

Abstract
Gephyrin was originally identified as a membrane-associated protein that is essential for the postsynaptic localization of receptors for the neurotransmitters glycine and GABAA. A sequence comparison revealed homologies between gephyrin and proteins necessary for the biosynthesis of the universal molybdenum cofactor (MoCo). Because gephyrin expression can rescue a MoCo-deficient mutation in bacteria, plants, and a murine cell line, it became clear that gephyrin also plays a role in MoCo biosynthesis. Human MoCo deficiency is a fatal disease resulting in severe neurological damage and death in early childhood. Most patients harbor MOCS1 mutations, which prohibit formation of a precursor, or carry MOCS2 mutations, which abrogate precursor conversion to molybdopterin. The present report describes the identification of a gephyrin gene (GEPH) deletion in a patient with symptoms typical of MoCo deficiency. Biochemical studies of the patient’s fibroblasts demonstrate that gephyrin catalyzes the insertion of molybdenum into molybdopterin and suggest that this novel form of MoCo deficiency might be curable by molybdate supplementation.
In mammals, molybdenum cofactor (MoCo) is essential for the activity of sulfite oxidase, xanthine dehydrogenase, and aldehyde oxidase (Johnson and Wadman 1995). Mutations affecting the MoCo biosynthetic pathway result in the simultaneous loss of all molybdoenzyme activities. The cerebral atrophy associated with MoCo deficiency (MIM 252150) is also observed in isolated sulfite oxidase deficiency (MIM 272300) and can be attributed exclusively to the loss of this enzyme. Both the isolated and the combined form are inherited as autosomal recessive traits, without any symptoms in heterozygous carriers, and they come to clinical attention because of untreatable neonatal seizures, with opisthotonos and facial dysmorphism. The combined form is found more often, with ∼100 cases known worldwide. We had examined 42 of these patients and had found mutations in MOCS1(MIM 603707) (Reiss et al. 1998b) or MOCS2 (MIM 603708) (Stallmeyer 1999a) in 40 cases (Reiss et al. 1998a, 1999; Reiss 2000).
We studied the last of three affected infants born to a Danish mother and father who were cousins. All three died in the neonatal period (day 12, 29, and 3, respectively), with symptoms identical to MoCo deficiency. Three other pregnancies of the mother resulted in two healthy sibs and one spontaneous abortion. There was no family history of genetic disease, and all three affected infants had a normal karyotype. The first infant was a boy, and the other two were girls. All showed hypotonia combined with hyperreflexia, as well as tonic-clonic convulsions. Fibroblasts of the third infant, the patient, were used to verify a MoCo deficiency by biochemical and in vitro complementation assays and to isolate DNA for genetic analysis. The study was approved by the ethics committee of the Medical Faculty of Göttingen. Complete sequencing of all MOCS1 and MOCS2 exons, plus adjacent splice-junction sequences, did not reveal a disease-causing mutation in the patient. The human MOCS3 cDNA and genomic sequence, encoding the molybdopterin synthase sulfurylase (Appleyard et al. 1998), were also found to be devoid of mutations in the patient’s DNA (data not shown).
We analyzed the GEPH gene (MIM 603930) for the receptor-clustering neuroprotein gephyrin (Kirsch et al 1973; Essrich et al. 1998; Kneussel and Betz 2000) as a candidate gene for the MoCo deficiency in this family. Gephyrin consists of two distinct protein domains separated by a linker sequence (fig. 1a). The N-terminal domain of gephyrin is homologous to the bacterial protein MogA, and the C-terminal domain is homologous to bacterial MoeA, both proteins being involved in the biosynthesis of MoCo (Stewart 1988). This architecture is shared by the Drosophila protein Cinnamon (Kamdar et al. 1994) and, with a reversed domain order, the plant protein Cnx1 (Stallmeyer et al. 1995). The bacterial moaB gene is also homologous to the 5′ region of the GEPH cDNA, as well as to the bacterial mogA gene. However, no bacterial mutant and no function is known for moaB. The moeA gene product is believed to activate molybdenum before its incorporation into molybdopterin (Hasona et al. 1998; Kuper et al. 2000). Bacterial mogA mutants can produce active MoCo in media supplemented with high concentrations of molybdate (Stewart and MacGregor 1982). This “molybdenum rescue” is also observed for the corresponding plant mutants (Stallmeyer et al. 1995) and the MoCo-deficient murine cell line L929 (Falciani et al. 1994).
Figure 1.

Gephyrin and MoCo biosynthesis. a, Schematic representation of gephyrin and homologous proteins. The domain symbols G and E correspond to the respective bacterial operons. b, Illustration of the last step in MoCo biosynthesis, the gephyrin-catalyzed insertion of molybdenum into molybdopterin.
Geph cDNA derived from rats could restore molybdenum-repairable plant mutants and bacterial mogA mutants, as well as L929 cells (Stallmeyer et al. 1999b). Likewise, Geph knockout mice showed the expected absence of synaptic glycine-receptor clustering and developed symptoms identical to those of MoCo deficiency (Feng et al. 1998). Therefore, gephyrin combines an evolutionarily novel function (neuroreceptor clustering) with a conserved old function (MoCo biosynthesis), the latter being essential for activation (E domain) and insertion (G domain) of molybdenum into molybdopterin, thereby forming the biologically active cofactor (fig. 1b).
The rat Geph cDNA sequence (Prior et al. 1992) was used to screen the nonredundant GenBank database for human cDNA sequences with the homology search program Advanced BLAST. A 4,193-bp cDNA clone derived from brain tissue contains the complete coding sequence for human gephyrin, with the start codon at position 1122 and the stop codon at position 3330. The nucleotide and amino acid sequence homologies are 93% and 100%, respectively. The human cDNA sequence was used to identify the genomic sequences in the HTGS (high throughout genomic sequences) library. A homology search revealed that the gene is located on chromosome 14 and consists of 22 exons covering a genomic region of ⩾375 kbp. These genomic sequences were used as primer targets to amplify all exons individually, plus each of their adjacent splice-signal sequences (table 1). Amplification of patient and control DNA revealed a deletion encompassing exons 2 and 3 in the patient (fig. 2a). This deletion was verified by reverse transcriptase PCR, in which exclusively truncated transcripts were detected (fig. 2b), followed by sequencing of the amplification products (fig. 3a). On the mRNA level, this deletion results in a frameshift after only 21 codons of the normal coding sequence. Therefore, neither of the two gephyrin domains (fig. 1) is expressed. Western blot analysis confirmed the complete absence of gephyrin protein in crude extracts of the patient’s fibroblasts, whereas the expression of at least two splice variants (Prior et al. 1992) can be seen in control cells (fig. 3b).
Table 1.
Oligonucleotides Used in Study of Patient with Gephyrin Gene Deletion
| Methoda | Primer | Sequence |
| EA: | ||
| E1 | G1F | 5′-CCTAGCTGTCGCGCTCTCCT-3′ |
| G1R | 5′-TCCCGAGGCCGCAGAGAAGG-3′ | |
| E2 | G2F | 5′-GGTCAGCAATAGCTTAAATG-3′ |
| G2R | 5′-CTCTTTTGAGAAAAGGAACAC-3′ | |
| E3 | G3F | 5′-GCATTCTGATGGTAATGGCA-3′ |
| G3R | 5′-ACCCCTCACCAAGATGCTAA-3′ | |
| E4 | G4F | 5′-GGGATGTTTTGAGCAAGCAG-3′ |
| G4R | 5′-CCATGATTAGTTTAATCCTTG-3′ | |
| E5 | G5F | 5′-GTGGGTTTTACTAGTCTGAC-3′ |
| G5R | 5′-ACAGTTCACCTAGCAAATGG-3′ | |
| E6 | G6F | 5′-CTTAATGTATTTAAACCGGGC-3′ |
| G6R | 5′-TTTGGCTCCCTAACTTTCAC-3′ | |
| E7 | G7F | 5′-CAGTTTGATTGCCACCATCT |
| G7R | 5′-TTACCTGTGGGTCCTTTAGG-3′ | |
| E8 | G8F | 5′-AAGGGGGTCTTGATTCTACA-3′ |
| G8R | 5′-CCCAGATTACTATAGAAGAGC-3′ | |
| E9 | G9F | 5′-ACCTCAGGAGCTTGCCCATT-3′ |
| G9R | 5′-AAGCTCTAGTTCAGCAGCCC-3′ | |
| E10 | G10F | 5′-GTCATTGCCACTTTTTAATCA-3′ |
| G10R | 5′-CAGGAAAACTGTGCATTAATG-3′ | |
| E11 | G11F | 5′-CAAGCACTCATGCCCATCTT-3′ |
| G11R | 5′-CAGTGCCTGATTATGTTTAAG-3′ | |
| E12 | G12F | 5′-CTTGTTCCATGCTGTAGGTC-3′ |
| G12R | 5′-TTCCACTAAACTGATAGGAGA-3′ | |
| E13 | G13F | 5′-CTTTTCCTTTGCAGCAGCAA-3′ |
| G13R | 5′-ACTGCCATAGGAACAACAGC-3′ | |
| E14 | G14F | 5′-TATCCTGGGCCTATCTGATG-3′ |
| G14R | 5′-GCCAGGGTTTCCTGAGTAAA-3′ | |
| E15 | G15F | 5′-CACTAAAGTTTCCCTCTGAG-3′ |
| G15R | 5′-CAACACAGAACATATGTCAG-3′ | |
| E16 | G16F | 5′-TATGCAACATTAACCTAATAG-3′ |
| G16R | 5′-ATGAGTATTCCAAAAACTCG-3′ | |
| E17 | G17F | 5′-GCCTATTAGTGAATAAGGCG-3′ |
| G17R | 5′-AGATGCCTACCAGACCACAG-3′ | |
| E18 | G18F | 5′-TCATTTAAAGTGTTGAAAGTC-3′ |
| G18R | 5′-CCCATATATGAGATAACAAGA-3′ | |
| E19 | G19F | 5′-AAAACACTGGAGTACTTAATG-3′ |
| G19R | 5′-CCAGAAAAAGGAAAGGAAAC-3′ | |
| E20 | G20F | 5′-TAGACAGACATAATTATTTGGC-3′ |
| G20R | 5′-TTAGGAAATCATATCCCTAAC-3′ | |
| E21 | G21F | 5′-GTCCACTGTATTCTTTGCAC-3′ |
| G21R | 5′-CAGGATAGGTGTCTAGGAA-3′ | |
| E22 | G22F | 5′-AGGGCCCAACTGTATACGCC-3′ |
| G22R | 5′-GCTTTCTCCTGCTGGTGACC-3′ | |
| RT-PCR: | ||
| RT | RT1 | 5′-CAGGGCCATCCCTGGTGCTT-3′ |
| PCR1 | G1F | 5′-CCTAGCTGTCGCGCTCTCCT-3′ |
| RT1 | 5′-CAGGGCCATCCCTGGTGCTT-3′ | |
| PCR2 | RT2 | 5′-TTCTCCCGGCTCCTGTCAGT-3′ |
| RT3 | 5′-TCGTGGTGCAAATCCTGTTC-3′ |
A = amplification; E = exon.
Figure 2.

Detection of a gephyrin gene deletion. a, PCR amplification of individual exons (1–22) from genomic DNA of a control (top) and the patient (bottom). DNA was isolated from fibroblasts with standard phenol/chloroform extraction methods. PCR (40 cycles at 94°C for 45 s, at 50°C–60°C for 45 s, and at 72°C for 2 min after an initial incubation of 15 min at 94°C) was done with HotStarTaq PCR master mix (Qiagen) and exon-flanking primers (table 1). b, Nested PCR cDNA amplification from exon 1 to exon 4 after reverse transcription. Total RNA was isolated from fibroblasts with an RNeasy minikit combined with QIAshredder columns (Qiagen). First-strand synthesis was done with the Omniscript RT kit (Qiagen), including RNasin (Promega) and a sequence-specific primer in a 20-μl volume. In a first PCR of 25 cycles, 10 μl of this reaction was used, with the conditions specified above, and 1 μl of first-round PCR product was the template for a second-round PCR with 40 cycles and two nested primers. B = blank (no template); C = healthy control; M = size marker (100-bp ladder) (New England Biolabs); P = patient.
Figure 3.
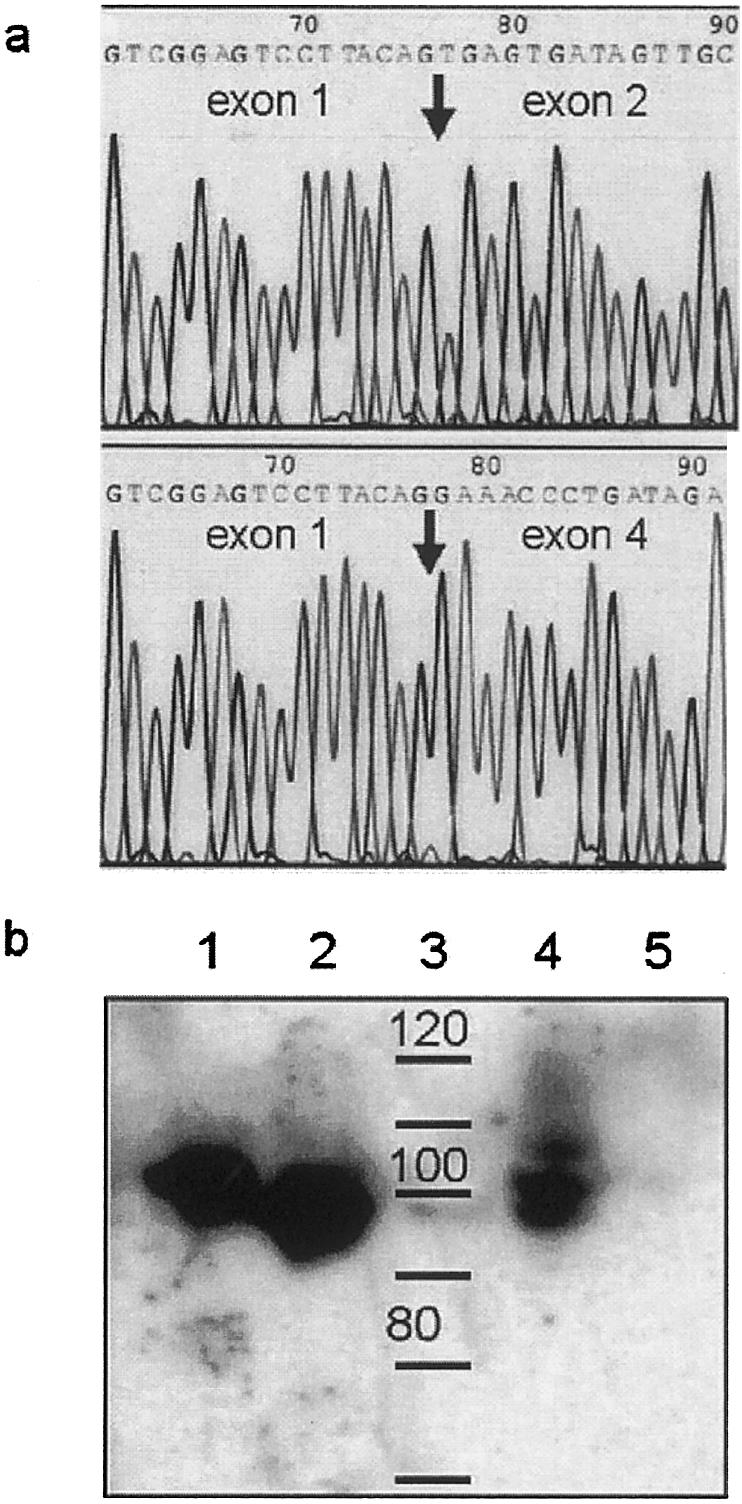
a, Sequence analysis of the RT-PCR product of control (top) and patient (bottom). Sequencing was done commercially (SeqLab) after purification of the amplification products with the QIA quick PCR purification kit (Qiagen) with the PCR primers and dideoxy fluorescent dye terminators (ABI). The transition from exon 1 to exon 2 or 4, respectively, is indicated by the arrow. b, Western blot analysis of gephyrin expression in crude protein extracts using a polyclonal anti-gephyrin antibody. Crude protein extracts were prepared from fibroblasts, separated by SDS-PAGE using a 7.5 % polyacrylamide gel, and blotted onto a polyvinyldiene fluoride membrane. Positive controls were recombinant rat Geph P1 and P2 splice forms expressed in Escherichia coli (G.S., unpublished data). A primary polyclonal antibody generated against recombinant rat gephyrin P1 protein was used (diluted in serum at 1:2000), followed by chemiluminescent detection (secondary antibody, Promega; ECL system, Amersham/Pharmacia). Lane 1, recombinant rat gephyrin P2 (20 ng); lane 2, recombinant rat gephyrin P1 (20 ng); lane 3, size marker (labeled in kDa); lane 4, positive control (18 μg); lane 5, patient with gephyrin gene deletion (20 μg).
Because of the presumed function of gephyrin in MoCo formation (fig. 1b), loss of this protein should result in the accumulation of molybdenum-free molybdopterin. We demonstrated the presence of the fluorescent oxidation product of molybdopterin (“form A”) (Johnson and Wadman 1995) by high-performance liquid chromatography analysis (HPLC) (fig. 4a). Biologically active molybdopterin and MoCo can be quantified by the in vitro nit-1 reconstitution assay, which is based on the transfer of MoCo to the nitrate reductase apoprotein of the Neurospora crassa nit-1 mutant (Nason et al. 1971). Depending on presence or absence of molybdate in the assay, it can be used for the detection of total molybdopterin (+Mo), including all forms of the pterin and MoCo (−Mo). HPLC analysis of the patient's cells shows an increased level of molybdopterin (fig. 4b), compared with control cells. Active MoCo could be detected only in control cells and in those patient cells that were supplemented with 1 mM molybdate (fig. 4b). This molybdate repair of the cofactor could also be demonstrated by the regained sulfite oxidase activity (fig. 4c).
Figure 4.

Biochemical characterization of fibroblasts with the gephyrin gene deletion. a, HPLC detection of the molybdopterin derivative “form A” in crude fibroblast extracts of a healthy control subject (top), a patient with a MOCS1 mutation as negative control (middle), and the patient with the gephyrin gene deletion (bottom). Molybdopterin was detected by conversion to its stable oxidation product form A-dephospho. Oxidation (1 mg protein per sample) with I2/KI, QAE-sephadex chromatography and HPLC analysis on a C18 reversed-phase column were performed as described elsewhere (Schwarz et al. 1997). b, nit-1 reconstitution assay for the detection of active MoCo (−Mo) and total molybdopterin, including both the metal-free and metal-loaded pterin (+Mo) of a control (left) and of the patient without (middle) and with (right) molybdate supplementation of the medium. c, Sulfite oxidase activity of control fibroblasts (left), the patient's fibroblasts grown on normal medium (middle) and the patient's fibroblasts grown on medium supplemented with molybdate (10 mM) (right). Neurospora crassa nit-1 extract was prepared as described elsewhere (Nason et al. 1971). The assay was performed in a 90-μl (+Mo) or a 180-μl (−Mo) volume containing 30 μl or 60 μl, respectively, of nit-1 extract freshly filtered on gel (Nick columns; Amersham/Pharmacia) in the presence of 2 mM reduced glutathione. Protein extracts from fibroblasts were prepared in 50 mM sodium phosphate, 200 mM NaCl, 5 mM EDTA (pH 7.2) and were added in various amounts (20–120 μl) to the nit-1 extract, according to the linear range of the assay. Total molybdopterin content was determined by performing the reconstitution in the presence of 10 mM sodium molybdate. MoCo was detected in the absence of external molybdate. Complementation was carried out anaerobically overnight at 4°C. After addition of 20 mM NADPH for 10 min, reconstituted NADPH-nitrate reductase activity was determined. One nit-1 unit is defined as the A540 of 1.0/30 min nitrate reductase reaction time. Sulfite oxidase activity was determined as described elsewhere (Johnson et al. 1991).
After the primary sequence of the receptor-associated gephyrin revealed homologies to MoCo biosynthetic enzymes, it appeared counterintuitive that a eukaryotic protein might have two such different functions as (i) a structural role in receptor clustering and (ii) a biosynthetic activity in MoCo formation. It has been suggested that two bacterial biosynthetic genes (mogA and moeA) have been fused during evolution to form a multidomain protein with a novel function (Kamdar et al. 1994; Stallmeyer et al. 1995; Feng et al. 1998). This has happened under persistence of the biosynthetic activities, whose abrogation, as shown here, results in a lethal malfunction. Moreover, the fusion of the same prokaryotic genes, in reversed orientation, in higher plants (Stallmeyer et al. 1995) (fig. 1a) demonstrates that there is a strong selective pressure for this convergent development in eukaryotes.
The gravity of the disease in the family described here is very similar to the serious phenotype seen in the Geph knockout mice (Feng et al. 1998). Such mice could be used to test whether MoCo-deficient patients with GEPH mutations could be treated successfully by administering high concentrations of molybdate. In attempts reported elsewhere, administration of an oral (Duran et al. 1978; Munnich et al. 1983; Endres et al. 1988) or an intravenous (Bamforth et al. 1990) molybdate supplementation to MoCo-deficient patients did not lead to any clinical or biochemical improvement. These patients, however, have not been examined genetically, and, on the basis of the observed mutation frequencies, it seems likely that they carried mutations in the “nonrepairable” genes MOCS1 or MOCS2 (Reiss 2000). The dysfunction of gephyrin as a receptor-clustering molecule in the postsynaptic membrane cannot be restored by these means. Hyperekplexia (MIM 149400), as a consequence of isolated receptor dysfunction, is a relatively mild neurological disorder, which can be ameliorated by pharmaceutical agents (Andrew and Owen 1997). These drugs could be combined with a molybdate therapy in gephyrin-deficient patients to treat the synaptic receptor dysfunction, as well as the biosynthetic deficiency. As in the other forms of MoCo deficiency, the most successful approaches may include prenatal treatment to prevent neurological damage.
Acknowledgments
Technical assistance by T. Otte is gratefully acknowledged. We thank U. Lenz and D. Schmalz for helpful advice and W. Engel for continued support. This study has been supported by the Deutsche Forschungsgemeinschaft (support to J.R., R.R.M., and G.S.) and the Fritz Thyssen Stiftung (support to R.R.M.).
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for rat gephyrin cDNA [accession number X66366] and for human sequences MOCS3 cDNA [accession number AF102544], MOCS3 genomic [accession number HS914P20], gephyrin cDNA [accession numbers AB037806 and AB272663], and gephyrin genomic [accession numbers AC021012.2, AL139295.2, AL117667.2, AL049835.3, AL159179.2, AL135978.2, and AL133241.2])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for MoCo deficiency [MIM 252150], sulfite oxidase deficiency [MIM 272300], MOCS1 [MIM 603707], MOCS2 [MIM 603708], GEPH [MIM 603930], and hyperekplexia [MIM 149400]
References
- Andrew M, Owen MJ (1997) Hyperekplexia: abnormal startle response due to glycine receptor mutations. Br J Psychiatry 170:106–108 [DOI] [PubMed] [Google Scholar]
- Appleyard MVCL, Sloan J, Kana’n JM, Heck IS, Kinghorn JR, Unkles SE (1998) The Aspergillus nidulanscnxF gene and its involvement in molybdopterin biosynthesis. J Biol Chem 273:14869–14876 [DOI] [PubMed] [Google Scholar]
- Bamforth FJ, Johnson JL, Davidson AGF, Wong LTK, Lockitch G, Applegarth DA (1990) Biochemical investigation of a child with molybdenum cofactor deficiency. Clin Biochem 23:537–542 [DOI] [PubMed] [Google Scholar]
- Duran M, Beemer FA, Heiden CVD, Korteland J, Bree PKD, Brink M, Wadman SK (1978) Combined deficiency of xanthine oxidase and sulphite oxidase: a defect of molybdenum metabolism or transport? J Inherit Metab Dis 1:175–178 [DOI] [PubMed] [Google Scholar]
- Endres W, Shin YS, Günther R, Ibel H, Duran M, Wadman SK (1988) Report on a new patient with combined deficiencies of sulphite oxidase and xanthine dehydrogenase due to molybdenum cofactor deficiency. Eur J Pediatr 148:246–249 [DOI] [PubMed] [Google Scholar]
- Essrich C, Lorez M, Benson JA, Fritschy JM, Luscher B (1998) Postsynaptic clustering of major GABAA receptor subtypes requires the γ2 subunit and gephyrin. Nat Neurosci 1:563–571 [DOI] [PubMed] [Google Scholar]
- Falciani F, Terao M, Goldwurm S, Ronchi A, Gatto A, Minoia C, Calzi ML, Salmona M, Cazzaniga G, Garattini E (1994) Molybdenum (VI) salts convert the xanthine oxidoreductase apoprotein into the active enzyme in mouse L929 fibroblastic cells. Biochem J 298:69–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Tintrup H, Kirsch J, Nichol MC, Kuhse J, Beth H, Sanes JR (1998) Dual requirement for gephyrin in glycine receptor clustering and molybdoenzyme activity. Science 282:1321–1324 [DOI] [PubMed] [Google Scholar]
- Hasona A, Ramesh MR, Shanmugam KT (1998) Physiological and genetic analyses leading to identification of a biochemical role for the moeA (molybdate metabolism) gene product of Escherichia coli. J Bacteriol 180:1466–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JL, Rajagopalan KV, Lanman JT, Schutgens RBH, Van Gennip AH, Sorensen P, Applegarth DA (1991) Prenatal diagnosis of molybdenum cofactor deficiency by assay of sulphite oxidase activity in chorionic villus samples. J Inherit Metab Dis 14:932–937 [DOI] [PubMed] [Google Scholar]
- Johnson JL, Wadman SK (1995) Molybdenum cofactor deficiency and isolated sulfite oxidase deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease. McGraw-Hill, New York, pp 2271–2283 [Google Scholar]
- Kamdar KP, Shelton ME, Finnerty V (1994) The Drosophila molybdenum cofactor gene Cinnamon is homologous to three Escherichia coli cofactor proteins and to the rat protein gephyrin. Genetics 137:791–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch J, Wolters I, Triller A, Betz H (1993) Gephyrin antisense oligonucleotides prevent glycine receptor clustering in spinal neurons. Nature 366:745–748 [DOI] [PubMed] [Google Scholar]
- Kneussel M, Betz H (2000) Receptors, gephyrin and gephyrin-associated proteins: novel insights into the assembly of inhibitory postsynaptic membrane specializations. J Physiol 525:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuper J, Palmer T, Mendel RR, Schwarz G (2000) Mutations in the molybdenum cofactor biosynthetic protein Cnx1G from Arabidopsis thaliana define functions for molybdopterin binding, molybdenum insertion, and molybdenum cofactor stabilization. Proc Natl Acad Sci USA 97:6475–6480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munnich A, Saudubray JM, Charpentier C, Ogier H, Coude FX, Frezal J (1983) Multiple molybdoenzyme deficiencies due to an inborn error of molybdenum cofactor metabolism: two additional cases in a new family. J Inherit Metab Dis Suppl 6:95–966230486 [Google Scholar]
- Nason A, Lee KY, Pan SS, Ketchum PA, Lamberti A, De Vries J (1971) In vitro formation of assimilatory reduced nicotinamide adenine dinucleotide phosphate: nitrate reductase from a Neurospora mutant and a component of molybdenum-enzymes. Proc Natl Acad Sci USA 68:3242–3246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior P, Schmitt B, Grenningloh G, Priballa I, Multhaup G, Beyreuther K, Maulet Y, Werner P, Langosch D, Kirsch J, Betz H (1992) Primary structure and alternative splice variants of gephyrin, a putative glycine receptor–tubulin linker protein. Neuron 8:1161–1170 [DOI] [PubMed] [Google Scholar]
- Reiss J (2000) Genetics of molybdenum cofactor deficiency. Hum Genet 106:157–163 [DOI] [PubMed] [Google Scholar]
- Reiss J, Christensen E, Kurlemann G, Zabot MT, Dorche C (1998a) Genomic structure and mutational spectrum of the bicistronic MOCS1 gene defective in molybdenum cofactor deficiency type A. Hum Genet 103:639–644 [DOI] [PubMed] [Google Scholar]
- Reiss J, Cohen N, Dorche C, Mandel H, Mendel RR, Stallmeyer B, Zabot MT, Dierks T (1998b) Mutations in a polycistronic nuclear gene associated with molybdenum cofactor deficiency. Nat Genet 20:51–53 [DOI] [PubMed] [Google Scholar]
- Reiss J, Cohen N, Stallmeyer B, Mendel RR, Dorche C (1999) The human molybdopterin synthase gene: genomic structure and mutations in molybdenum cofactor deficiency type B. Am J Hum Genet 64:706–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz G, Boxer DH, Mendel RR (1997) Molybdenum cofactor biosynthesis: the plant protein Cnx1 binds molybdopterin with high affinity. J Biol Chem 272:26811–26814 [DOI] [PubMed] [Google Scholar]
- Stallmeyer B, Drugeon G, Reiss J, Haenni AL, Mendel RR (1999a) The human molybdopterin synthase gene: identification of a bicistronic transcript with overlapping reading frames. Am J Hum Genet 64:698–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallmeyer B, Nerlich A, Schiemann J, Brinkmann H, Mendel RR (1995) Molybdenum cofactor biosynthesis: the Arabidopsis thaliana cDNA cnx1 encodes a multifunctional two-domain protein homologous to a mammalian neuroprotein, the insect protein Cinnamon and three Escherichia coli proteins. Plant J 8:751–762 [DOI] [PubMed] [Google Scholar]
- Stallmeyer B, Schulze J, Schwarz G, Nerlich A, Reiss J, Kirsch J, Mendel RR (1999b) The neurotransmitter receptor-anchoring protein gephyrin reconstitutes molybdenum cofactor biosynthesis in bacteria, plants, and mammalian cells. Proc Natl Acad Sci USA 96:1333–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart V (1988) Nitrate respiration in relation to facultative metabolism in Enterobacteria. Microbiol Rev 52:190–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart V, MacGregor CH (1982) Nitrate reductase in Escherichia coli K-12: involvement of chlC, chlE, and chlG loci. J Bacteriol 151:788–799 [DOI] [PMC free article] [PubMed] [Google Scholar]