H-J Lüdecke
H-J Lüdecke
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
1,
J Schaper
J Schaper
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
2,
P Meinecke
P Meinecke
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
4,
P Momeni
P Momeni
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
1,
S Groß
S Groß
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
1,
D von Holtum
D von Holtum
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
1,
H Hirche
H Hirche
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
3,
M J Abramowicz
M J Abramowicz
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
5, *,
B Albrecht
B Albrecht
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
1,
C Apacik
C Apacik
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
6, †,
H-J Christen
H-J Christen
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
7,
U Claussen
U Claussen
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
9,
K Devriendt
K Devriendt
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
10,
E Fastnacht
E Fastnacht
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
11,
A Forderer
A Forderer
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
12,
U Friedrich
U Friedrich
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
13,
T H J Goodship
T H J Goodship
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
14,
M Greiwe
M Greiwe
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
15,
H Hamm
H Hamm
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
16,
R C M Hennekam
R C M Hennekam
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
17,
G K Hinkel
G K Hinkel
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
18,
M Hoeltzenbein
M Hoeltzenbein
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
19,
H Kayserili
H Kayserili
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
20,
F Majewski
F Majewski
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
21,
M Mathieu
M Mathieu
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
22,
R McLeod
R McLeod
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
23,
A T Midro
A T Midro
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
24,
U Moog
U Moog
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
25,
T Nagai
T Nagai
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
26,
N Niikawa
N Niikawa
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
27,
K H Ørstavik
K H Ørstavik
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
28,
E Plöchl
E Plöchl
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
29,
C Seitz
C Seitz
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
16,
J Schmidtke
J Schmidtke
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
8,
L Tranebjærg
L Tranebjærg
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
30,
M Tsukahara
M Tsukahara
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
31,
B Wittwer
B Wittwer
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
32,
B Zabel
B Zabel
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
33,
G Gillessen-Kaesbach
G Gillessen-Kaesbach
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
1,
B Horsthemke
B Horsthemke
1Institut für Humangenetik, 2Zentralinstitut für Röntgendiagnostik, Kinderradiologie, and 3Institut für Medizinische Informatik, Biometrie und Epidemiologie, Universitätsklinikum, Essen; 4Abteilung für Medizinische Genetik, Altonaer Kinderkrankenhaus, Hamburg; 5Services de Génétique médicale, Hôpital Erasme, Brussels; 6Kinderzentrum München, Munich; 7Kinderkrankenhaus auf der Bult, and 8Institut für Humangenetik, Medizinische Hochschule, Hannover; 9Institut für Humangenetik und Anthropologie, Jena, Germany; 10Center for Human Genetics, Universitaire Ziekenhuizen, Leuven; 11Zentrum für Kinder- und Jugendmedizin, Kreiskrankenhaus Lüdenscheid, Lüdenscheid, Germany; 12Praxis für Kinderheilkunde, Fulda, Germany; 13Institute of Human Genetics, University of Aarhus, The Bartholin Building, Aarhus, Denmark; 14Department of Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, Great Britain; 15Institut für Humangenetik, Universitätsklinikum, Lübeck, Germany; 16Klinik und Poliklinik für Haut- und Geschlechtskrankheiten, Universität, Würzburg, Germany; 17Department of Clinical Genetics and Pediatrics, University of Amsterdam, Amsterdam; 18Institut für Medizinische Genetik, Technische Universität, Dresden; 19Institut für Humangenetik, Ernst-Moritz-Arndt-Universität, Greifswald, Germany; 20Institute of Child Health, Division of Medical Genetics, University of Istanbul, Istanbul; 21Institut für Humangenetik und Anthropologie, Heinrich-Heine-Universität, Düsseldorf; 22Unité de Génétique Clinique, Centre Hospitalier Universitaire, Amiens, France; 23Department of Medical Genetics, Alberta Children's Hospital, Calgary; 24Department of Clinical Genetics, Medical University Bialystok, Bialystok, Poland; 25Stichting Klinische Genetica, Maastricht; 26Department of Pediatrics, Dokkyo University School of Medicine, Koshigaya Hospital, Koshigaya, Japan; 27Department of Human Genetics, University School of Medicine, Nagasaki; 28Avdeling for Medisinsk Genetikk, Ullevål Sykehus, Oslo; 29Klinische Genetik, St.-Johanns-Spital, Salzburg; 30Department of Medical Genetics, University Hospital, Regionsykehuset I Tromsø, Tromsø, Norway; 31School of Allied Health Sciences, Yamaguchi University, Yamaguchi, Japan; 32Institut für Humangenetik, Westfälische Wilhelms-Universität, Münster, Germany; and 33Kinderklinik und Poliklinik, Klinikum der Johannes Gutenberg-Universität, Mainz, Germany
1



 d(Left/Right)
d(Left/Right) = mean length deviation of the 19 tubular bones of the left or right hand.
= mean length deviation of the 19 tubular bones of the left or right hand. scores) are expressed as multiples of SDs. The mean values and SDs had been established for American-born individuals of European ancestry by Garn et al. (1972). The calculations and plots of profiles, the comparison of the MCPPs from different individuals, and the calculation of Pearson's correlation coefficient (r) were done with the computer program ANTRO (release 4.83E; Hosenfeld et al. 1991). In some cases we encountered problems with the length measurements because of bowing of metacarpals and axial deviations at the interphalangeal joints, which lead to a distorted projection of the bones on the film.
scores) are expressed as multiples of SDs. The mean values and SDs had been established for American-born individuals of European ancestry by Garn et al. (1972). The calculations and plots of profiles, the comparison of the MCPPs from different individuals, and the calculation of Pearson's correlation coefficient (r) were done with the computer program ANTRO (release 4.83E; Hosenfeld et al. 1991). In some cases we encountered problems with the length measurements because of bowing of metacarpals and axial deviations at the interphalangeal joints, which lead to a distorted projection of the bones on the film.

 ) of the remaining 75 patients was 1.41 SD below the average height of the respective population (with the standard deviation s=1.15 for single values). Although the average values for the seven patients with a chromosomal abnormality (
) of the remaining 75 patients was 1.41 SD below the average height of the respective population (with the standard deviation s=1.15 for single values). Although the average values for the seven patients with a chromosomal abnormality (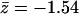 SD; s=1.38) and the six patients in whom we could not identify a mutation (
SD; s=1.38) and the six patients in whom we could not identify a mutation (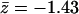 SD; s=1.25) are also in this range, we did not include them in our calculations, because effects of other genes cannot be excluded.
SD; s=1.25) are also in this range, we did not include them in our calculations, because effects of other genes cannot be excluded.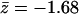 SD; s=1.29) than that of the 34 children (
SD; s=1.29) than that of the 34 children (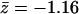 SD; s=0.96).
SD; s=0.96).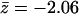 SD; s=1.42) than the 53 patients with nonsense mutations (
SD; s=1.42) than the 53 patients with nonsense mutations (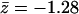 SD; s=1.06). The heights of children with missense mutations (
SD; s=1.06). The heights of children with missense mutations (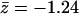 SD; s=0.96; n=5) are comparable to those with nonsense mutations (
SD; s=0.96; n=5) are comparable to those with nonsense mutations (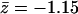 SD; s=0.98; n=29). However, when only the adult patients with missense (
SD; s=0.98; n=29). However, when only the adult patients with missense (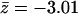 SD; s=1.28; n=4) or nonsense (
SD; s=1.28; n=4) or nonsense (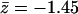 SD; s=1.15; n=24) mutations are compared, a statistically significant (P<.05) difference can be seen, although the ranges for patients with missense mutations (−1.7 to −4.6 SD) and patients with nonsense mutations (0 to −3.3 SD) overlap. Finally, comparison of the average height of adult patients with missense mutations (
SD; s=1.15; n=24) mutations are compared, a statistically significant (P<.05) difference can be seen, although the ranges for patients with missense mutations (−1.7 to −4.6 SD) and patients with nonsense mutations (0 to −3.3 SD) overlap. Finally, comparison of the average height of adult patients with missense mutations (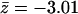 SD; s=1.28; n=4) with the average height of children with missense mutations (
SD; s=1.28; n=4) with the average height of children with missense mutations (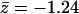 SD; s=0.97; n=5) proves (P<.05) that growth retardation in TRPS is a progressive process.
SD; s=0.97; n=5) proves (P<.05) that growth retardation in TRPS is a progressive process.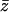 ) of all 19 tubular bones of a hand as a measure of the severity of brachydactyly (table 1). The X-rays of the youngest patient, individual II-1 of family 6820, did not show CSEs, and MCPP analysis could not disclose brachydactyly. The MCPPs of the remaining 43 patients disclosed a spectrum of affected bones, ranging from only two middle phalanges bilaterally in patient 10378 (
) of all 19 tubular bones of a hand as a measure of the severity of brachydactyly (table 1). The X-rays of the youngest patient, individual II-1 of family 6820, did not show CSEs, and MCPP analysis could not disclose brachydactyly. The MCPPs of the remaining 43 patients disclosed a spectrum of affected bones, ranging from only two middle phalanges bilaterally in patient 10378 (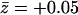 /+0.7), with hemizygosity for TRPS1, to nearly uniform shortness of all 19 bones of both hands, as in patient 13242 (
/+0.7), with hemizygosity for TRPS1, to nearly uniform shortness of all 19 bones of both hands, as in patient 13242 (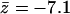 at age 17 years), in whom TRPS III was diagnosed (Vilain et al. 1999). Figure 4A shows examples of individual pattern profiles. With one exception (case 13287), we found that patients with a missense mutation had, on average, shorter metacarpals than did those with a nonsense or the splice-site mutation. Therefore, we calculated mean pattern profiles for either the 35 patients with a heterozygous nonsense (and the splice-site) mutation (“mean TRPS I”; fig. 4B) or the four patients with a heterozygous missense mutation (“mean TRPS III”). Mean pattern profiles were calculated from the left-hand MCPPs, except for patient 11049, for whom only the right-hand X-ray was available. For reasons explained above, we did not include patient G1298 (II-1), patients with chromosomal abnormalities, or patients with an unknown genetic defect in this analysis. We compared the mean MCPPs by calculating the product moment correlation (Pearson's r; Poznanski et al. 1972) with the mean Poznanski profile and with each other. The mean TRPS I profile correlates better with mean Poznanski (r=0.68; P<.01) than mean TRPS III does (r=0.47; P<.05), but mean TRPS I is significantly different from mean TRPS III (r=0.27). Like the difference between the mean heights of patients with TRPS I or TRPS III, the difference between the pattern profiles “mean TRPS I” and “mean TRPS III” proves that patients with a missense mutation tend to be more severely affected than patients with nonsense mutations. However, MCPP analysis cannot be recommended as an unequivocal diagnostic tool in TRPS. For example, we found nonsymmetrical changes in 3 of 30 patients (patients 6820 (I-1), 13365, and 13568), for whom X-rays of both hands were available. In patient 13365 (I-1), who has the recurrent R531X mutation, the discrepancy between hands was so extreme that the left-hand MCPP correlated with the mean TRPS I MCPP (r=0.73 [P<.001] vs. r=0.37; no correlation with the mean TRPS III), whereas the right-hand MCPP would suggest TRPS III (r=0.64 [P<.01] vs. r=0.5 [P<.05] for TRPS I). Furthermore, we found, for example, different MCPPs in a mother (individual II-2) and her daughter (individual III-1) in case 13333 (r=0.38, no correlation, fig. 4C) and in unrelated female patients of the same age with identical missense mutations, patients 13287 and 13307 (r=0.19; no correlation; not shown).
at age 17 years), in whom TRPS III was diagnosed (Vilain et al. 1999). Figure 4A shows examples of individual pattern profiles. With one exception (case 13287), we found that patients with a missense mutation had, on average, shorter metacarpals than did those with a nonsense or the splice-site mutation. Therefore, we calculated mean pattern profiles for either the 35 patients with a heterozygous nonsense (and the splice-site) mutation (“mean TRPS I”; fig. 4B) or the four patients with a heterozygous missense mutation (“mean TRPS III”). Mean pattern profiles were calculated from the left-hand MCPPs, except for patient 11049, for whom only the right-hand X-ray was available. For reasons explained above, we did not include patient G1298 (II-1), patients with chromosomal abnormalities, or patients with an unknown genetic defect in this analysis. We compared the mean MCPPs by calculating the product moment correlation (Pearson's r; Poznanski et al. 1972) with the mean Poznanski profile and with each other. The mean TRPS I profile correlates better with mean Poznanski (r=0.68; P<.01) than mean TRPS III does (r=0.47; P<.05), but mean TRPS I is significantly different from mean TRPS III (r=0.27). Like the difference between the mean heights of patients with TRPS I or TRPS III, the difference between the pattern profiles “mean TRPS I” and “mean TRPS III” proves that patients with a missense mutation tend to be more severely affected than patients with nonsense mutations. However, MCPP analysis cannot be recommended as an unequivocal diagnostic tool in TRPS. For example, we found nonsymmetrical changes in 3 of 30 patients (patients 6820 (I-1), 13365, and 13568), for whom X-rays of both hands were available. In patient 13365 (I-1), who has the recurrent R531X mutation, the discrepancy between hands was so extreme that the left-hand MCPP correlated with the mean TRPS I MCPP (r=0.73 [P<.001] vs. r=0.37; no correlation with the mean TRPS III), whereas the right-hand MCPP would suggest TRPS III (r=0.64 [P<.01] vs. r=0.5 [P<.05] for TRPS I). Furthermore, we found, for example, different MCPPs in a mother (individual II-2) and her daughter (individual III-1) in case 13333 (r=0.38, no correlation, fig. 4C) and in unrelated female patients of the same age with identical missense mutations, patients 13287 and 13307 (r=0.19; no correlation; not shown).
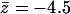 ) with involvement of metacarpals II–V. The A919T missense mutation exchanges a nonpolar to a polar amino acid within the Zn2+-chelating domain of the zinc finger, which probably destabilizes the finger structure by an impairment of Zn2+ binding. The A919T mutation was found in a family from Japan (case 13915, individuals I-1 and II-2) and in two sporadic patients from Belgium (case 13287) and Turkey (case 13307). For the Japanese patients, the diagnosis of Ruvalcaba syndrome had been proposed (Sugio and Kajii 1984) but was corrected to TRPS III 10 years later (Kajii et al. 1994). The three elder patients with the A919T mutation have moderate short stature but remarkably short hands. On average, adult patients with TRPS1 missense mutations are smaller than those with nonsense mutations but are not as small as patients with the contiguous gene syndrome TRPS II, in whom the deletion of the EXT1 gene, which is also involved in bone development, exerts an additive effect (average height
) with involvement of metacarpals II–V. The A919T missense mutation exchanges a nonpolar to a polar amino acid within the Zn2+-chelating domain of the zinc finger, which probably destabilizes the finger structure by an impairment of Zn2+ binding. The A919T mutation was found in a family from Japan (case 13915, individuals I-1 and II-2) and in two sporadic patients from Belgium (case 13287) and Turkey (case 13307). For the Japanese patients, the diagnosis of Ruvalcaba syndrome had been proposed (Sugio and Kajii 1984) but was corrected to TRPS III 10 years later (Kajii et al. 1994). The three elder patients with the A919T mutation have moderate short stature but remarkably short hands. On average, adult patients with TRPS1 missense mutations are smaller than those with nonsense mutations but are not as small as patients with the contiguous gene syndrome TRPS II, in whom the deletion of the EXT1 gene, which is also involved in bone development, exerts an additive effect (average height 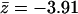 SD; n=20; Langer et al. 1984).
SD; n=20; Langer et al. 1984).