

Abstract
Directed evolution is a technique that allows for protein engineering without prior knowledge. Continuous directed evolution employs gene-specific hypermutation tied to functional selection within a single cell, enabling a broad search of sequence space for gene variants with improved or novel functions. However, currently available techniques for continuous directed evolution can be inflexible or laborious to establish. To address this issue, we present a modular toolkit for deaminase-fused viral RNA polymerase continuous directed evolution, based on Golden Gate assembly. We include an alternative RNA polymerase from phage SP6 and show that it can introduce gene-specific mutations. This work builds on the available repertoire of synthetic biology techniques, brings accessibility and versatility to directed evolution, and enables researchers to build custom and complex plasmids for their own evolutionary campaigns.
Keywords: continuous directed evolution, Golden Gate, toolkit, synthetic biology
Graphical Abstract
Graphical Abstract.

Introduction
Protein engineering has led to new medicines and improved industrial processes [1]. A key aspect of this field is the enhancement of enzymes for industrial applications. Creating improved enzymes has the potential to assist in addressing societal challenges from development of green chemistry to more efficient conversion of lignocellulosic biomass into useful products [2–4]. Due to the complexity of protein-structure–function relationships, engineering methods often rely on experimental diversification followed by screening, known as directed evolution. Classical approaches involve iterative cycles of in vitro genetic diversification and selection for altered function [5–9]. However, this can be time-consuming and labour-intensive. Continuous directed evolution (CDE) systems have been developed to overcome these limitations and involve in vivo mutation and selection of target sequences, with protein function coupled to a measurable phenotype, such as growth or fluorescence [9–15]. Each system has advantages and limitations [11], but ease of use and adaptability to a variety of host systems are likely to be important for adoption. One of the simplest approaches involves mutating a gene of interest using a deaminase-fused viral RNA polymerase [16–19]. All the machinery is contained within a plasmid, requiring only cloning the gene of interest into a target vector. This approach can be used for any organism in which T7 can be used as an expression system, such as cyanobacteria [20, 21], yeast [19, 22], or mammalian and plant cells [22, 23].
However, current T7-based systems are tailored to specific organisms, and are based on conventional restriction digest cloning, which presents a barrier to implementation in new species. To address this, we constructed a standardized modular cloning (MoClo) assembly library based on Golden Gate assembly (GG). GG uses type IIS restriction enzymes for single-step, ordered, multipart assembly [24], the utility of which is reflected by the number of existing assembly standards and toolkits for a variety of organisms [25–34], enabling part sharing across laboratories. Our toolkit uses the syntax of the Plant Golden Gate MoClo toolkit [27], dictating the 5′ and 3′ overhangs of Level 0 parts, and is therefore compatible with existing Plant MoClo-based toolkits, which will enable use in bacterial and photosynthetic systems.
In addition to the conventionally used T7 polymerase, our toolkit contains an alternative viral RNA polymerase from Salmonella enterica phage SP6 [35] to allow for greater flexibility. The kit enables users to assemble custom vectors for directed evolution, varying mutators, promoters, and genes of interest streamlining the process of setting up directed evolution experiments.
Materials and methods
Strains and growth conditions
For cloning, Escherichia coli 5-alpha (NEB, #C2987) was grown in liquid Terrific Broth at 37°C shaking at 250 rpm, and selected on Lysogeny Broth (LB) agar 1.5% (w/v) plates, supplemented with appropriate antibiotics (kanamycin, 50 μg/ml, spectinomycin, 100 μg/ml, ampicillin, 100 μg/ml, and chloramphenicol, 34 μg/ml) at 37°C. For evolution experiments, E. coli BL21 (NEB, #C2530) or 5-alpha were plated in LB agar supplemented with 100 μg/ml ampicillin, and 50 μg/ml kanamycin or 34 μg/ml chloramphenicol, and 0.2% arabinose to allow for expression.
Assembly of plasmids
Oligonucleotides were purchased from Twist Bioscience, GenScript and Integrated DNA Technologies (Supplementary Table S1). Parts were domesticated, removing internal BbsI, BsaI, and SapI restriction sites by polymerase chain reaction (PCR) with Q5 (NEB, #M0492) using overhang primers, and cloned into the universal acceptor vector, pL0R. Spectinomycin resistance artificial stop codon mutants were created by performing two mutagenic PCRs per plasmid followed by Gibson Assembly [36, 37].
Plasmids were generated by Golden Gate Assembly, following Plant MoClo syntax [27, 38–40] and using the Universal Loop assembly system [33, 34]. Briefly, a mixture of 1.5 nM of each insert and 0.75 nM backbone was assembled with 0.5 U/μl BsaI-HF-v2 (NEB, #R3733), or 0.5 U/μl SapI (NEB, #R0569) and 10 U/μl T4 DNA ligase (NEB, #M0202) in 1X T4 DNA ligase buffer and BSA (0.05 mg/ml, for BsaI reactions) or 1X rCutSmart (for SapI reactions). Reactions were cycled 25 times at 37°C for 3 min, 16°C for 4 min, then 50°C for 20 min and 80°C for 20 min (Supplementary Text S1). The plasmid product was transformed into E. coli via CaCl2-heat shock transformation, extracted with ZymoPure kit (Zymo Research, #D4211) according to the manufacturer’s instructions, and checked by whole plasmid sequencing (Plasmidsaurus, USA). Plasmids used are given in Supplementary Table S2.
pL0R, pCAo-1,2,3,4, and pCAe-1,2,3,4 were a gift from Christopher Dupont and can be accessed via Addgene (#1000000238 [34]). In addition, the CyanoGate Kit (#1000000146 [38]), pJUMP26-1A and pJUMP27-1A (#126973 and #126974 [41]), and MoClo-YTK (#1000000061 [26]) were purchased from Addgene.
Evolution experiments
Plasmids were cotransformed into E. coli by electroporation using 0.2 cm cuvettes (V = 2.5 kV, BioRad # 1652100), and plated in appropriate media. Colonies were picked into 3 ml LB with appropriate antibiotics and grown to saturation. This was passage 0 (P0). This culture was diluted 1:1000 in 3 ml of fresh LB supplemented with 0.2% 0.22-μm-filter-sterilized arabinose, kanamycin, and ampicillin, and grown to saturation (~12 h). This was a single passage and was repeated up to 14 times. At the end of each passage, serial dilutions were made and plated in selective (spectinomycin 100 μg/ml) and non-selective LB. Each individual evolving culture was counted as one biological replicate (n = 3–4).
Suppressor frequency was calculated according to Equation (1):
 |
(1) |
according to Seo et al. (2023) [42] and the log10(suppressor frequency) was calculated for graphing purposes. Populations with no colonies in selective plates were graphed as log10(suppressor frequency) = −9 for visualization purposes. To determine mutation count, colonies were randomly picked from selective plates. Each colony from a plasmid configuration was one biological replicate (n = 8–10). They were grown in LB with spectinomycin (100 μg/ml), plasmids were extracted, and target sequences were amplified by PCR with Q5 (NEB, #M0492) using primers F.JUMP27.GOI_bc_n.seq and R.JUMP27.GOI_bc_n.seq (Supplementary Table S1), with a Tm of 65°C and an extension time of 45 s due to the amplicon length of 1.3 kb, where n was a barcode at the 5′ and 3′ ends of the primer. The PCR amplicons were size verified by gel electrophoresis and purified using Monarch PCR purification kit (NEB #T1130) according to the manufacturer’s instructions, pooling 16 PCRs at a time (Supplementary Table S1 ), and sequenced by Plasmidsaurus Premium PCR (fragmentation-free linear amplicon sequencing, see Supplementary Text S2), or Sanger sequenced wherever indicated (Roy J. Carver Biotechnology Center). The sequences were analysed with a simplified MAPLE pipeline [43] (Supplementary Text S2) and aligned to the unmutated plasmid sequence. Mutation count was calculated by noting the mutation frequencies at given nucleotide positions across the amplicon. A position was counted as mutated if its mutation frequency was higher than the background Nanopore error rate to account for plasmid heterogeneity (Supplementary Text S2).
Statistical analysis
Data were processed and analysed in R 4.4.0 and RStudio 2024.12.0 + 467. Graphs were generated using ggplot2, Viridis, and ggpubR [44–46]. Data were checked for normality by Shapiro–Wilk test. Statistical tests were performed on mutation count data with each sequence and significance assessed by the Kruskal–Wallis test followed by the post hoc Dunn’s test. All raw fluctuation assay colony count data, mutation count data, and statistical tests can be found in the source data file.
Results and discussion
The toolkit was created by separating the main components of the current gold standard deaminase-based CDE tool, eMutaT7transition [42] into Level 0 parts. eMutaT7transition contains two arabinose-inducible chimeric T7 RNAP fusions, with an adenine or cytosine deaminase (TadA8E or PmCDA1, respectively), as well as a highly expressed uracil-N-glycosylase inhibitor (UGI). In addition, we included in the toolkit a T7-responsive promoter, T7mut2, with an optimized ribosome binding sequence [21], an additional inducible promoter, and variants of the RNAP coding sequences that connect with and without the need for deaminases, as control parts (Fig. 1; Supplementary Table S3).
Figure 1.
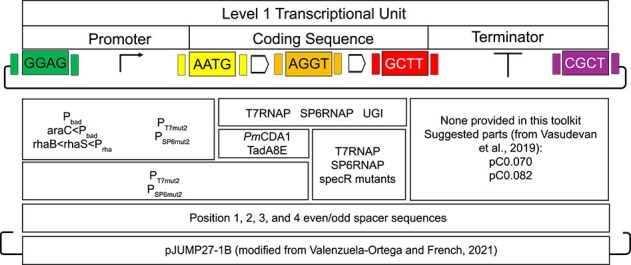
A Golden Gate toolkit for VRNAP CDE. The basic architecture for this toolkit enables users to rapidly build plasmids using Golden Gate assembly and modular cloning (MoClo) that enables continuous directed evolution of specified genes. Each included part is given in the box corresponding to the overhangs marked in the upper part of the figure. Part versions with different overhangs are included in their respective boxes. The multigene assembly aspect allows users to include in a single plasmid other genes of interest that might be required for the evolution to take place.
Because T7 RNA polymerase (RNAP) is used as an expression system, CDE with MutaT7-style tools is incompatible when T7 is used for the expression of other genes, which could be important when chaperones or other proteins need to be expressed to enable evolution schemes. To address this, we synthesized SP6 RNAP from SP6 phage, which is also used for transcription and synthesis of nucleic acids owing to its high promoter specificity and processivity [47] and created an SP6-specific promoter, SP6mut2, modifying the T7 binding sequence from T7mut2 for recognition by SP6.
To construct higher order assemblies, the Loop assembly standard and toolkit were employed [33, 34]. In brief, Level 1 constructs represent individual transcriptional units assembled from a variable number of compatible parts, while Level 2 is assembled from four Level 1’s, Level 3 is assembled from four Level 2’s, and so forth. This setup allows our toolkit to be fully compatible with laboratories established with Plant MoClo or other toolkits, while enabling efficient (Supplementary Fig. S3; Supplementary Table S4) multigene assemblies with few vectors and expanding potential organism applicability due to the Loop vectors and standard.
To test SP6 and the toolkit, we chose a stop codon reversal fluctuation assay. A library of spectinomycin resistance (SpecR) stop codon mutants (Supplementary Table S2 and Supplementary Text S3) was created. The selection scheme was tested with eMutaT7transition [42] and the mutant library to verify the assay’s selection and tool’s function. Colonies appeared for all stop codon mutant-carrying populations except for S36* (Supplementary Fig. S1). The tool was able to introduce between one and eight mutations across colonies sequenced without any obvious mutational bias (Supplementary Fig. S2), and all sequenced colonies having reverted the stop codon. Therefore, we focused on reversion in one of the mutants, L13*, which was chosen for all subsequent analyses of mutators. It was cloned under SP6mut2 into our target plasmid: a low copy (SC101-nonTS) backbone, pJUMP27 [41], modified to contain AmpR to enable compatibility with mutator plasmids made in the Loop standard.
We re-assembled eMutaT7transition using GG parts to validate the modularity of the tool and created eMutaSP6 with the same concept but with SP6 RNAP instead (Fig. 2A–G). Our mutators were constructed to be arabinose-inducible (PBAD originating from Cyanogate kit [38]), contain UGI, a DNA repair inhibitor, expressed from the promoter J23104, a strong constitutive synthetic E. coli promoter, into a p15 backbone plasmid pJUMP26 [41]. Using this approach, Level 3 constructs can be made and verified in 10 days.
Figure 2.

Characterization of continuous directed evolution mutator plasmids in BL21. (A–G) Schematic of constructs used in each evolution experiment and log (suppressor frequency) of independently evolving populations among the different constructs at select passages, P0–P14 (24 h between each shown timepoint) (n = 3 biological replicates). p15 indicates p15 backbone (low copy) and pCA indicates pCAMBIA backbone (high copy). Origin of replication is indicated by point colour and induction by shape. H—Number of mutations found in the gene of interest at the final timepoint for the indicated constructs. Each data point corresponds to an individual colony picked from a selective plate. Significance calculated using Kruskal–Wallis test followed by a post hoc Dunn’s test (n = 10 biological replicates) with the Bonferroni correction. RNAP = RNA polymerase, PmCDA1 = Petromyzon marinus cytidine deaminase 1, TadA8E = evolved E. coli adenine deaminase, UGI = uracil-N-glycosylase inhibitor.
During vector assembly, we observed a high frequency of aberrant plasmids when using two or more spacer parts, leading to concatemers, multi-backbone or multi-marker plasmids. This was especially prevalent in Level 2 assemblies. We hypothesized that this may result from recombination due to the identical sequences of the Loop kit spacer. We created new spacers, based on ~ 200 bp regions of GFP and other parts that should not lead to unwanted effects. When using these, we no longer observed recombination events, suggesting that GG assembly efficiency can be improved by avoiding part repetition [34] (Supplementary Fig. S3; Supplementary Table S4).
We tested eMutaSP6 in the stop codon reversal fluctuation assay. We observed an increase in colony number between P0 and P2, which continued to increase up to P14 (Fig. 2C). The same effect could be seen for the reassembled eMutaT7. Additionally, we hypothesized that higher plasmid copy number would lead to an increase in mutation rate, so we cloned this same construct into pCAo-1, containing the pBR322 origin of replication [33], but were unable to discern a difference in the suppressor frequency patterns between the two origins of replication (Fig. 2B).
We constructed negative controls that lacked the fused deaminases and the mutator altogether, as well as controls with swapped promoters (Fig. 2C–G). These controls showed colony count increases, but only in later passages (Fig. 2C–G). This could have been due to genomic mutations [48]. Finally, we used eMutaT7transition [42] as a positive control (Fig. 2A), which showed a similar pattern to that of our constructs.
During testing of the finalized constructs, we noticed colonies appearing in mutator-carrying lines at P0 prior to inducer exposure (Supplementary Fig. S4), which showed mutations in the gene (Supplementary Fig. S5). We observed that the template that had been used for making our PBAD part lacked critical regions, including the regulator CDS of araC, a transcription start site, and a canonical ribosome binding site [38]. We addressed this by modifying our part to include araC and the canonical elements [49], and also creating another version lacking araC for stacking inducible mutators. We also include in the toolkit the tighter-repression, rhamnose-inducible promoter PRHAB [50] that did not need domestication and whose functionality was verified in an unrelated experiment (Supplementary Fig. S6). Through the toolkit, these plasmid modifications only took around 2 weeks to implement, highlighting the value of streamlined assembly methods and the importance of part verification. Throughout the pipeline, cloning efficiency was high throughout (0.869–1.0 white colony frequency, Supplementary Table S4) similar to previous reports [34], and the overall colony count was reflective of acceptor vector copy number.
To assess mutation induction, the gene of interest in colonies from selective plates was investigated following the end of the evolution experiment. The target region was amplified containing the gene of interest and ~ 300 bp up and downstream. We reasoned that this would be sufficient to assess whether off-target mutations were common given the reported frequency of eMutaT7transition [42]. Sequencing the target amplicon of eight randomly picked colonies from the final passage showed that both eMutaSP6 and reassembled eMutaT7 were able to introduce mutations to the gene of interest (Fig. 2H), but not to the extent of eMutaT7transition. This could potentially be explained by the different degrees of UGI expression, which has been shown to influence the rate of mutation accumulation [42] (Supplementary Fig. S7).
However, mutations were also present in controls, suggesting that resistant colonies in those lines, or in mutator-carrying lines, may be linked to random mutagenesis stemming from metabolically burdening of mutation-prone BL21 cells with the maintenance of two plasmids [48] (Fig. 2H). In all mutator-carrying lines but not all sequenced colonies, one to two mutations occurring in the terminator (TrrnB) could be observed (Supplementary Table S5), that were not present in the control lines. Our amplicon was too short to determine whether the terminator was able to prevent further read-through mutations beyond it. Previous studies have shown that genomic off-target mutation is rare with RNAP-based mutators [16, 17, 25, 42, 51], but it is possible that read-through and consequent mutation of the plasmid backbone could lead to detrimental effects for plasmid maintenance and subsequent evolution. Further optimization could be performed by testing different terminators to reduce the risk of read-through, as terminator strength has been well characterized in E. coli [52]. An alternative approach could be to use concatenated terminators [16].
The same constructs were tested in a different chassis, 5-alpha E. coli, for a shorter evolution. This strain is engineered to produce and preserve DNA, which we reasoned could lead to lower innate mutation. No colonies appeared in any control line (Supplementary Fig. S8A) at the 24-h time point, P2, contrary to the observed colonies in control lines at P2 in BL21 (Fig. 2B). On the other hand, colonies appeared in our construct-carrying lines (Supplementary Fig. S8A), but to a lesser degree than what had been observed in BL21 across all lines. Sequencing of resistant colonies in the final passage revealed mutations present in mutator-carrying lines (Supplementary Fig. S8B). This suggests that low innate mutation is likely to be important in CDE experiments.
The target plasmid hosting the gene of interest contains a pSC101 origin of replication. While low copy (typically maintained at around four to five copies per cell [53]), each copy could accumulate unique mutations. Analysis of the mutation frequencies in both the BL21 and 5-alpha experiments revealed that, when occurring, stop codon reversal swept the population, appearing at frequencies of or near 1.0, suggesting that this mutation was necessary for spectinomycin resistance re-evolution. For lines carrying mutators, increased mutation frequency (between 10% and 100% of reads) above background Nanopore error rate was sometimes detectable at other nucleotide positions, depending on demultiplexing efficiency. This is possibly attributed to a heterogeneous plasmid population within a colony at the time of picking or emerging during outgrowth. In CDE, it is of interest to be able to differentiate causal mutations from those which are neutral. As shown, one possible way of doing so is observing convergent evolution across multiple independent populations, although this is not always feasible when selection schemes or setups are costlier or more elaborate than ours. An alternative would be to integrate the gene of interest into the E. coli chromosome, so it exists in a single copy in each cell [54]. However, it is also conceivable that multiple copies could be beneficial for evolution, as it could be important as a mechanism to buffer selection survival with suboptimal solutions while traversing fitness landscapes in search of global optima. Further experimentation is required to assess the relative importance of each possibility in evolution campaigns.
Overall, these results indicate that deaminase-fused SP6 functions as an alternative to T7 in RNAP-based CDE. It can specifically introduce mutations to a gene of interest under the SP6 promoter, allowing for T7-driven protein expression in tandem with CDE, albeit with an apparent lower rate of mutation despite previous reports suggesting higher processivity [47]. Consistent with previous reports [18] our results highlight the importance of tuning UGI expression for optimal mutation rates and selection of an appropriate chassis to separate mutations and background.
In addition, the toolkit enables researchers to create customizable directed evolution plasmids with the necessary evolution and ancillary selection parts, and uniquely needed parts can be added easily, which increases this technique’s applicability. Furthermore, it could also permit more complex schemes, such as the parallel evolution of two genes, outcome-dependent sequential evolution, or porting to other organisms, although these remain to be tested. This work is a step towards a more accessible and versatile synthetic biology arsenal to tackle challenges of the 21st century.
Supplementary Material
Acknowledgements
We thank the members of the Burgess, the Brooks, and Leonelli labs for useful discussions. We thank the Roy J. Carver Biotechnology Center for their help with Sanger sequencing.
Contributor Information
Ignacio Sparrow Muñoz, Department of Plant Biology, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA; Institute for Genomic Biology, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA; Center for Advanced Bioenergy and Bioproducts Innovation, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA.
Steven J Burgess, Department of Plant Biology, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA; Institute for Genomic Biology, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA; Center for Advanced Bioenergy and Bioproducts Innovation, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA.
Author contributions
Ignacio Sparrow Muñoz (Data curation, Formal analysis, Investigation, Methodology, Writing—original draft, Writing—review & editing), Steven Burgess (Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing—original draft, Writing—review & editing)
Conflict of interest: The authors have filed a provisional patent application.
Funding
I.S.M. and S.J.B. were supported by the Center for Advanced Bioenergy and Bioproducts Innovation (CABBI) at the University of Illinois and supported by U.S. Department of Energy, Office of Science, Biological and Environmental Research Program under Award Number DE-SC0018420.
Data and material availability
All supplementary data, including raw fluctuation assay data, mutation count data, primers, and code are available at SYNBIO online and at doi: 10.6084/m9.figshare.28570430 as a ZIP file. Some of these data are presented additionally in the provided supplementary information file for ease of access. Please note that this DOI has been reserved for publication and will not be published until we receive final confirmation of the patent.
The ZIP file contains the following:
Novel nucleic acid sequences (created plasmids) are provided in the GenBank format and are annotated.
PCR primers are included in the file primers.csv, including information on the purpose of the primer (pair), template used, and reference where available.
Sequencing is deposited in SRA under accession XXX.
R script for analysis and graphing of fluctuation assay data and mutation count data.
All parts in the toolkit will be made available at Addgene XXXXX as a 33-part kit once patent confirmation is received.
References
- 1. Arnold FH. Innovation by evolution: bringing new chemistry to life (nobel lecture). Angew Chem Int Ed 2019;58:14420–6. 10.1002/anie.201907729 [DOI] [PubMed] [Google Scholar]
- 2. Lee Y, Khanna M, Chen L et al. Quantifying uncertainties in greenhouse gas savings and abatement costs with cellulosic biofuels. Eur Rev Agric Econ 2023;50:1659–84. 10.1093/erae/jbad036 [DOI] [Google Scholar]
- 3. Deshavath NN, Woodruff W, Singh V. Sustainable strategies to achieve industrial ethanol titers from different bioenergy feedstocks: scale-up approach for better ethanol yield. Sustain Energy Fuels 2024;8:3386–98. 10.1039/D4SE00520A [DOI] [Google Scholar]
- 4. Joshi G, Pandey JK, Rana S et al. Challenges and opportunities for the application of biofuel. Renew Sust Energ Rev 2017;79:850–66. 10.1016/j.rser.2017.05.185 [DOI] [Google Scholar]
- 5. Arnold FH. Design by directed evolution. Acc Chem Res 1998;31:125–31. 10.1021/ar960017f [DOI] [Google Scholar]
- 6. McLure RJ, Radford SE, Brockwell DJ. High-throughput directed evolution: a golden era for protein science. Trends Chem 2022;4:378–91. 10.1016/j.trechm.2022.02.004 [DOI] [Google Scholar]
- 7. Molina RS, Rix G, Mengiste AA et al. In vivo hypermutation and continuous evolution. Nat Rev Methods Primers 2022;2:1–22. 10.1038/s43586-022-00119-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rix G, Liu CC. Systems for in vivo hypermutation: a quest for scale and depth in directed evolution. Curr Opin Chem Biol 2021;64:20–6. 10.1016/j.cbpa.2021.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang Y, Xue P, Cao M et al. Directed evolution: methodologies and applications. Chem Rev 2021;121:12384–444. 10.1021/acs.chemrev.1c00260 [DOI] [PubMed] [Google Scholar]
- 10. You L, Arnold FH. Directed evolution of subtilisin E in Bacillus subtilis to enhance total activity in aqueous dimethylformamide. Protein Eng 1996;9:77–83. 10.1093/protein/9.1.77 [DOI] [PubMed] [Google Scholar]
- 11. García-García JD, Van Gelder K, Joshi J et al. Using continuous directed evolution to improve enzymes for plant applications. Plant Physiol 2022;188:971–83. 10.1093/plphys/kiab500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rix G, Watkins-Dulaney EJ, Almhjell PJ et al. Scalable continuous evolution for the generation of diverse enzyme variants encompassing promiscuous activities. Nat Commun 2020;11:5644. 10.1038/s41467-020-19539-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ravikumar A, Arzumanyan GA, Obadi MKA et al. Scalable, continuous evolution of genes at mutation rates above genomic error thresholds. Cell 2018;175:1946–1957.e13. 10.1016/j.cell.2018.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuchner O, Arnold FH. Directed evolution of enzyme catalysts. Trends Biotechnol 1997;15:523–30. 10.1016/S0167-7799(97)01138-4 [DOI] [PubMed] [Google Scholar]
- 15. Packer MS, Liu DR. Methods for the directed evolution of proteins. Nat Rev Genet 2015;16:379–94. 10.1038/nrg3927 [DOI] [PubMed] [Google Scholar]
- 16. Moore CL, Papa LJ, Shoulders MD. A processive protein chimera introduces mutations across defined DNA regions in vivo. J Am Chem Soc 2018;140:11560–4. 10.1021/jacs.8b04001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mengiste AA, Wilson RH, Weissman RF et al. Expanded MutaT7 toolkit efficiently and simultaneously accesses all possible transition mutations in bacteria. Nucleic Acids Res 2023;51:e31. 10.1093/nar/gkad003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Park H, Kim S. Gene-specific mutagenesis enables rapid continuous evolution of enzymes in vivo. Nucleic Acids Res 2021;49:e32. 10.1093/nar/gkaa1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cravens A, Jamil OK, Kong D et al. Polymerase-guided base editing enables in vivo mutagenesis and rapid protein engineering. Nat Commun 2021;12:1579. 10.1038/s41467-021-21876-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shono C, Ariyanti D, Abe K et al. A green light-regulated T7 RNA polymerase gene expression system for cyanobacteria. Mar Biotechnol 2021;23:31–8. 10.1007/s10126-020-09997-w [DOI] [PubMed] [Google Scholar]
- 21. Jin H, Lindblad P, Bhaya D. Building an inducible T7 RNA polymerase/T7 promoter circuit in Synechocystis sp. PCC6803. ACS Synth Biol 2019;8:655–60. 10.1021/acssynbio.8b00515 [DOI] [PubMed] [Google Scholar]
- 22. Hobl B, Hock B, Schneck S et al. Bacteriophage T7 RNA polymerase-based expression in Pichia pastoris. Protein Expr Purif 2013;92:100–4. 10.1016/j.pep.2013.09.004 [DOI] [PubMed] [Google Scholar]
- 23. Chen H, Liu S, Padula S et al. Efficient, continuous mutagenesis in human cells using a pseudo-random DNA editor. Nat Biotechnol 2020;38:165–8. 10.1038/s41587-019-0331-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Engler C, Kandzia R, Marillonnet S. A one pot, one step, precision cloning method with high throughput capability. PLoS One 2008;3:e3647. 10.1371/journal.pone.0003647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Larroude M, Park Y-K, Soudier P et al. A modular Golden Gate toolkit for Yarrowia lipolytica synthetic biology. Microb Biotechnol 2019;12:1249–59. 10.1111/1751-7915.13427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee ME, DeLoache WC, Cervantes B et al. A highly characterized yeast toolkit for modular, multipart assembly. ACS Synth Biol 2015;4:975–86. 10.1021/sb500366v [DOI] [PubMed] [Google Scholar]
- 27. Engler C, Youles M, Gruetzner R et al. A Golden Gate modular cloning toolbox for plants. ACS Synth Biol 2014;3:839–43. 10.1021/sb4001504 [DOI] [PubMed] [Google Scholar]
- 28. Chiasson D, Giménez-Oya V, Bircheneder M et al. A unified multi-kingdom Golden Gate cloning platform. Sci Rep 2019;9:10131. 10.1038/s41598-019-46171-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu D, Schandry N, Lahaye T. A modular toolbox for Golden-Gate-based plasmid assembly streamlines the generation of Ralstonia solanacearum species complex knockout strains and multi-cassette complementation constructs. Mol Plant Pathol 2018;19:1511–22. 10.1111/mpp.12632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bonturi N, Pinheiro MJ, de Oliveira PM et al. Development of a dedicated Golden Gate Assembly Platform (RtGGA) for Rhodotorula toruloides. Metab Eng Commun 2022;15:e00200. 10.1016/j.mec.2022.e00200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pryor JM, Potapov V, Kucera RB et al. Enabling one-pot Golden Gate assemblies of unprecedented complexity using data-optimized assembly design. PLoS One 2020;15:e0238592. 10.1371/journal.pone.0238592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bird JE, Marles-Wright J, Giachino A. A user’s guide to Golden Gate cloning methods and standards. ACS Synth Biol 2022;11:3551–63. 10.1021/acssynbio.2c00355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pollak B, Cerda A, Delmans M et al. Loop assembly: a simple and open system for recursive fabrication of DNA circuits. New Phytol 2019;222:628–40. 10.1111/nph.15625 [DOI] [PubMed] [Google Scholar]
- 34. Pollak B, Matute T, Nuñez I et al. Universal loop assembly: open, efficient and cross-kingdom DNA fabrication. Synth Biol 2020;5:ysaa001. 10.1093/synbio/ysaa001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Conrad T, Plumbom I, Alcobendas M et al. Maximizing transcription of nucleic acids with efficient T7 promoters. Commun Biol 2020;3:1–8. 10.1038/s42003-020-01167-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gibson DG, Young L, Chuang R-Y et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 2009;6:343–5. 10.1038/nmeth.1318 [DOI] [PubMed] [Google Scholar]
- 37. Olszakier S, Berlin S. A simplified Gibson assembly method for site directed mutagenesis by re-use of standard, and entirely complementary, mutagenesis primers. BMC Biotechnol 2022;22:10. 10.1186/s12896-022-00740-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vasudevan R, Gale GAR, Schiavon AA et al. CyanoGate: a modular cloning suite for engineering cyanobacteria based on the plant MoClo syntax. Plant Physiol 2019;180:39–55. 10.1104/pp.18.01401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Werner S, Engler C, Weber E et al. Fast track assembly of multigene constructs using Golden Gate cloning and the MoClo system. Bioengineered 2012;3:38–43. 10.4161/bbug.3.1.18223 [DOI] [PubMed] [Google Scholar]
- 40. Weber E, Engler C, Gruetzner R et al. A modular cloning system for standardized assembly of multigene constructs. PLoS One 2011;6:e16765. 10.1371/journal.pone.0016765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Valenzuela-Ortega M, French C. Joint universal modular plasmids (JUMP): a flexible vector platform for synthetic biology. Synth Biol (Oxf) 2021;6:ysab003. 10.1093/synbio/ysab003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Seo D, Koh B, Eom G et al. A dual gene-specific mutator system installs all transition mutations at similar frequencies in vivo. Nucleic Acids Res 2023;51:e59. 10.1093/nar/gkad266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rix G, Williams RL, Hu VJ et al. Continuous evolution of user-defined genes at 1 million times the genomic mutation rate. Science 2024;386:eadm9073. 10.1126/science.adm9073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wickham H. ggplot2. Cham: Springer International Publishing; 2016. 10.1007/978-3-319-24277-4 [DOI] [Google Scholar]
- 45. Kassambara A. ggpubr: “ggplot2” Based Publication Ready Plots 2023. https://cran.r-project.org/web/packages/ggpubr/index.html
- 46. Garnier S, Ross N, Camargo AP, Rudis Bob, Woo K. Sjmgarnier/viridisLite: viridisLite 0.4.0 (Pre-CRAN Release) 2021. 10.5281/zenodo.4678327 [DOI]
- 47. Stump WT, Hall KB. SP6 RNA polymerase efficiently synthesizes RNA from short double-stranded DNA templates. Nucleic Acids Res 1993;21:5480–4. 10.1093/nar/21.23.5480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Csörgő B, Fehér T, Tímár E et al. Low-mutation-rate, reduced-genome Escherichia coli: an improved host for faithful maintenance of engineered genetic constructs. Microb Cell Factories 2012;11:11. 10.1186/1475-2859-11-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Guzman LM, Belin D, Carson MJ et al. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 1995;177:4121–30. 10.1128/jb.177.14.4121-4130.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jensen SI, Lennen RM, Herrgård MJ et al. Seven gene deletions in seven days: fast generation of Escherichia coli strains tolerant to acetate and osmotic stress. Sci Rep 2015;5:17874. 10.1038/srep17874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mengiste AA, McDonald JL, Nguyen Tran MT et al. MutaT7GDE: a single chimera for the targeted, balanced, efficient, and psrocessive installation of all possible transition mutations in vivo. ACS Synth Biol 2024;13:2693–701. 10.1021/acssynbio.4c00316 [DOI] [PubMed] [Google Scholar]
- 52. Chen Y-J, Liu P, Nielsen AAK et al. Characterization of 582 natural and synthetic terminators and quantification of their design constraints. Nat Methods 2013;10:659–64. 10.1038/nmeth.2515 [DOI] [PubMed] [Google Scholar]
- 53. Thompson MG, Sedaghatian N, Barajas JF et al. Isolation and characterization of novel mutations in the pSC101 origin that increase copy number. Sci Rep 2018;8:1590. 10.1038/s41598-018-20016-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee JW, Gyorgy A, Cameron DE et al. Creating single-copy genetic circuits. Mol Cell 2016;63:329–36. 10.1016/j.molcel.2016.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Garnier S, Ross N, Camargo AP, Rudis Bob, Woo K. Sjmgarnier/viridisLite: viridisLite 0.4.0 (Pre-CRAN Release) 2021. 10.5281/zenodo.4678327 [DOI]