

Abstract
Background
Bardet–Biedl syndrome is a rare autosomal recessive disease occurring due to a ciliopathic genetic defect. It is caused by mutations in genes encoding proteins vital for the BBSome complex. This complex is essential for ciliary function and cellular signaling. It has multisystem involvement and presents with a variety of phenotypes.
Case Presentation
A 30-year-old adult male patient, Indian by ethnicity, presented with a 2-week history of ascites and dyspnea. The ascitic fluid analysis confirmed abdominal tuberculosis. However, the patient showed other symptoms and signs of a syndromic nature. The patient has been entirely blind since the age of 9 years, with confirmed retinitis pigmentosa. The other complaints were progressive weight gain and cognitive impairment. Examination showed central obesity, almond-shaped eyes, moon-shaped face, and hexadactyly in the left lower limb. Liver functional tests, renal function tests, lipid profile, and ultrasonography of the abdomen were abnormal. Beales diagnostic criteria confirmed Bardet–Biedl syndrome. The patient was treated for abdominal tuberculosis, and psychosocial support and nutritional counseling were provided.
Conclusion
Effective treatment of Bardet–Biedl syndrome requires genetic counseling and a personalized care plan that includes a multidisciplinary team, regular monitoring, and supportive services such as neuropsychological and psychiatric care and family support. This case also increases clinicians’ awareness of the presentation of Bardet–Biedl syndrome and the diagnosis in settings without advanced diagnostic modalities.
Keywords: Bardet–Biedl syndrome, BBS, Beales diagnostic criteria, Case report
Introduction:
Bardet–Biedl syndrome (BBS) is a rare autosomal recessive disease commonly found in consanguineous marriages [1]. It occurs due to a ciliopathic genetic defect. BBS is caused by mutations in over 25 genes, primarily BBS1, BBS10, and BBS2, which encode proteins vital for the BBSome complex. This complex is essential for ciliary function and cellular signaling [2, 3]. The syndrome has a frequency of 1 in 160,000 [4]. The disease typically appears in the first decade of life, with vision problems being the first symptom. The other major features of this syndrome include cone-rod dystrophy, obesity, polydactyly, learning disabilities, hypogonadism in male individuals, renal anomalies, cardiovascular anomalies, nystagmus, speech disorders, developmental delay, and ataxia. Additionally, endocrine abnormalities, such as type 2 diabetes mellitus, are observed in many patients [5, 6].
Treatment of BBS requires a multidisciplinary approach focusing on early diagnosis and supportive care. This includes renal monitoring, weight management, and treatment of endocrine- and vision-related complications [5, 6]. The prognosis of the condition depends on the time of diagnosis and the extent of renal involvement.
We present a case of a 30-year-old male patient, Indian by ethnicity, who presented with abdominal tuberculosis (TB) and was incidentally diagnosed with BBS on the basis of his clinical presentation and laboratory results. By presenting this case, we aim to raise clinician awareness and facilitate prompt diagnosis, particularly in environments lacking sophisticated diagnostic tools.
Case Presentation
A 30-year-old male patient, Indian by ethnicity, born out of a non-consanguineous marriage, presented to the emergency department with abdominal distension, pedal edema, and dyspnea for 2 weeks. However, the patient showed other symptoms of a syndromic nature. He has been entirely blind since the age of 9 years, with the deterioration of his vision characterized as benign and progressive. His fundoscopy confirmed retinitis pigmentosa (Fig. 1). He complained of progressively increasing weight. He was also unable to pursue formal education owing to cognitive difficulties and visual impairment. The physical examination showed pedal edema, almond-shaped eyes, moon-shaped face, and hexadactyly in the left lower limb (Figs. 2, 3). Body mass index (BMI) was 34.2, indicating a state of general obesity. His nervous system examination was normal. He was diagnosed with BBS on the basis of diagnostic criteria. Table 1 lists the major criteria for which the patient was positive. However, a genetic profile could not be obtained due to the patient’s unavailability and low socioeconomic status. The patient denied any family history of similar symptoms.
Fig. 1.
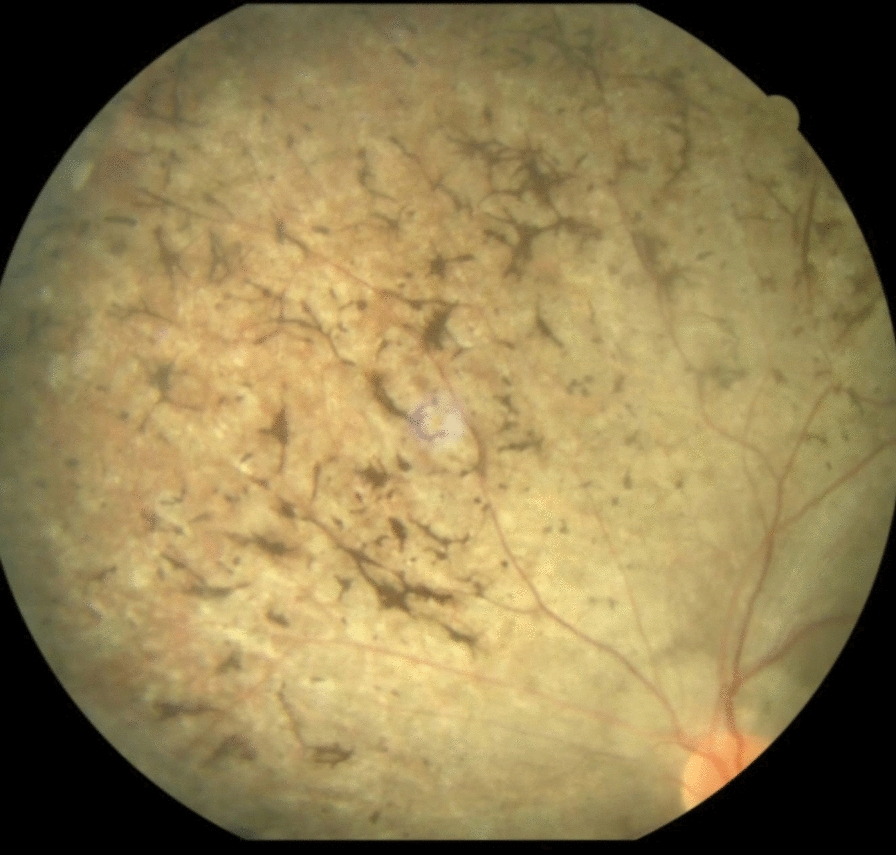
Fundoscopy image showing retinitis pigmentosa
Fig. 2.

Central obesity in the patient
Fig. 3.
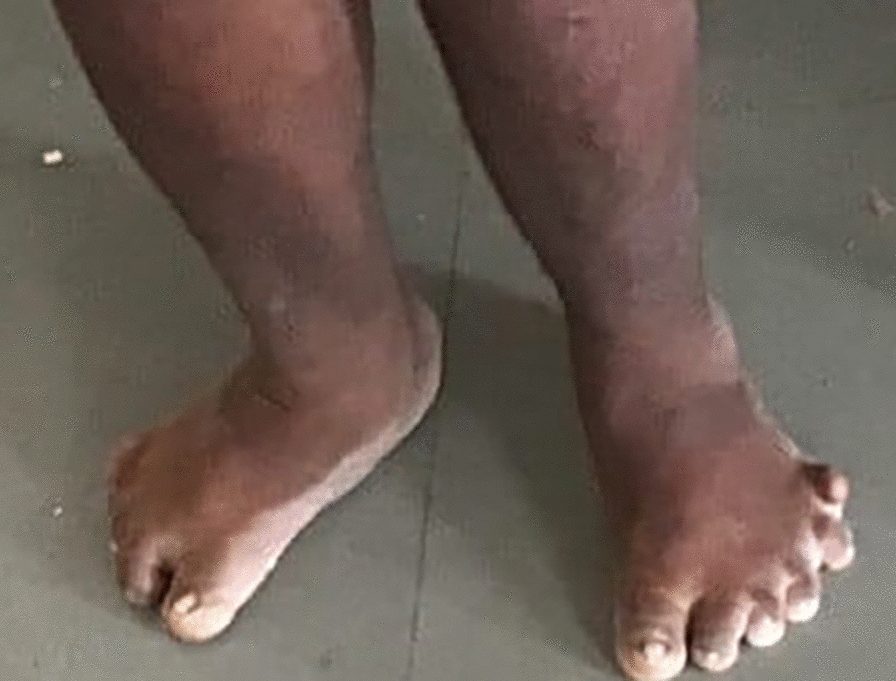
Polydactyly in the lower limbs
Table 1.
Beales diagnostic criteria
| Major criteria | |
|---|---|
| Rod cone dystrophy | + |
| Obesity | + |
| Postaxial polydactyly | + |
| Renal anomalies or renal dysfunction | + |
| Cognitive impairment | + |
An abdominal ultrasound showed grade 3 gross ascites along with a 12.5 cm liver with surface irregularities suggestive of chronic parenchymal liver disease. It also revealed a 2.5 cm renal cyst in the right kidney. Ascitic fluid analysis confirmed abdominal TB, though the patient never had symptoms of TB; the results are presented in Table 2. The patient had elevated creatinine levels, abnormal liver function tests, and abnormal lipid profile. The laboratory results are presented in Tables 2, 3, and 4.
Table 2.
Ascitic fluid analysis
| Test | Value |
|---|---|
| Color | Yellow hazy fluid |
| Albumin | 2.1 g/dL |
| Protein | 4.39 g/dL |
| Sugar | 96 mg/dL |
| Cell count | 1200 cells/cumm |
| Neutrophils | Nil |
| Lymphocytes | 86% |
| Eosinophils | Nil |
| Macrophages | 10% |
| Mesothelial cells | 4% |
| ADA | 102.6 U/l |
| Microscopy: | Smear is composed of sheets of lymphocytes, macrophages, neutrophils, and mesothelial cells. There is no evidence of malignancy in the smears studied |
Table 3.
Liver, renal, and thyroid function tests
| Test | Result | Reference range |
|---|---|---|
| Total bilirubin | 1.072 mg/dL | 0.2–1.2 mg/dL |
| Direct bilirubin | 0.72 mg/dL | Up to 0.3 mg/dL |
| AST (SGOT) | 34.9 U/L | 5–40.0 U/L |
| ALT (SGPT) | 13.0 U/L | 5–40.0 U/L |
| Albumin | 2.73 g/dL | 3.2–5.5 g/dL |
| Globulins | 5.7 g/dL | 1.8–3.4 g/dL |
| Total proteins | 8.43 g/dL | 6–8.3 g/dL |
| Alkaline phosphatase | 68 U/L | 40–129 U/L |
| Random glucose | 173.1 mg/dL | 70–140 mg/dL |
| Urea | 44.9 mg/dL | 10–45 |
| Creatinine | 2.26 mg/dL | 0.4–1.4 |
| Sodium | 132 mmol/L | 136–149 |
| Potassium | 2.70 mmol/L | 3.5–5.3 |
| Chloride | 86.3 mmol/L | 98–111 |
| T3 | 107.1 ng/dL | 81–178 |
| T4 | 10.05 μg/dL | 4.5–12.5 |
| TSH | 0.400 μIU/ml | 0.4–4.2 |
| Cholesterol | 255 mg/dL | < 200 mg/dL |
| Triglycerides | 172 mg/dL | < 150 mg/dL |
| HDL cholesterol | 63 mg/dL | > 60 mg/dL |
| LDL cholesterol | 148 mg/dL | 60–130 mg/dL |
Table 4.
Complete blood counts
| Test | Value | Reference range |
|---|---|---|
| Hemoglobin | 11.1 g/dL | 13.2 to 16.6 g/dL |
| Total leukocyte count | 6440 cells/cumm | 3400–9600 cells/cumm |
| Differential leukocyte count | ||
| Neutrophils | 61.4% | 40–70% |
| Lymphocytes | 25.5% | 20–40% |
| Eosinophils | 2.2% | 0–5% |
| Monocytes | 10.6% | 0–10% |
| Basophils | 0.3% | 0–1% |
| Platelet count | 102,000 lakhs/cumm | 135,000–317,000 lakhs/cumm |
| Total RBC count | 4.00 million/cumm | 4.35–5.65 million/cumm |
| Hematocrit value (Hct) | 32.6% | 38.3–48.6% |
| Mean corpuscular volume (MCV) | 81.5 fL | 76–100 fL |
| Mean corpuscular hemoglobin (MCH) | 27.8 Pg | 27–32 Pg |
| Mean corpuscular hemoglobin concentration (MCHC) | 34.0% | 32–35% |
| R.D.W. | 14.4% | 12–14% |
The patient was prescribed diuretics and was kept on a low-sodium diet to resolve edema and gross ascites. He was corrected for hypokalemia with potassium chloride before therapy. He was initiated on antitubercular therapy to address the abdominal TB. Therapeutic paracentesis and oxygen administration were provided to reduce the abdominal distension and dyspnea. Psychosocial support was offered to help him and his family deal with his intellectual handicap and general condition, while nutritional advice was given to help control his weight. To monitor his response to treatment, routine checkups have been scheduled, with an emphasis on electrolyte levels, weight, and abdominal imaging.
Discussion
The pathophysiology of BBS is thought to be caused by or involve at least 28 distinct genes. The condition’s underlying genetics are complicated. Numerous protein products derived from these genes support the integrity and functionality of the BBSome, a structure essential for the structure and function of primary cilia [7]. The BBS phenotype develops gradually over the first 10 years of life. Consequently, the majority of patients are diagnosed in late childhood or early adulthood [8]. The average age at diagnosis was 9 years [9]; however, this patient presented at the age of 30 years, which is unusual.
Early diagnosis facilitates anticipatory guidance and can be particularly beneficial in preventing or mitigating the onset of obesity. Whole-exome sequencing is currently the first diagnostic test used for patients with numerous congenital abnormalities. However, this patient has been treated in a limited setting, making genetic sequencing not possible. Therefore, Beales criteria are used to diagnose the patient. The diagnosis of BBS requires at least four major features or three major features and two minor features [9]. This patient had five out of six major criteria and one minor criterion, confirming BBS. Table 5 presents the major and minor Beales criteria.
Table 5.
Beales criteria
| Major criteria | |
|---|---|
| Rod cone dystrophy | |
| Obesity | |
| Postaxial polydactyly | |
| Hypogonadism | |
| Renal anomalies or renal dysfunction | |
| Cognitive impairment |
| Minor criteria | |
|---|---|
| Speech delay, dental anomalies, brachydactyly, genitourinary anomalies, diabetes, ataxia, syndactyly, developmental delay, congenital heart disease, anosmia or hyposmia, hepatic fibrosis |
Obesity is estimated to affect 72–86% of people with BBS [10]. In general, birth weight lies within the normal range, and weight gain frequently starts in the first few months and years of life [11]. Childhood obesity has typically been characterized as having a broad and nonspecific distribution. However, by adulthood, the trunk and proximal portions of the limbs exhibit the most evident adiposity. This patient presented in adulthood and was unable to provide a childhood history. However, the most significant sign of BBS is still rod-cone dystrophy, which is present in 90–100% of cases. In BBS, visual impairment has always started early, usually between the ages of 8 and 9, and by the third decade, 98% of patients had lost all of their vision. This patient was completely blinded by the age of 9 years from retinitis pigmentosa. Myopia, astigmatism, strabismus, cataracts, color abnormalities, and retinal degeneration are among the additional visual characteristics [12].
In total, 30% of patients also have liver disease, which is defined as abnormalities in liver imaging and/or high levels of plasma transaminase enzymes; other gastrointestinal abnormalities, such as Hirschsprung’s disease, celiac disease, inflammatory bowel disease, anal stenosis, and other anatomic anomalies, are more common than one might expect in the general population. Patients also had sensorineural hearing loss and hormonal manifestations such as metabolic syndrome, diabetes mellitus, hypothyroidism, and polycystic ovary syndrome [13]. Children with BBS often experience developmental delays in all domains, including gross motor, fine motor, and speech/language. Yet, most of them eventually reach significant developmental milestones, such as walking and talking [14]. They may experience a delay in reaching developmental milestones due to mental retardation or eyesight impairment. This case has suffered from cognitive impairment, leading to difficulty in completing his education.
In addition, 25% of people with BBS die by the age of 44 years, indicating significantly lower survival and longevity. The most frequent and common cause of death is renal failure. A study found that chronic uremia accounted for 38% of deaths, and renal impairment was present in 38% of patients at the time of death [15]. This patient had normal kidney sizes on ultrasound but had a renal cyst in the right kidney. He also had elevated creatinine levels, indicating renal manifestations of BBS. With the early onset of blindness, obesity, hypertension, and diabetes mellitus, Bardet–Biedl syndrome has a poor prognosis.
BBS is treated symptomatically and includes routine screening and monitoring for associated conditions such as obesity, renal abnormalities, and vision problems. Patients benefit from nutritional counseling and weight-management strategies. Ophthalmologic care for vision issues and nephrology evaluation for kidney abnormalities in adults is suggested [16]. Patients may receive surgical interventions for specific complications such as polydactyly, scoliosis, and urogenital malformations.
Conclusion
BBS is a rare genetic syndrome characterized by multisystem involvement and presenting with a variety of phenotypes. Genetic counseling and a comprehensive care plan tailored to the patient’s needs, incorporating a multidisciplinary team of experts, regular monitoring, and supportive services such as neuropsychological care and family support, are essential for effectively treating patients with BBS. In the coming years, preclinical research is expected to yield more targeted treatments and a deeper understanding of the pathophysiology of BBS. This case report highlights the importance of clinical diagnostic criteria, such as Beales criteria, in identifying BBS in resource-limited settings where genetic testing is not readily available. Despite the absence of genetic evaluation, a comprehensive clinical diagnosis enabled the timely diagnosis and management of the patient. Timely diagnosis and proper counseling of BBS are essential, as they can significantly improve the patient’s outcome and ultimately enhance the prognosis.
Acknowledgements
None
Author contributions
Author AA collected the data and participated in drafting the article, ASSM drafted the article, and the rest of the authors participated in writing the manuscript. All authors read and approved the final manuscript.
Funding
No external funding was received.
Data availability
Not applicable.
Declarations
Ethics approval and consent to participate
Not applicable in our institution.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that there are no competing interests regarding the publication of this paper.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Haq FU, Riaz A, Ullah I, Ali N, Khan I, Ikram J, Maqbool A. Understanding Bardet-Biedl syndrome: unveiling the complexities of this rare genetic disorder and its systematic review to identify its various variants with genetic analysis. World J Adv Res Rev. 2024;22(1):088–96. 10.30574/wjarr.2024.22.1.1073. [Google Scholar]
- 2.Ece Solmaz A, Onay H, Atik T, Aykut A, Cerrah Gunes M, Ozalp Yuregir O, Bas VN, Hazan F, Kirbiyik O, Ozkinay F. Targeted multi-gene panel testing for the diagnosis of Bardet Biedl syndrome: Identification of nine novel mutations across BBS1, BBS2, BBS4, BBS7, BBS9, BBS10 genes. Eur J Med Genet. 2015;58(12):689–94. 10.1016/j.ejmg.2015.10.011. [DOI] [PubMed] [Google Scholar]
- 3.Forsyth R, Gunay-Aygun M. Bardet-Biedl Syndrome Overview. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews®. Seattle (WA): University of Washington Seattle; 2003. p. 1993–2025. [PubMed] [Google Scholar]
- 4.Ankleshwaria C, Prajapati B, Parmar S, Rathod V, Patel H, Dhorajiya D, Chavda N, Parmar K, Pathan F, Chauhan M. Bardet-Biedl syndrome presenting in adulthood. Indian J Nephrol. 2022;32(6):633–6. 10.4103/ijn.ijn_320_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kute VB, Vanikar AV, Gumber MR, Patel HV, Shah PR, Patil SB, Trivedi HL. Bardet-biedl syndrome: a rare cause of chronic kidney disease. Indian J Clin Biochem. 2013;28(2):201–5. 10.1007/s12291-012-0275-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arora E, Fuks A, Meyer J, Chervenak J. Prenatal diagnosis of Bardet Biedl syndrome: a case report. Radiol Case Rep. 2022;18(1):326–30. 10.1016/j.radcr.2022.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tomlinson JW. Bardet-Biedl syndrome: a focus on genetics, mechanisms and metabolic dysfunction. Diabetes Obes Metab. 2024;26(Suppl 2):13–24. 10.1111/dom.15480. [DOI] [PubMed] [Google Scholar]
- 8.Forsythe E, Beales PL. Bardet-Biedl syndrome. Euro J Human Gen : EJHG. 2013;21(1):8–13. 10.1038/ejhg.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999;36(6):437–46. [PMC free article] [PubMed] [Google Scholar]
- 10.Rooryck C, Lacombe D. Le syndrome de Bardet-Biedl [Bardet-Biedl syndrome]. Ann Endocrinol. 2008;69(6):463–71. 10.1016/j.ando.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 11.Pomeroy J, Krentz AD, Richardson JG, Berg RL, VanWormer JJ, Haws RM. Bardet-Biedl syndrome: weight patterns and genetics in a rare obesity syndrome. Pediatr Obes. 2021;16(2): e12703. 10.1111/ijpo.12703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Priya S, Nampoothiri S, Sen P, Sripriya S. Bardet-Biedl syndrome: genetics, molecular pathophysiology, and disease management. Indian J Ophthalmol. 2016;64(9):620–7. 10.4103/0301-4738.194328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Melluso A, Secondulfo F, Capolongo G, Capasso G, Zacchia M. Bardet-Biedl syndrome: current perspectives and clinical outlook. Ther Clin Risk Manag. 2023;19:115–32. 10.2147/TCRM.S338653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forsyth R, Gunay-Aygun M. Bardet-Biedl Syndrome Overview. In: Adam MP, editor. GeneReviews®. Seattle: University of Washington; 2003. [PubMed] [Google Scholar]
- 15.O’Dea D, Parfrey PS, Harnett JD, Hefferton D, Cramer BC, Green J. The importance of renal impairment in the natural history of Bardet-Biedl syndrome. Am J Kidney Dis. 1996;27(6):776–83. 10.1016/s0272-6386(96)90513-2. [DOI] [PubMed] [Google Scholar]
- 16.Dollfus H, Lilien MR, Maffei P, et al. Bardet-Biedl syndrome improved diagnosis criteria and management: inter European Reference Networks consensus statement and recommendations. Eur J Hum Genet. 2024;32:1347–60. 10.1038/s41431-024-01634-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.