

Abstract
Using linkage analysis, we identified a novel dominant locus, DFNA25, for delayed-onset, progressive, high-frequency, nonsyndromic sensorineural hearing loss in a large, multigenerational United States family of Czech descent. On the basis of recombinations in affected individuals, we determined that DFNA25 is located in a 20-cM region of chromosome 12q21-24 between D12S327 (centromeric) and D12S84 (telomeric), with a maximum two-point LOD score of 6.82, at recombination fraction .041, for D12S1030. Candidate genes in this region include ATP2A2, ATP2B1, UBE3B, and VR-OAC. DFNA25 may be the human ortholog of bronx waltzer (bv).
Severe-to-profound sensorineural hearing impairment occurs in 1 of 1,000 births in the United States (Morton 1991). For nearly four decades, it has been recognized that >50% of cases of sensorineural hearing impairment in children have a genetic etiology (Fraser 1964). In ∼70% of these cases, the hearing impairment is described as “nonsyndromic,” because it is not associated with other clinical manifestations (Bergstrom et al. 1971; Konigsmark and Gorlin 1976). The genetic heterogeneity of this disorder has been well established, with >30 dominant hearing loss loci mapped to date (Hereditary Hearing Loss home page). These loci are numbered sequentially, on the basis of time of discovery, and carry the prefix “DFNA.” At present, 11 autosomal dominant genes have been cloned (Hereditary Hearing Loss home page).
Autosomal dominant nonsyndromic hearing loss (ADNSHL) typically has a delayed (postlingual) onset, and the hearing impairment becomes progressively worse with age (Van Camp et al. 1997). The distinction between presbycusis (hearing loss that commonly occurs with aging) and ADNSHL is often difficult when the phenotype involves hearing loss in the high frequencies. In addition, environmental noise exposure may cause hearing loss, particularly affecting thresholds at 4,000 Hz.
The Institutional Review Board of the University of Michigan Medical School approved the study, and appropriate informed consent was obtained from all subjects. A pedigree was constructed on the basis of information obtained from questionnaires and interviews. The clinical evaluation included an interview/questionnaire, photography, and pure-tone audiometry and tympanometry.
All participants aged >27 mo underwent audiologic testing by certified audiologists. Pure-tone air-conduction thresholds were obtained at 500, 1,000, 2,000, 4,000, and 6,000 Hz for each ear. When air-conduction thresholds were >20 dB hearing level (HL), bone-conduction thresholds were obtained at 2,000 and 4,000 Hz, and tympanometry was performed.
To discriminate between hearing loss caused by the DFNA25 locus and presbycusis or noise-induced hearing loss, thresholds were compared with those of an unselected control population (Robinson 1988). For pediatric patients, thresholds were compared with data obtained from unselected children (Haapaniemi 1996). Patients were designated “affected” if they had a sensorineural hearing impairment that was worse than the 90th percentile of sex- and age-matched subjects in the control population (including those with presbycusis and/or noise exposure) for at least four thresholds. Those with a history of noise exposure were not excluded from the affected group if the above criteria were met. Affected children were included in the linkage analysis, whereas those aged <20 years without sensorineural hearing loss were considered to have indeterminate affection status. Family members aged >20 years with normal hearing or with hearing loss that was not worse than the 90th percentile by these criteria were deemed “unaffected.”
Peripheral lymphocytes obtained from the proband were examined by the Cytogenetics Laboratory at the University of Michigan Hospital, using standard karyotyping techniques. Venous blood samples were obtained from consenting family members. For young family members and those from whom blood could not be drawn, buccal-cell samples were obtained. Genomic DNA was prepared from leukocytes or buccal cells according to standard methods.
The DNA samples underwent a genome scan by the National Heart, Lung, and Blood Institute’s Mammalian Genotyping Service (MGS). The genome scan used the Marshfield Screening Set 9, consisting of 387 polymorphic markers, at 10-cM average spacing, with average heterozygosity of 73% (Yuan et al. 1997). The MGS required that the known dominant loci (DFNA1-20) be excluded by linkage analysis prior to submission of the samples. After the genome scan, genotyping of family samples was performed as described elsewhere (Lesperance et al. 1995) for 4 markers in the Marshfield Screening Set 9 (D12S1064, D12S1300, PAH, and D12S2070) and for 10 markers selected from the Marshfield chromosome 12 map (D12S1684, D12S2077, D12S327, D12S1051, D12S1063, D12S1607, D12S1030, D12S84, D12S1583, and D12S1341) (Broman et al. 1998).
Alleles were sized by genotyping of individuals 1331-01 and 1331-02 from the CEPH pedigrees (CEPH/Généthon). The marker allele frequencies were determined by genotyping 19 unrelated spouses in the pedigree. To avoid overstated evidence for linkage caused by underestimation of marker allele frequencies, the allele frequency for the linked allele was not allowed to be <.1. The standard model for analysis assumed a disease allele frequency (GF) of .00001, with 0% phenocopy rate (PC) and penetrance (P) of 90%. Four additional models were tested: GF=.00001, PC=.001, P=90%; GF=.01, PC=.001, P=90%; GF=.01, PC=0, P=90%; and GF=.00001, PC=0, P=1%. The two-point LOD scores, recombination fraction (θ), and maximum LOD scores (Zmax) were then calculated by means of the MLINK and ILINK programs from the LINKAGE package (Lathrop et al. 1984).
The pedigree, demonstrating 51 family members informative for linkage analysis, is shown in figure 1. Clinical evaluation did not reveal any evidence of syndromic features associated with the hearing impairment, and karyotype results were normal. Several affected and unaffected family members had a history of noise exposure. Affected family members had a nonsyndromic, slowly progressive, sensorineural hearing impairment. The frequencies most commonly affected were ⩾2,000 Hz. Twenty family members were deemed affected, 9 were spouses of affected family members, and 22 were unaffected. The age at onset appeared to vary within the family. Ten affected family members either suspected hearing loss prior to age 20 years or had such hearing loss diagnosed during this study. The remaining 10 affected family members had hearing loss diagnosed in the third through sixth decades of life.
Figure 1.

Haplotype analysis of DFNA25 family with nonsyndromic high-frequency sensorineural hearing impairment. Circles and squares denote females and males, respectively. Diagonal lines through circles and squares indicate deceased individuals, and inferred alleles are in parentheses. Blackened symbols indicate affected persons, whereas unblackened symbols indicate unaffected persons. Haplotypes for chromosome 12 markers are indicated at the left of the figure, with obligate recombination events indicated. The haplotype assumed to carry the disease allele is indicated by the black bar, with other shadings representing the other haplotypes. Lines in haplotypes represent indeterminate recombination events.
The calculated allele frequencies were comparable to those reported in the CEPH and Cooperative Human Linkage Center genotype databases, although some of the alleles in our population were not found in these genotype databases. No evidence of linkage was observed for 383 of 387 markers tested in the genome screen. Linkage was first detected to a polymorphic marker within intron 3 of the phenylalanine hydroxylase gene (PAH [MIM 261600]), chromosome 12q21-q24, which ultimately yielded Zmax 6.24 at θ=.05.
The haplotype analysis demonstrated evidence of reduced penetrance (fig. 1). Individuals III:9 and III:10 are siblings tested at ages 61 and 60 years, respectively, but neither had hearing loss that met criteria for affected status. Both had a history of noise exposure, with III:10 having two thresholds that were worse than or near the 90th percentile cutoff and III:9 having only one significantly abnormal threshold (75th percentile). In summary, although both siblings inherited a significant part of the haplotype common to the affecteds, neither's hearing is worse than would be expected from presbycusis or noise exposure alone. Thus, the penetrance was not assumed to be complete at any age in this family, and recombination events in unaffected individuals were not used to define the DFNA25 interval.
Individual III:11 was classified as affected on the basis of our criteria. However, only D12S1684 indicates that III:11 definitely inherited the allele common to the affecteds, since marker D12S1064 is uninformative. III:11 reported a significant history of occupational noise exposure and was first diagnosed with hearing loss at age 37 years. He has worn bilateral hearing aids since that time, and his hearing is generally more severely affected than that of other family members of similar age. All three of his children have normal hearing. Since individual III:11 essentially lacks the haplotype shared by the affecteds, we conclude that he is most likely a phenocopy. However, on the basis of his phenotype, assigned prior to the genetic analysis, he is classified as affected in the LOD score calculations.
Two affected recombinant individuals (III:19 and III:22) and their offspring define the centromeric end of the interval. Obligate recombination events have occurred between D12S327 and D12S1051 in individuals III:19, IV:13, IV:15, and V:1. A second recombination event can be inferred at marker D12S1684 for individual IV:15. III:22 and IV:21 are affected individuals with obligate recombination events between D12S2077 and D12S327. A second recombination event has occurred between D12S1063 and D12S1607 in IV:20, who had normal hearing when tested at age 31 years. IV:24 has inherited the haplotype common to the affecteds, except for a recombination event excluding D12S1684; when tested at age 27 years, she was considered unaffected, since she had only one abnormal threshold.
The telomeric end of the interval is defined by individual III:13, an affected with a recombination event excluding markers D12S84, D12S1583, D12S1341, and D12S2070. His daughter, IV:10, inherited the same portion of the affecteds’ haplotype; she is 37 years old with normal hearing. Affected individual III:20 and his affected offspring IV:18 are nonrecombinant for D12S84 but are recombinant for D12S1583, D12S1341, and D12S2070. IV:16 inherited the same recombinant haplotype as her parent III:20; IV:16 is 45 years old, with normal hearing, except for one abnormal threshold. A recombination event occurred between D12S327 and D12S1051 in individual IV:17, who had normal hearing when tested at age 40 years. Multiple affected individuals (III:3, III:4, III:7, IV:3, IV:4, IV:23, and V:2) inherited the same recombinant chromosome excluding markers D12S1341 and D12S2070. In summary, the interval defined by affected recombinants at D12S327 (centromeric) and D12S84 (telomeric) spans 20 cM in 12q21-24 (fig. 2).
Figure 2.

Ideogram of chromosome 12q, indicating analyzed markers in bands 12q21-24 and DFNA25 interval. Distances between markers were obtained from the Marshfield Center for Medical Genetics. The blackened portions of the bars indicate informative recombination events. The number of affected individuals with each type of crossover is indicated at the bottom of each bar.
LOD scores for 14 markers are shown in table 1. Under the standard model, a maximum two-point LOD score of 6.82 was observed at θ=.041 for D12S1030. A LOD score of 5.04 was observed at θ=0 for D12S1051, since the affected parent (II:3) of the affected recombinant III:11 is presumed to be homozygous for the linked allele. Otherwise, LOD scores at θ=0 tended to be negative for all markers, because of the effects of classifying a likely phenocopy (III:11) as affected and unaffected family members carrying the linked allele. Zmax and the maximum-likelihood estimate of θ (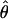 ) did not change significantly under the different models tested, with the exception of a lower Zmax under the model with penetrance of 1% (data not shown).
) did not change significantly under the different models tested, with the exception of a lower Zmax under the model with penetrance of 1% (data not shown).
Table 1.
Two-Point LOD Scores for Chromosome 12 Markers, Calculated under a Model of 90% Penetrance, Disease Allele Frequency .00001, and Phenocopy Rate 0
| LOD Score at θ = |
|||||||||
| Locus | 0 | .01 | .05 | .1 | .2 | .3 | .4 | 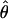 |
Zmax |
| D12S1684 | −14.94 | −2.99 | .17 | 1.42 | 2.13 | 1.86 | 1.02 | .212 | 2.14 |
| D12S1064 | 3.24 | 3.27 | 3.28 | 3.17 | 2.68 | 1.97 | 1.07 | .033 | 3.29 |
| D12S2077 | −5.50 | −2.35 | −.09 | .80 | 1.27 | 1.01 | .46 | .202 | 1.27 |
| D12S327 | −3.68 | 1.67 | 2.93 | 3.23 | 2.94 | 2.12 | 1.01 | .113 | 3.24 |
| D12S1051 | 5.04 | 4.96 | 4.62 | 4.18 | 3.26 | 2.26 | 1.17 | 0 | 5.04 |
| D12S1300 | .39 | 2.99 | 3.66 | 3.75 | 3.26 | 2.30 | 1.06 | .088 | 3.76 |
| D12S1063 | −2.49 | 3.52 | 4.16 | 4.22 | 3.67 | 2.66 | 1.34 | .081 | 4.24 |
| D12S1607 | −4.06 | 3.72 | 4.26 | 4.23 | 3.57 | 2.50 | 1.17 | .068 | 4.29 |
| PAH | 1.64 | 5.83 | 6.24 | 6.04 | 5.02 | 3.55 | 1.76 | .05 | 6.24 |
| D12S1030 | −1.18 | 6.51 | 6.81 | 6.51 | 5.33 | 3.75 | 1.86 | .041 | 6.82 |
| D12S84 | −1.89 | 4.52 | 5.55 | 5.58 | 4.78 | 3.45 | 1.75 | .076 | 5.63 |
| D12S1583 | −1.99 | 2.43 | 4.10 | 4.40 | 3.93 | 2.86 | 1.46 | .103 | 4.40 |
| D12S1341 | −14.70 | −1.372 | 1.79 | 2.78 | 3.02 | 2.38 | 1.28 | .166 | 3.08 |
| D12S2070 | −14.79 | −2.41 | .89 | 2.01 | 2.45 | 1.96 | .97 | .184 | 2.46 |
It has been increasingly recognized that genotype does not completely predict phenotype, even for classic “simple” Mendelian traits such as the autosomal dominant hearing loss segregating in this family (Dipple and McCabe 2000). Phenotypes may vary widely between individuals with different mutations in the same gene and even between individuals in the same family who have inherited the identical mutation. It is hypothesized that other genetic factors (such as nonallelic polymorphisms) or environmental influences can modulate the effect of a genetic mutation on the phenotype. Restricting the definition of affected status to those most severely affected and/or those with early onset may facilitate linkage analysis by enriching for those who have the disorder because of the genetic factor.
However, in this family, two individuals with hearing loss consistent with presbycusis (III:9 and III:10, aged 61 and 60 years, respectively) carry a significant portion of the haplotype shared by the affecteds. In addition, a 45-year-old woman (IV:16) who is presumed to have inherited the mutated gene has essentially normal hearing. Because some affected family members first became aware of their hearing loss as late as the sixth decade of life, retests of unaffected individuals several years from now may eventually reveal the onset of hearing loss. It is interesting to note that examples of incomplete penetrance appear to be clustered within certain sibships (III:9 and III:10; IV:16 and IV:17 and cousin IV:20), suggesting that other genetic factors or “modifier genes” may influence the expression of the phenotype. Conversely, once the responsible gene is identified, it is also possible that III:9 and III:10 will be found to carry the same mutation as is carried by other affected family members. If that is the case, the phenotype of the underlying mutation overlaps with presbycusis, and our criteria for affected status may have been too restrictive.
Database searches revealed numerous genes in the region that are known to be expressed in the ear. More than 30 expressed sequence tags map within the DFNA25 candidate-gene interval (Schuler et al. 1996; Skvorak et al. 1999; Unigene electronic updates). The proximal portion of the interval corresponds to mouse chromosome 10, whereas distal 12q24 corresponds to mouse chromosome 5. No mouse mutant with nonsyndromic deafness has yet been mapped to that interval on mouse 10; however, bronx waltzer (bv), mapping to mouse 5, may be a model for DFNA25 (Bussoli et al. 1997).
ATP2A2 [MIM 108740] and ATP2B1 [MIM 108731] both encode calcium channels and map to the DFNA25 interval (Olson et al. 1991; Sakuntabhai et al 1999). A related gene, Atp2b2 [MIM 108733], also known as Pmca2, encodes a plasma membrane calcium–ATPase type 2 pump known to be mutated in deafwaddler (dfw) mice (Street et al. 1998). Mutations in ATP2A2 cause Darier–White disease (DAR [MIM 124200]), a dermatologic disorder (Sakuntabhai et al. 1999). Although deafness is not part of the Darier phenotype, this gene’s role is thought to be analogous to that of connexin 26 (GJB2 [MIM 121011]) and connexin 31 (GJB3 [MIM 603324]) in intercellular communication between epidermal cells. GJB2 and GJB3 are known causes of nonsyndromic deafness as well as dermatologic disorders (Kelsell et al. 1997; Richard et al. 1998; Xia et al. 1999).
Two genes expressed in the basilar papilla of the chick are known to map to BACs (Human Genome Sequencing Center, Baylor College of Medicine) in the DFNA25 interval. UBE3B is a novel E3 ubiquitin ligase identified by upregulation of its mRNA immediately after noise exposure in the chick (Gong et al. 1996) and is known to be expressed in human fetal cochlea (Lomax et al., in press). VR-OAC (vanilloid receptor–related osmotically activated channel) is a novel calcium channel gene (Liedtke et al. in press).
The human ortholog of a novel zinc finger gene zfOC1, cloned from a guinea pig organ of Corti library, has been mapped to 12q24.3 (Rivolta et al. 1996). Other genes that map unambiguously within the interval include aldehyde dehydrogenase 2 (ALDH2 [MIM 100650]), B-cell translocation gene 1 (BTG1 [109580]), decorin (DCN [MIM 125255]), insulin-like growth factor 1 (IGF1 [MIM 147440]), leukotriene A4 hydrolase (LTA4H [MIM 151570]), mast cell growth factor (MGF [MIM 184745]) phenylalanine hydroxylase (PAH [MIM 261600], spinocerebellar ataxia 2 (SCA2 [MIM 183090]), and thioredoxin reductase 1 (TXNRD1 [MIM 601112]).
In summary, a novel locus for nonsyndromic sensorineural hearing impairment, DFNA25, has been mapped to 12q21-24. Because the phenotype is similar to presbycusis, this locus should be considered as a candidate region for presbycusis in the general population.
Acknowledgments
We thank the family for their participation and Michael Boehnke for critical review and helpful comments. This work was supported in part by the University of Michigan Phoenix Memorial Project, by grant K23 DC00161 from the National Institutes on Deafness and Other Communication Disorders (to M.M.L.), by grant M01RR00042 from the National Institutes of Health to the General Clinical Research Center at the University of Michigan, and by the NHLBI Mammalian Genotyping Service.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- Baylor College of Medicine, http://dot.imgen.bcm.tmc.edu:9331/
- Centre d’Etude du Polymorphisme Humain (CEPH/Genéthon), http://www.cephb.fr
- Center for Medical Genetics, Marshfield Medical Research Foundation, http://research.marshfieldclinic.org/genetics/ (for the order and genetic distances of loci on 12q)
- Cooperative Human Linkage Center, http://lpg.nci.nih.gov/CHLC
- Hereditary Hearing Loss Homepage, http://dnalab-www.uia.ac.be/dnalab/hhh/
- Morton Cochlear EST Database, http://hearing.bwh.harvard.edu/mappedcochlearests.htm
- NCBI/Unigene, http://www.ncbi.nlm.nih.gov/UniGene/
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for ALDH2 [MIM 100650], ATP2A2 [MIM 108740], ATP2B1 [MIM 108731], ATP2B2 [MIM 108733], BTG1 [MIM 109580], DAR [MIM 124200], DCN [MIM 125255], GJB2 [MIM 121011], GJB3 [MIM 603324], IGF1 [MIM 147440], LTA4H [MIM 151570], MGF [MIM 184745], PAH [MIM 261600], SCA2 [MIM 183090], and TXNRD1 [MIM 601112])
References
- Bergstrom L, Hemenway WG, Downs MP (1971) A high risk registry to find congenital deafness. Otolaryngol Clin North Am 4:369–399 [PubMed] [Google Scholar]
- Broman KW, Murray JC, Sheffield VC, White RL, Weber JL (1998) Comprehensive human genetic maps: individual and sex-specific variation in recombination. Am J Hum Genet 63:861–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussoli TJ, Kelly A, Steel KP (1997) Localization of the bronx waltzer (bv) deafness gene to mouse chromosome 5. Mamm Genome 8:714–717 [DOI] [PubMed] [Google Scholar]
- Dipple KM, McCabe ERB (2000) Phenotypes of patients with “simple” Mendelian disorders are complex traits: thresholds, modifiers, and systems dynamics. Am J Hum Genet 66:1729–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser GR (1964) Profound childhood deafness. J Med Genet 1:118–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong T-W, Hegeman AD, Shin JJ, Adler JH, Raphael Y, Lomax MI (1996) Identification of genes expressed after noise exposure in the chick basilar papilla. Hear Res 96:20–32 [DOI] [PubMed] [Google Scholar]
- Haapaniemi JJ (1996) The hearing threshold levels of children at school age. Ear Hear 17:469–477 [DOI] [PubMed] [Google Scholar]
- Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, Mueller RF, Leigh IM (1997) Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 387:80–83 [DOI] [PubMed] [Google Scholar]
- Konigsmark BW, Gorlin RJ (1976) Genetic hearing loss with no associated abnormalities. In: Konigsmark BW, Gorlin RJ (eds) Genetic and metabolic deafness. W. B. Saunders Co., Philadelphia, pp 7–23 [Google Scholar]
- Lathrop GM, Lalouel JM, Julier C, Ott J (1984) Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci USA 81:3443–3446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesperance MM, Hall JW, Bess FH, Fukushima K, Jain PK, Ploplis B, San Agustin TB, Skarka H, Smith RJH, Wills M, Wilcox ER (1995) A gene for autosomal dominant nonsyndromic heredity hearing impairment maps to 4p16.3. Hum Mol Genet 4:1967–1972 [DOI] [PubMed] [Google Scholar]
- Liedtke W, Choe Y, Martí-Renom MA, Bell AM, Denis CS, Šali A, Hudspeth AJ, Friedman JM, Heller S (2000) Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 103:525–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomax MI, Huang L, Cho Y, Gong T-WL, Altschuler RA (2000) Differential display and gene arrays to examine auditory plasticity. Hear Res 147:293–302 [DOI] [PubMed] [Google Scholar]
- Morton NE (1991) Genetic epidemiology of hearing impairment. Ann N Y Acad Sci 630:16–31 [DOI] [PubMed] [Google Scholar]
- Olson S, Wang MG, Carafoli E, Strehler EE, McBride OW (1991) Localization of two genes encoding plasma membrane Ca2+-transporting ATPases to human chromosomes 1q25-32 and 12q21-23. Genomics 9:629–641 [DOI] [PubMed] [Google Scholar]
- Richard G, Smith LE, Bailey RA, Itin P, Hohl D, Epstein EH, DiGiovanna JJ, Compton JG, Bale SJ (1998) Mutations in the human connexin gene GJB3 cause erythrokeratodermia variabilis. Nat Genet 20:366–369 [DOI] [PubMed] [Google Scholar]
- Rivolta MN, Negrini C, Wilcox ER (1996) A novel zinc finger gene preferentially expressed in the retina and the organ of Corti localizes to human chromosome 12q24.3. Biochim Biophys Acta 1306:127–132 [DOI] [PubMed] [Google Scholar]
- Robinson DW (1988) Threshold of hearing as a function of age and sex for the typical unscreened population. Br J Audiol 22:5–20 [DOI] [PubMed] [Google Scholar]
- Sakuntabhai A, Ruiz-Perez V, Carter S, Jacobsen N, Burge S, Monk S, Smith M, Munro CS, O’Donovan M, Craddock N, Kucherlapati R, Rees JL, Owen M, Lathrop GM, Monaco AP, Strachan T, Hovnanian A (1999) Mutations in ATP2A2, encoding a Ca2+ pump, cause Darier disease. Nat Genet 21:271–277 [DOI] [PubMed] [Google Scholar]
- Schuler GD, Boguski MS, Stewart EA, Stein LD, Gyapay G, Rice K, White RE, et al (1996) A gene map of the human genome. Science 274:540–546 [PubMed] [Google Scholar]
- Skvorak AB, Weng Z, Yee AJ, Robertson NG, Morton CC (1999) Human cochlear expressed sequence tags provide insight into cochlear gene expression and identify candidate genes for deafness. Hum Mol Genet 8:439–452 [DOI] [PubMed] [Google Scholar]
- Street VA, McKee-Johnson JW, Fonseca RC, Tempel BL, Noben-Trauth K (1998) Mutations in a plasma membrane Ca2+-ATPase gene cause deafness in deafwaddler mice. Nat Genet 19:390–394 [DOI] [PubMed] [Google Scholar]
- Van Camp G, Willems PJ, Smith RJ (1997) Nonsyndromic hearing impairment: unparalleled heterogeneity. Am J Hum Genet 60:758–764 [PMC free article] [PubMed] [Google Scholar]
- Xia JH, Liu CY, Tang BS, Pan Q, Huang L, Dai HP, Zhang BR, Xie W, Hu DX, Zheng D, Shi XL, Wang DA, Xia K, Yu KP, Liao XD, Feng Y, Yang YF, Xiao JY, Xie DH, Huang JZ (1998) Mutations in the gene encoding gap junction protein β-3 associated with autosomal dominant hearing impairment. Nat Genet 20:370–373 [DOI] [PubMed] [Google Scholar]
- Yuan B, Vaske D, Weber JL, Beck J, Sheffield VC (1997) Improved set of short-tandem-repeat polymorphisms for screening the human genome. Am J Hum Genet 60:459–460 [PMC free article] [PubMed] [Google Scholar]