [DOI] [PubMed] [Google Scholar]
- [228].Pinheiro I, Margueron R, Shukeir N, Eisold M, Fritzsch C, Richter FM, et al. (2012). Prdm3 and Prdm16 are H3K9me1 methyltransferases required for mammalian heterochromatin integrity. Cell, 150:948-960. [DOI] [PubMed] [Google Scholar]
- [229].Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell, 119:941-953. [DOI] [PubMed] [Google Scholar]
- [230].Metzger E, Wissmann M, Yin N, Müller JM, Schneider R, Peters AH, et al. (2005). LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature, 437:436-439. [DOI] [PubMed] [Google Scholar]
- [231].Fang R, Barbera AJ, Xu Y, Rutenberg M, Leonor T, Bi Q, et al. (2010). Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol Cell, 39:222-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, et al. (2006). Histone demethylation by a family of JmjC domain-containing proteins. Nature, 439:811-816. [DOI] [PubMed] [Google Scholar]
- [233].He J, Kallin EM, Tsukada Y, Zhang Y (2008). The H3K36 demethylase Jhdm1b/Kdm2b regulates cell proliferation and senescence through p15(Ink4b). Nat Struct Mol Biol, 15:1169-1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J, et al. (2006). JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell, 125:483-495. [DOI] [PubMed] [Google Scholar]
- [235].Brauchle M, Yao Z, Arora R, Thigale S, Clay I, Inverardi B, et al. (2013). Protein complex interactor analysis and differential activity of KDM3 subfamily members towards H3K9 methylation. PLoS One, 8:e60549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Kim SM, Kim JY, Choe NW, Cho IH, Kim JR, Kim DW, et al. (2010). Regulation of mouse steroidogenesis by WHISTLE and JMJD1C through histone methylation balance. Nucleic Acids Res, 38:6389-6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Klose RJ, Yamane K, Bae Y, Zhang D, Erdjument-Bromage H, Tempst P, et al. (2006). The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature, 442:312-316. [DOI] [PubMed] [Google Scholar]
- [238].Fodor BD, Kubicek S, Yonezawa M, O'Sullivan RJ, Sengupta R, Perez-Burgos L, et al. (2006). Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev, 20:1557-1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, et al. (2006). The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature, 442:307-311. [DOI] [PubMed] [Google Scholar]
- [240].Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, et al. (2006). Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell, 125:467-481. [DOI] [PubMed] [Google Scholar]
- [241].Christensen J, Agger K, Cloos PA, Pasini D, Rose S, Sennels L, et al. (2007). RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell, 128:1063-1076. [DOI] [PubMed] [Google Scholar]
- [242].Yamane K, Tateishi K, Klose RJ, Fang J, Fabrizio LA, Erdjument-Bromage H, et al. (2007). PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol Cell, 25:801-812. [DOI] [PubMed] [Google Scholar]
- [243].Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, et al. (2007). The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell, 128:1077-1088. [DOI] [PubMed] [Google Scholar]
- [244].Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, et al. (2007). UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature, 449:731-734. [DOI] [PubMed] [Google Scholar]
- [245].Hong S, Cho YW, Yu LR, Yu H, Veenstra TD, Ge K (2007). Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc Natl Acad Sci U S A, 104:18439-18444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Yokoyama A, Okuno Y, Chikanishi T, Hashiba W, Sekine H, Fujiki R, et al. (2010). KIAA1718 is a histone demethylase that erases repressive histone methyl marks. Genes Cells, 15:867-873. [DOI] [PubMed] [Google Scholar]
- [247].Liu W, Tanasa B, Tyurina OV, Zhou TY, Gassmann R, Liu WT, et al. (2010). PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature, 466:508-512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Wen H, Li J, Song T, Lu M, Kan PY, Lee MG, et al. (2010). Recognition of histone H3K4 trimethylation by the plant homeodomain of PHF2 modulates histone demethylation. J Biol Chem, 285:9322-9326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Amendola PG, Zaghet N, Ramalho JJ, Vilstrup Johansen J, Boxem M, Salcini AE (2017). JMJD-5/KDM8 regulates H3K36me2 and is required for late steps of homologous recombination and genome integrity. PLoS Genet, 13:e1006632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Neustadtl AL, Winston CN, Parsadanian M, Main BS, Villapol S, Burns MP (2017). Reduced cortical excitatory synapse number in mice is associated with increased calcineurin activity. Neuroreport, 28:618-624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Yin J, Reiman EM, Beach TG, Serrano GE, Sabbagh MN, Nielsen M, et al. (2020). Effect of ApoE isoforms on mitochondria in Alzheimer disease. Neurology, 94:e2404-e2411. [DOI] [PMC free article] [PubMed] [Google Scholar]