

Abstract
Acheiropodia is an autosomal recessive developmental disorder presenting with bilateral congenital amputations of the upper and lower extremities and aplasia of the hands and feet. This severely handicapping condition appears to affect only the extremities, with no other systemic manifestations reported. Recently, a locus for acheiropodia was mapped on chromosome 7q36. Herein we report the narrowing of the critical region for the acheiropodia gene and the subsequent identification of a common mutation in C7orf2—the human orthologue of the mouse Lmbr1 gene—that is responsible for the disease. Analysis of five families with acheiropodia, by means of 15 polymorphic markers, narrowed the critical region to 1.3 cM, on the basis of identity by descent, and to <0.5 Mb, on the basis of physical mapping. Analysis of C7orf2, the human orthologue of the mouse Lmbr1 gene, identified a deletion in all five families, thus identifying a common acheiropodia mutation. The deletion was identified at both the genomic-DNA and mRNA level. It leads to the production of a C7orf2 transcript lacking exon 4 and introduces a premature stop codon downstream of exon 3. Given the nature of the acheiropodia phenotype, it appears likely that the Lmbr1 gene plays an important role in limb development.
Introduction
Acheiropodia (MIM 200500) is a unique condition presenting with bilateral congenital amputations of the upper and lower extremities and with aplasia of the hands and feet (Toledo and Saldanha 1969). It is distinguished from other hemimelias by a specific pattern of malformations, consisting of a complete amputation of the distal epiphysis of the humerus, amputation of the distal part of the tibial diaphysis, and aplasia of the radius, ulna, fibula and of the carpal, metacarpal, tarsal, metatarsal, and phalangeal bones (Toledo and Saldanha 1969, 1972). There appears to be little variability of expression (Grimaldi et al. 1983); however, in some affected individuals an ectopic bone (Bohomoletz bone) has been found at the distal end of the humerus (Toledo and Saldanha 1969). This severely handicapping condition appears to affect only the extremities, with no other systemic manifestations reported. With the exception of two affected siblings in Puerto Rico, the reported cases are of Brazilian origin (Kruger and Kumar 1994). The incidence of acheiropodia in Brazil has been estimated to be ∼1/250,000 births (Freire-Maia et al. 1975a). Acheiropodia is inherited as an autosomal recessive trait, and heterozygotes are phenotypically normal (Freire-Maia et al. 1975a). The vast majority of affected individuals are the offspring of consanguineous matings. The etiology of acheiropodia has remained obscure. Earlier studies ruled out abnormalities in glycosaminoglycan metabolism (Mourao et al. 1977). In a recent report, a locus for acheiropodia was mapped on chromosome 7q36 (Escamilla et al. 2000), to a region that overlaps with the preaxial polydactyly (PPD [MIM 174500]) and triphalangeal thumb loci (MIM 190605) (Heutink et al. 1994; Tsukurov et al. 1994). Herein we report the narrowing of the critical region for the acheiropodia gene and the subsequent identification of a common mutation in C7orf2 (GenBank accession number AF107454), the human orthologue of the mouse Lmbr1 gene responsible for the disease.
Subjects and Methods
Clinical Phenotype
The family panel comprises five families with acheiropodia, each consisting of an affected individual and his or her unaffected relatives. The families originate from the states of Sao Paulo (families 1 and 3–5) and Rio Grande do Sul (family 2) in Brazil (fig. 1). The phenotype of the proband in family 1 was characterized by truncation of the distal humeri, absence of the Bohomoletz bone, truncation of the tibiae, and absence of the fibulae. The phenotype of the proband in family 2 has been described elsewhere and is defined by bilateral truncations of the distal humeri and the tibiae, absence of the fibulae, and presence of the Bohomoletz bone at the distal aspect of the humeri (Lemos Silveira and Freire-Maia 1998). The phenotype of the proband in family 3 is similar to that of family 1. Pedigree 4 has been described elsewhere (Toledo and Saldanha 1969). The affected individuals have no feet, hands, or forearms. Some of the affected individuals possess one digit implanted into each upper extremity, either unilaterally or bilaterally (fig. 2). The phenotype of the proband in family 5 is characterized by the presence of small fingerlike appendages, bilaterally, which, on x-ray have been recognized as the Bohomoletz bone.
Figure 1.

Pedigrees of panel of families with acheiropodia that were used in this study
Figure 2.
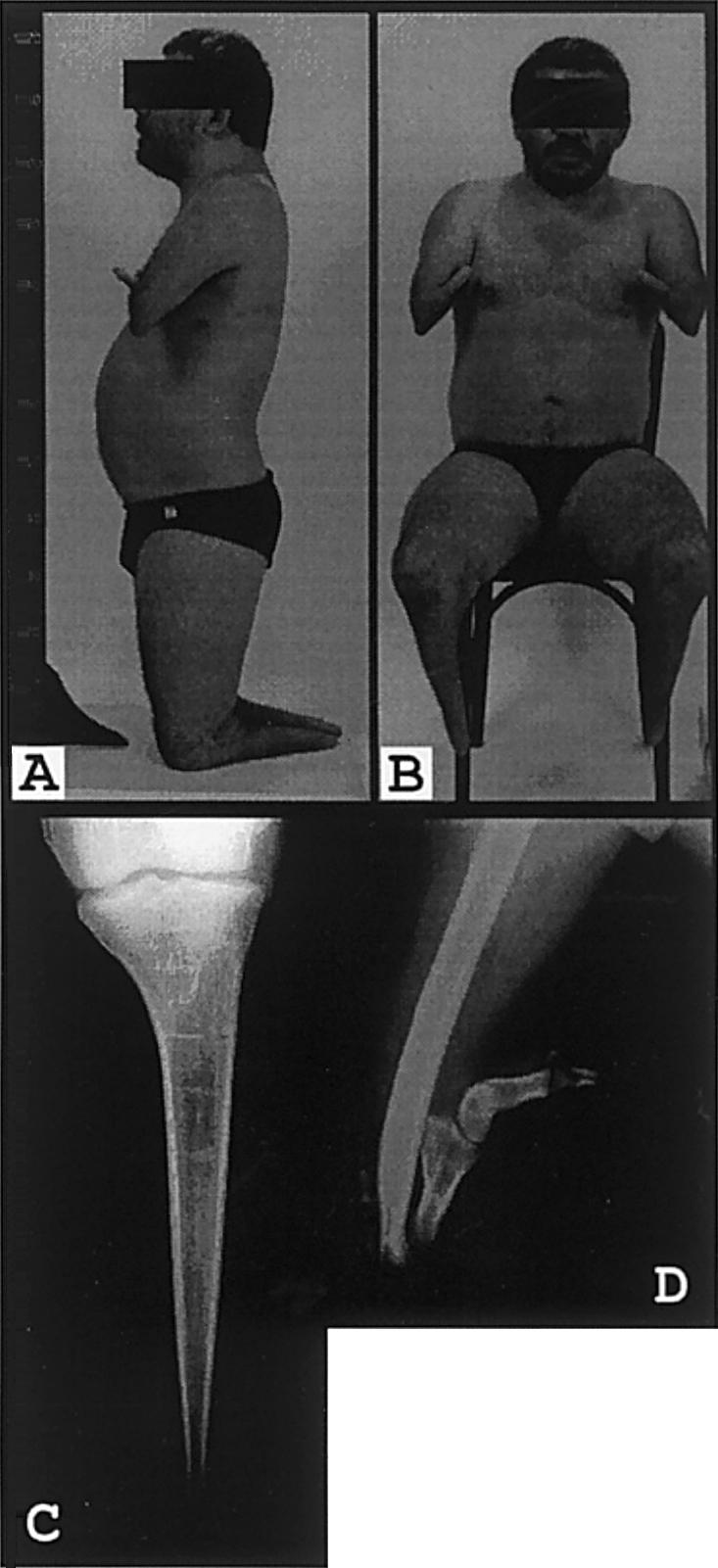
Affected individual (IV-18) from family 4. Note the small fingerlike appendages at the end of the arm, which are present bilaterally (A and B). C, Radiograph showing tapered amputation of the distal tibia. The proximal tibial epiphysis is well preserved. D, Radiograph showing dysplastic distal humerus articulating with a rudimentary forearm composed of three dysplastic long bones.
Haplotype Analysis
Peripheral blood from individuals affected with acheiropodia and from their relatives was taken after informed consent was obtained, and genomic DNA was extracted. The DNA was genotyped for 15 polymorphic markers from the 7q36 region, by means of radioactive PCR amplification followed by denaturing PAGE. Conditions for PCR reactions were as follows: 10 ng of genomic DNA, 0.2 mM of each dNTP, 1×PCR buffer (Boehringer), 1 μM nonlabeled primer, 0.1 μM [32P]– end-labeled primer, and 0.2 U of Taq polymerase were incubated at 97°C for 20 s, 55°C for 30 s, and 72°C for 30 s, for 30 cycles. Sequences of the primers used for amplification have been published elsewhere (The Genome Database). Allele sizes for the markers in the acheiropodia critical region, used for estimation of the age and ancestry of the acheiropodia mutation, were determined, and they were numbered according to designations used by The Genome Database.
Mutation Analysis
Multiplex PCR
Simultaneous amplification primer C7orf2-35 (5′-tgt tgg ctg tta tca ccc aga aaa t-3′) with primer C7orf2-36 (5′-gag aat tga tga ctt cag aat cag-3′) (exon 3), primer C7orf2-93 (5′-ttt gat gac ttt cgt gtt cat tg-3′) with primer C7orf2-94 (5′-gac cca aat cgt tta ctg gag ga-3′) (exon 4), and primer C7orf2-95 (5′-ttc cca ata ggt aca agt tac aat g-3′) with primer C7orf2-96 (5′-tgc ttt ttc tag ttc aat gtg aat g-3′) (exon 5) (Zguricas et al. 1999) was performed under standard conditions. In brief, 10 ng of genomic DNA, 0.2 mM of each dNTP, 1 μM of each primer, and 0.2 U of Taq polymerase, in 1×PCR buffer (Boehringer), were incubated at 97°C for 20 s, 62°C for 30 s, and 72°C for 30 s, for 30 cycles. PCR products were resolved on an agarose gel and were visualized by staining with ethidium bromide.
Reverse Transcriptase–PCR (RT-PCR)
Total RNA was extracted from whole blood from individuals affected with acheiropodia and from their parents, according to existing protocols. Ten micrograms of total RNA were subjected to reverse transcription using random hexamers and a first-strand–synthesis kit (Gibco BRL). The resulting cDNA was then PCR amplified by primers C7orf2-F3 (5′-gca tga agg agg atg gaa gg-3′) and C7orf2-R1 (5′-tga gtg ctg aag cta ccc aca-3′), under the conditions described above, which produced a 521-bp fragment that spans exons 1–6 of the cDNA.
Sequence Analysis
RT-PCR product from unaffected and affected individuals was column purified (Qiagen) and was sequenced by means of an internal primer (C7orf2-F2 [5′-agt cca cga tat gtt tcc ttc-3′]). Sequencing reactions were performed by means of a Beckman Coulter sequencing kit for automated sequencing, under the conditions recommended by the manufacturer. Products from the sequencing reaction were separated and then were analyzed on a Beckman Coulter model SEQ 2000 automated sequencer.
Results
Fine Mapping of the Acheiropodia Locus
To facilitate the identification of the gene responsible for this disorder, we refined the acheiropodia critical region by haplotype analysis of the five families with acheiropodia. Genotyping of these families for a series of 15 highly polymorphic 7q36 markers enabled us to compute, by using the GENEHUNTER package (Kruglyak et al. 1996), a LOD score of 4.04 for the three families (families 1, 2, and 4; see fig. 1) in which the offspring was the result of a known consanguineous mating. Furthermore, a long region of homozygosity was observed in affected individuals from all five families (fig. 3) , and these regions overlapped in identical alleles at D7S3037–D7S3036, suggesting that the most likely region for the acheiropodia locus can be reduced to the interval between D7S550 and D7S2465 (fig. 3). Thus, analysis of this family panel reduced the acheiropodia critical region from 8.4 cM (Escamilla et al. 2000) to 1.3 cM, on the basis of identity by descent (fig. 4) (Kruglyak et al. 1995; Ianakiev et al. 2000). The physical map of the region (see the Integrated Chromosome 7 Database [The Chromosome 7 Project]) suggests that it is wholly contained within a single YAC (c655a11) and that the size of the region is <0.5 Mb (fig. 4).
Figure 3.

Haplotypes derived by analysis of the five nuclear families. The putative ancestral haplotype shared by all affected individuals is boxed.
Figure 4.

Map of the 7q36 region containing the acheiropodia locus. Distances (in cM) between markers are shown. The acheiropodia critical region derived by Escamilla et al. (2000) is represented the white vertical bar, and the critical region identified in the present study is represented by the hatched vertical bar. A physical map of the D7S550-D7S2465 minimal critical region is shown, including the c655a11 YAC that spans the region and the positions of three BAC clones (RP11 332e22, RP11 580k21, and RP5 982e9) that contain the C7orf2 gene. Graphic representations of both the C7orf2 gene and the Δexon4 acheiropodia mutation are shown below the physical map; sizes (in kb) of introns are shown above representation of the C7orf2 gene, and exon designations are shown below the representation of the Δexon4 acheiropodia mutation.
Genomic Structure of C7orf2
Physical and transcriptional mapping of the PPD locus on chromosome 7q36, which partially overlaps with the acheiropodia critical region, identified three transcripts, designated “C7orf2,” “C7orf3,” and “C7orf4” (Heus et al. 1999). C7orf2 is the human orthologue of the mouse Lmbr1 gene and encodes a putative receptor, whereas C7orf3 and C7orf4 encode proteins of unknown function that are not expressed in the developing limb (Heus et al. 1999; Clark et al. 2000; M.J.v.B. and P.H., unpublished data). The mouse Lmbr1 gene shows striking alterations of expression in the limbs of Hemimelic extra toes (Hx) mice (Clark et al. 2000). The Hx mutation causes hemimelia of the radius and tibia and preaxial polydactyly on both forelimbs and hind limbs (Knudsen and Kochhar 1981). The involvement of the mouse Lmbr1 gene in a limb phenotype, as well as its genomic location, suggest that C7orf2 is a good candidate gene for acheiropodia.
The 17 exons of C7orf2 encompass ∼200 kb of genomic DNA (M.J.v.B. and P.H., unpublished data). All splice sites follow the consensus (5′ AG/N, 3′ N/GT), except for the 3′ splice site of exon 2, which uses the less common N/GC. The C7orf2 gene encodes a 1,470-nucleotide open reading frame preceded by a 176-nucleotide 5′UTR and followed by a 3,203-nucleotide 3′UTR (M.J.v.B. and P.H., unpublished data). The open reading frame predicts a 490-amino-acid protein containing nine putative transmembrane domains and a coiled-coil domain, which has led to the speculation that it might be located in the plasma membrane and might be a receptor protein. The predicted protein is 95% identical to the mouse Lmbr1 protein (Clark et al. 2000).
Mutation Analysis of C7orf2
To test the possibility that mutations in the C7orf2 gene are responsible for acheiropodia, genomic DNA from affected individuals from each of the five families with acheiropodia was amplified by sets of primers for all 17 exons of the C7orf2 gene. All exons amplified successfully, with the exception of exon 4—which consistently failed to amplify in affected individuals, in contrast to its reproducible amplification in both obligate carriers and normal controls. Multiplex PCR with primer sets for exons 3–5 clearly demonstrated the lack of exon 4 in affected individuals but not in obligate carriers (fig. 5A)
Figure 5.

A, Results of multiplex PCR analysis of genomic DNA. For each of the five families with acheiropodia, results of analysis of exons 3–5 of the C7orf2 gene, both in affected individuals (lanes A) and in one of their obligate-carrier parents (lanes C), are shown. Lane M, Size markers and positions of bands from each of the three exons (which are identified to the right of the gel). B, Results of RT-PCR analysis of total RNA, using primers spanning exons 1–6 from an affected individual and obligate-carrier parents in family 4 and from a normal control individual. Lane M, Size markers and sizes of RT-PCR fragments (which are denoted to the right of the gel). C, Results of sequence analysis of cDNA from an affected individual in family 4 and from a normal control individual. The sequence from the normal individual shows the exon 3/exon 4 and exon 4/exon 5 boundaries. The sequence from the affected individual shows the exon 3/exon 5 boundary created by the Δexon4 mutation.
To determine the status of exon 4 at the RNA level, total RNA from an affected individual, obligate carriers, and normal controls was analyzed by RT-PCR using primers spanning exons 1–6. RNA from normal controls and obligate carriers produced the expected 521-bp fragment; however, RNA from the individual with acheiropodia produced a 381-bp fragment, 140 bp smaller than the normal fragment and exactly the size of exon 4. RNA from the obligate-carrier parents of the affected individual produced both the 521-bp fragment and the 381-bp fragment, consistent with their heterozygous state (fig. 5B). Analysis by RT-PCR of RNA from a sample of 16 normal individuals from an ethnically mixed population showed only the normal-size fragment (data not shown).
Sequence analysis of the fragments produced by RT-PCR confirmed the lack of exon 4 of C7orf2 mRNA in individuals with acheiropodia (fig. 5C). Sequencing of the 381-bp fragment showed a clean transition from exon 3 to exon 5 in acheiropodia individuals (fig. 5C). In contrast, sequencing of the 521-bp fragment from normal controls showed the expected sequence for exons 3–5. Thus, the acheiropodia mutation leads to the production of a C7orf2 transcript lacking exon 4 of the gene and introduces a frameshift leading to a premature stop codon in exon 6. It therefore appears to be a true null mutation, consistent with the lack of any phenotypic effect in heterozygotes.
PCR-based analysis of genomic DNA from individuals with acheiropodia, by sets of primers spaced throughout introns 3 and 4, delineated the deletion's boundaries as being 1.2–2.5 kb 5′ of exon 4 and 2.7–3.5 kb 3′ of exon 4. These data indicate a deletion of 4–6 kb (data not shown).
Discussion
After the initial mapping of acheiropodia to 7q36 (Escamilla et al. 2000), we attempted to confirm and refine the mapping of the acheiropodia locus, by studying an affected individual from each of five Brazilian families. Confirmation was straightforwardly achieved, since three of the five individuals were the offspring of known consanguineous marriages. We computed a LOD score of 4.04 in these families—well in excess of the statistical proof required to confirm significant linkage. Furthermore, in each of the five affected individuals, homozygosity was observed in no fewer than eight consecutive microsatellite markers. Within these overlapping regions of homozygosity, we observed a two-marker haplotype that was shared identically among all these patients and that was not seen in homozygous form in any unaffected individuals in these families. The rarity of certain alleles strongly indicates that this segment, which spans <1.3 cM, was inherited by descent from a single ancestor. Interestingly, this 1.3-cM region is completely contained within the 8.4-cM region implicated by Escamilla et al.'s (2000) report, and the shared alleles near this region match the shared haplotype in that study. This observation strongly suggests that all the cases in the two studies share homozygosity for a common haplotype and confirms the expectation that a single common ancestral mutation (Freire-Maia 1975b) is the cause of nearly all cases of acheiropodia in Brazil.
The length (∼1.3 cM) of the 7q36 region shared in all five patients in the present study suggests a relatively recent age (31 generations) for this common mutation; however, because only five individuals (assumed to represent only five independent paths to the ancestor, with very recent consanguinity causing duplication) were examined, simulations indicated that a mutation arising in a common ancestor anywhere from 5 to 95 generations ago would also be consistent with this extent of haplotype sharing. If we include in this analysis the families studied by Escamilla et al. (2000), simulations indicate a 20-generation-old mutation, with a 95% confidence interval of 4–60 generations.
Analysis of the C7orf2 gene identified a common acheiropodia mutation in all five unrelated affected individuals studied. C7orf2 originally had been identified as one of the candidate genes for PPD (Heus et al. 1999) and is the human orthologue of the mouse Lmbr1 gene. Lmbr1 maps to the candidate interval defined for the mouse Hx and Hammertoe (Hm) mutants. These are generally considered to be the mouse homologous phenotypes for PPD and complex polysyndactyly, respectively, suggesting C7orf2 and Lmbr1 as candidate genes for these phenotypes. Although no coding-sequence alterations or genomic rearrangements of C7orf2 were detected in five unrelated families with PPD (M.J.v.B. and P.H., unpublished data) and no mutations were found in Hx or Hm mice, Lmbr1 expression has been found to be altered in developing Hx limbs (Clark et al. 2000). Lmbr1 is ubiquitously expressed, including the developing limbs, but in the Hx mutant the level of expression is significantly reduced during a specific period of limb development (E10.5–E12.5) (Clark et al. 2000). This aberrant expression coincides with the ectopic expression of Shh at the anterior aspect of the apical ectodermal ridge, as opposed to its normal expression, which restricted to the posterior aspect (Riddle et al. 1993). Shh is one of the key signaling molecules involved in specification of the antero-posterior and proximo-distal axes of the limb. A possible explanation for the observed ectopic expression of Shh in the Hx mutant could be that C7orf2/Lmbr1 acts as a repressor of Shh. Whether this effect occurs through direct interaction with Shh or is mediated by other proteins remains to be clarified. Given the nature of the acheiropodia and Hx phenotypes, it is clear that C7orf2/Lmbr1 plays a crucial role in distal-limb formation and outgrowth. Dissection of the function of this gene will foster our understanding of the processes involved in pattern formation during embryogenesis.
Acknowledgments
The authors express their gratitude to the families for participating in this study. This work was supported in part by the Coles Family Foundation (support to P.T.), the Dutch Organisation for Scientific Research (NWO) (support to P.H.), and the Stichting Klinische Genetica Rotterdam (support to P.H.). S.P.A.T. is a CNPq Researcher supported by grant 300346-82-4. The authors thank H. van der Linde for technical assistance.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- Chromosome 7 Project, The, http://www.genet.sickkids.on.ca/chromosome7 (for the Integrated Chromosome 7 Database)
- GenBank Overview, http://www.ncbi.nlm.nih.gov/Genbank/GenbankOverview.html (for C7orf2 [accession number AF107454])
- Genome Database, The, http://gdbwww.gdb.org (for allele sizes and amplification sequences)
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for acheiropodia [MIM 200500] and triphalangeal thumb locus [MIM 190605])
References
- Clark RM, Marker PC, Kingsley DM (2000) A novel candidate gene for mouse and human preaxial polydactyly with altered expression in limbs of hemimelic extra-toes mutant mice. Genomics 67:19–27 [DOI] [PubMed] [Google Scholar]
- Escamilla MA, DeMille MC, Benavides E, Roche E, Almasy L, Pittman S, Hauser J, Lew DF, Freimer B, Whittle MR (2000) A minimalist approach to gene mapping: locating the gene fora acheiropodia by homozygosity analysis. Am J Hum Genet 66:1995–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire-Maia A, Freire-Maia N, Morton NE, Azevedo ES, Quelce-Salgado A (1975a) Genetics of acheiropodia (the handless and footless families of Brazil). VI. Formal genetic analysis. Am J Hum Genet 27:521–527 [PMC free article] [PubMed] [Google Scholar]
- Freire-Maia A, Li WH, Maruyama T (1975b) Genetics of acheiropodia (the handless and footless families of Brazil) VII. Population dynamics. Am J Hum Genet 27:665–675 [PMC free article] [PubMed] [Google Scholar]
- Grimaldi A, Masiero D, Richieri-Costa A, Freire-Maia A (1983) Variable expressivity of the acheiropodia gene. Am J Med Genet 16:631–634 [DOI] [PubMed] [Google Scholar]
- Heus HC, Hing A, van Baren MJ, Joosse M, Breedveld GJ, Wang JC, Burgess A, Donnis-Keller H, Berglund C, Zguricas J, Scherer SW, Rommens JM, Oostra BA, Heutink P (1999) A physical and transcriptional map of the preaxial polydactyly locus on chromosome 7q36. Genomics 57:342–351 [DOI] [PubMed] [Google Scholar]
- Heutink P, Zguricas J, van Oosterhout L, Breedveld GJ, Testers L, Sandkuijl LA, Snijders PJ, Weissenbach J, Lindhout D, Hovius SE (1994) The gene for triphalangeal thumb maps to the subtelomeric region of chromosome 7q. Nat Genet 6:287–292 [DOI] [PubMed] [Google Scholar]
- Ianakiev P, Kilpatrick MW, Daly MJ, Zolindaki A, Bagley D, Beighton G, Beighton P, Tsipouras P (2000) Localization of an acromesomelic dysplasia on chromosome 9 by homozygosity mapping. Clin Genet 57:278–283 [DOI] [PubMed] [Google Scholar]
- Knudsen TB, Kochhar DM (1981) The role of morphogenetic cell death during abnormal limb-bud outgrowth in mice heterozygous for the dominant mutation hemimelia-extra toe (Hmx). J Embryol Exp Morphol Suppl 65:289–307 [PubMed] [Google Scholar]
- Kruger LM, Kumar A (1994) Acheiropody: a report of two cases. J Bone Joint Surg 76A:1557–1560 [DOI] [PubMed] [Google Scholar]
- Kruglyak L, Daly MJ, Lander ES (1995) Rapid multipoint linkage analysis of recessive traits in nuclear families including homozygosity mapping. Am J Hum Genet 56:519–517 [PMC free article] [PubMed] [Google Scholar]
- Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES (1996) Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58:1347–1363 [PMC free article] [PubMed] [Google Scholar]
- Lemos Silveira E, Freire-Maia A (1998) Acheiropodia: new cases from Brazil. Clin Genet 54:256–257 [DOI] [PubMed] [Google Scholar]
- Mourao PAS, Toledo SPA, Dietrich CP (1977) Urinary mucopolysaccharides in acheiropodia. Acta Genet Med Gemellol (Roma) 26:92–94 [DOI] [PubMed] [Google Scholar]
- Riddle RD, Johnson RL, Laufer E, Tabin C (1993) Sonic hedgehog mediates the polarizing activity of the ZPA. Cell 75:1401–1416 [DOI] [PubMed] [Google Scholar]
- Toledo SPA, Saldanha PH (1969) A radiological and genetic investigation of acheiropody in a kindred including six cases. J Genet Hum 17:81–94 [PubMed] [Google Scholar]
- ——— (1972) Further data on acheiropody. J Genet Hum 20:253–258 [PubMed] [Google Scholar]
- Tsukurov O, Boehmer A, Flynn J, Nicolai JP, Hamel BC, Traill S, Zaleske D, Mankin HJ, Yeon H, Ho C (1994) A complex bilateral polysyndactyly disease locus maps to chromosome 7q36. Nat Genet 6:282–286 [DOI] [PubMed] [Google Scholar]
- Zguricas J, Heus H, Morales-Peralta E, Breedveld G, Kuyt B, Mumcu EF, Bakker W, Akarsu N, Kay SP, Hovius SE, Heredero-Baute L, Oostra BA, Heutink P (1999) Clinical and genetic studies on 12 preaxial polydactyly families and refinement of the localization of the gene responsible to a 1.9 cM region on chromosome 7q36. J Med Genet 36:32–40 [PMC free article] [PubMed] [Google Scholar]