

Abstract
3-Methylcrotonylglycinuria is an inborn error of leucine catabolism and has a recessive pattern of inheritance that results from the deficiency of 3-methylcrotonyl-CoA carboxylase (MCC). The introduction of tandem mass spectrometry in newborn screening has revealed an unexpectedly high incidence of this disorder, which, in certain areas, appears to be the most frequent organic aciduria. MCC, an heteromeric enzyme consisting of α (biotin-containing) and β subunits, is the only one of the four biotin-dependent carboxylases known in humans that has genes that have not yet been characterized, precluding molecular studies of this disease. Here we report the characterization, at the genomic level and at the cDNA level, of both the MCCA gene and the MCCB gene, encoding the MCCα and MCCβ subunits, respectively. The 19-exon MCCA gene maps to 3q25-27 and encodes a 725-residue protein with a biotin attachment site; the 17-exon MCCB gene maps to 5q12-q13 and encodes a 563-residue polypeptide. We show that disease-causing mutations can be classified into two complementation groups, denoted “CGA” and “CGB.” We detected two MCCA missense mutations in CGA patients, one of which leads to absence of biotinylated MCCα. Two MCCB missense mutations and one splicing defect mutation leading to early MCCβ truncation were found in CGB patients. A fourth MCCB mutation also leading to early MCCβ truncation was found in two nonclassified patients. A fungal model carrying an mccA null allele has been constructed and was used to demonstrate, in vivo, the involvement of MCC in leucine catabolism. These results establish that 3-methylcrotonylglycinuria results from loss-of-function mutations in the genes encoding the α and β subunits of MCC and complete the genetic characterization of the four human biotin-dependent carboxylases.
Introduction
3-Methylcrotonylglycinuria (MCG [MIM 210200]) is an inborn error of leucine catabolism and results from isolated, biotin-insensitive deficiency of 3-methylcrotonyl-CoA carboxylase (MCC; E.C.6.4.1.4), the enzyme converting 3-methylcrotonoyl-CoA to 3-methylglutaconyl-CoA (Sweetman and Williams 1995). This mitochondrial enzyme is one of the four biotin-dependent carboxylases known in humans (Wolf 1995), together with acetyl CoA carboxylase (ACC), pyruvate carboxylase (PC), and propionyl CoA carboxylase (PCC). In marked similarity to PCC, MCC is a hetero-oligomeric enzyme consisting of α and β subunits. Both PCCα and MCCα contain a covalently bound biotin moiety, as well as binding sites for two of the substrates, HCO3− and ATP, whereas the β subunits contain the binding site for the acyl-CoA substrate (Lau et al. 1980; Lamhonwah et al. 1986; Song et al. 1994; Sweetman and Williams 1995).
Patients with MCG, which is inherited as an autosomal recessive trait, show a highly variable clinical phenotype, ranging from asymptomatic to severe. In the latter phenotype, episodes of acute metabolic decompensation can lead to coma, lethargy, and death (Sweetman and Williams 1995). The metabolic phenotype characterizing MCC deficiency is the elevated excretion of the diagnostic compounds 3-methylcrotonylglycine or 3-hydroxyisovaleric acid and the presence of abnormally elevated blood or urine levels of 3-hydroxyisovalerylcarnitine (C5-OH), as determined by tandem mass spectrometry (MS/MS) (van Hove et al. 1995; Gibson et al. 1998). The recent introduction of neonatal screening of organic acidurias by MS/MS has revealed MCG as an unexpectedly frequent disorder (Naylor and Chace 1999; Ranieri et al. 2000a; Roscher et al. 2000; Smith et al. 2000; Wilcken et al. 2000). Despite the exponentially increasing genomic information available, the genes for human MCC subunits have not yet been characterized, precluding molecular studies of this disease.
Here we report the cDNA sequence and the structure of the human MCCA and MCCB genes, encoding the MCC α and β subunits, respectively. We also report, for both genes, the identification of mutations leading to MCG. Finally, we use a fungal model for MCG carrying an MCCA null allele, to demonstrate, in vivo, the specific involvement of MCC in leucine catabolism.
Patients, Material, and Methods
Patients
Seven patients have been included in this study. All eight probands showed an MCC activity of <10% of that in the controls, in extracts of lymphoblasts or cultured skin fibroblasts, and showed elevated urinary excretion of the diagnostic compounds 3-methylcrotonylglycine or 3-hydroxyisovaleric acid. Probands 15468 (Spain), 15469 (Argentina), and 15765 (United States) presented, at <6 mo of age, with a severe episode of metabolic decompensation with hypoglycemia, hyperammonemia, hypocarnitinemia, and metabolic acidosis. Proband 15766 (United States) was diagnosed at 4 years of age. Probands 15626 and 15628 were two clinically asymptomatic female adults belonging to the Amish/Mennonite population who came to clinical attention after their children were suspected to be obligate carriers (Gibson et al. 1998).
Expressed-Sequence Tags (ESTs), DNA Sequencing, and Chromosomal Localization of MCCA and MCCB
Human IMAGE ESTs were obtained from Incyte Genomics. Aspergillus nidulans ESTs were obtained from the Fungal Genetics Stock Centre. DNA sequencing was performed with a dye-terminator cycle-sequencing kit (PE Biosystems) and an ABI PRISM 377 automatic sequencer. The MCCA and MCCB genes were mapped by radiation hybrid (RH) mapping in the Stanford Human Genome Center G3 panel and the Genebridge 4 (Radiation Hybrid Mapping database) panel (Gyapay et al. 1996; Stewart et al. 1997) (Research Genetics). Primers are shown in table 1.
Table 1.
Primers for Amplification of MCCA and MCCB Sequences
| Forward Primer(5 ′→3′) | Reverse Primer(5 ′→3′) | Amplified Target | PCR-Fragment Size(bp) |
|
MCCA |
|||
| TGGAATTTCTAGTAAGGCAAGCa | AAGTTGTGTTTCTCTTGCCTTCa | Exon 9 | 219 |
| ACCATTGAATGAAGACAGCGGb | GTGCTGGGACTACAGGCTTGb | Exon 10 | 306 |
| CATAAGAAACACTATTCTATTGCCb | AATATGCTAGACAGTTCTGTAAGb | Exon 11 | 316 |
| AATCAGACAAAAGGGAATCGGa | TAATTAGTGACCCAAATGCATGa | Exon 19 | 194 |
|
MCCB |
|||
| GCTATATTTTGGATTAAGTATTATGc | TACAAGACTGTCTAATAAAGTACc | Exon 3 | 245 |
| ATCTCTTAAATTCTCTCTCAATGc | CAAACTATTTGCATTTTACCAGCc | Exon 5 | 226 |
| GCTGTCATTGATACAAGTTTCCc | CATCCCAGAGTACCTAATTCGc | Exon 6 | 269 |
| CTATAGAAGTCACTGCATGAGCTGc | TGTTCTGCATATTTAATTTCATCGc | Exon 7 | 219 |
| GAAAATTAACACATGTAGTAGCCa | TATATTAATCATCTCAATAATACAGa | Exon 17 | 300 |
| AGGCAGCCAATGTGTTGGCd | CTTTATTAGGAAAACATTTGATAGd | 3′ UTR | 2,100 |
Used only in RH mapping.
Used both in mutation screening and/or SSCP analysis with human genomic DNA as template and in RH mapping.
Used only in mutation screening and/or SSCP analysis with human genomic DNA as template.
Used to amplify the alternative long 3′ UTR of the MCCB gene; the template here was human liver cDNA.
FISH Mapping
Metaphase chromosomes from human peripheral blood lymphocytes were prepared as described elsewhere (Moorhead et al. 1960). Slides were treated with 2 × SSC at 37°C for 30 min, were dehydrated, were denatured for 2 min at 70°C in 70% formamide, 2 × SSC pH 7, and then were dehydrated again. One microgram of the human MCCA plasmid artificial chromosome (PAC) RPCIP-4 615H13 (Resource Center/Primary Database RZPD) was labeled, by nick-translation, with SpectrumGreen-dUTP (Vysis) and was coprecipitated with 5 mg of human Cot-1 DNA (Vysis). DNA was resuspended in 50% formamide, 2 × SSC, 15% dextran sulfate, were heat-denatured for 10 min at 70°C, and were hybridized to slides for 24 h at 37°C. Slides were washed for 5 min in 1× SSC at 60°C. Chromosomes were counterstained with 125 ng of DAPI/ml. Images were visualized by use of a Nikon Eclipse E400 fluorescence microscope and were analyzed by SmartCapture software (Vysis).
Cell Culture, Complementation, and Biotin-Labeling Assays
Fibroblast and lymphoblast cell lines obtained from patients were confirmed to be deficient in MCC, which was assayed in extracts by measurement of the incorporation of [14C]-HCO3− into acid-precipitable material, as described elsewhere (Gibson et al. 2000). Cells were cultured, according to standard procedures, in minimal essential medium (MEM) (Life Technologies) supplemented with 1% glutamine, 10% fetal calf serum, and antibiotics. For complementation experiments, cells from two different lines were plated together at an equal density, and cell fusion was induced in MEM (without serum) in the presence of 50% PEG 4000 for 1 min. Cells were washed to remove residual polyethylene glycol 4000 and were cultured for a further 24 h under standard conditions. Then the cells were incubated in MEM with 15% fetal calf serum and 0.1 mM [1-14C]-isovalerate (10 μCi/μmol; American Radiolabeled Chemicals). Complementation was monitored by measurement of the incorporation of labeled isovalerate into macromolecules. Fibroblast cell lines deficient in the leucine degradation–pathway enzyme isovaleryl-CoA dehydrogenase were used as positive controls complementing MCC-deficient cell lines. All assays were done in triplicate, with standard deviations, which, in all cases, were within 10% of the mean value. SDS-PAGE analysis of [3H]-biotin labeled mitochondrial carboxylases was as described elsewhere (Lamhonwah et al. 1983).
Immunohistochemical Localization of hMCCβ in Mitochondria
A His-tagged (at its N-terminus) hMCCβ protein containing residues 33–563 was expressed in BL21 Escherichia coli cells and was purified by Ni2+ affinity chromatography. The purified protein was used to raise a mouse polyclonal antiserum. This antiserum detects, in western blot analysis, a liver protein with the expected electrophoretic mobility for human MCCβ. Cultured human fibroblasts were fixed with 4% paraformaldehyde, were treated with 0.5% Triton X-100 (in PBS), and were incubated, for 1 h at 37°C, with mouse anti-MCCβ antiserum (diluted 1:100 in 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.2% Tween 20, and 3% bovine serum albumin). Anti-mouse biotinylated secondary antibody and Alexa 488–conjugated streptavidin were from DAKO and Molecular Probes, respectively. Immunostaining was analyzed by means of a confocal imaging system.
Identification of Mutations in Patients with MCG
All mutations reported here were identified at both the genomic level and the cDNA level. To identify mutations at the mRNA level, reverse transcriptase–PCR (RT-PCR) was performed with the Thermoscript RT-PCR System (Life Technologies), with mRNA (1 μg) isolated from fibroblasts of patients and controls. The entire coding sequence of the MCCA gene and the MCCB gene was amplified in seven or six overlapping fragments, respectively, and subsequently was sequenced. For genomic characterization, exons and flanking intronic sequences were PCR-amplified by means of primers designed to be specific to the intronic genomic sequences (table 1). Both sequencing of PCR-amplified fragments and SSCP analysis were essentially as described elsewhere (Beltrán Valero de Bernabé et al. 1998). To rule out the possibility that missense mutations are frequent polymorphisms in North American or Spanish populations, we built a control sample including >100 chromosomes, using anonymous donors from Madrid and New York.
Aspergillus Techniques
A λEMBL4 library was screened with fungal EST candidates (Fungal Genetics Stock Center) for genes encoding MCCα or MCCβ. The cloned inserts in two partially overlapping positive clones were used to reconstruct a 14-kb region containing mccA, mccB, and a fungal isovaleryl-CoA dehydrogenase gene. Intron positions were determined by comparison with the cDNA sequence. A linear fragment containing genomic flanking sequences upstream (1.04 kb) and downstream (1.35 kb) of the mccA open reading frame in which the complete mccA coding region between the initiation and stop codons had been replaced by an argB+ allele was used to transform an arginine-requiring methG1 argB2 biA1 strain to arginine prototrophy. Transformed colonies were purified in minimal medium lacking arginine and were analyzed by Southern blot analysis using mccA- and argB-specific probes to select a number of independent transformants carrying the expected replacement of the resident mccA gene by the argB+ allele. All these ΔmccA transformants were phenotypically indistinguishable and were unable to grow on leucine. An argB+ transformant in which the argB+ allele had replaced the resident mutant argB2 allele was used as an isogenic mccA+ control. Growth tests were performed as described elsewhere (Fernández-Cañón and Peñalva 1995a), with each amino acid used, at 30 mM, as sole carbon source. Plates were incubated for 5 d at 37°C.
Results
Identification and Characterization of Human MCCA and MCCB cDNAs
The cDNA sequence of MCCA (GenBank accession number AF310972 [GenBank Overview]) (fig. 1a), encoding human MCCα (hMCCα), was assembled by a combination of dbEST (Expressed Sequence Tags database) searching and conventional cDNA screening of cDNA libraries. TBLASTN (BLAST) searches of the human dbEST database, with the Arabidopsis thaliana MCCα amino acid sequence (Song et al. 1994) used as a probe, identified incomplete (at the N-terminal coding region) uterus (GenBank accession number AA134548 [GenBank Overview]; 1.24 kb, excluding polyA) and adrenal carcinoma (AA605162 [GenBank Overview]; 1.74 kb) EST clones. Sequencing of these clones revealed a predicted protein with a biotin-binding site (León del Río and Gravel 1994) whose amino acid sequence differed from those of PCCα and other biotin-dependent carboxylases. It therefore fulfilled the criteria expected of MCCα. The largest clone was used as a [32P]-labeled probe in a hybridization screening of a human liver cDNA library, which failed to identify clones carrying the translation start codon but which yielded sequences extending farther upstream of codon 218 (the most N-terminal codon in the probe). A further dbEST search with these sequences identified a muscle EST (AW410916 [GenBank Overview]) that provided the missing 5′ region of the transcript containing the initiator methionine codon, as predicted on the basis of multiple-sequence alignment (data not shown) with Arabidopsis thaliana and Aspergillus nidulans (see below) MCCα polypeptides.
Figure 1.

cDNA nucleotide sequence and intron-exon organization of human MCCA and MCCB. A and B, Nucleotide sequence of MCCA (A) and MCCB (B) cDNAs, with deduced translation sequences. Red arrowheads indicate the position of the introns. C, Intron-exon organization of the MCCA gene (top) and the MCCB (bottom) gene. D, MCCβ protein localizing in mitochondria. A mouse polyclonal antiserum raised against histidine-tagged MCCβ purified from bacteria was used to reveal the mitochondrial localization of MCCβ, in immunohistochemical localization experiments with cultured fibroblasts.
hMCCα contains 725 residues and includes the tetrapeptide 679-AMKM-682 matching the consensus binding sequence for the essential biotin cofactor (León del Río and Gravel 1994), predicted to be attached, via an amide bond, to the ε-amino group of Lys681 (fig. 1a). Residues 50–502 of human MCCα show 48% identity to the biotin-carboxylating polypeptide of Escherichia coli ACC (Waldrop et al. 1994) and include the sequence 209-GGGGKGMR-216, which is strongly conserved among biotin-carboxylating enzymes. The crystal structure of bacterial ACC (Thoden et al. 2000) revealed that this peptide makes a major contribution to the ATP-binding pocket. hMCCα shows ∼42% identity to its fungal homologue (see below) and ∼39% identity to hPCCα.
A human MCCB cDNA, encoding MCCβ, was initially identified by a search of the dbEST, with the human PCCβ amino acid sequence used as a probe. TBLASTN searches of human dbEST identified a candidate clone (GenBank accession number AA465612 [GenBank Overview]) for MCCB. Although this clone is not currently available, the translation product of the deposited sequence identified an Aspergillus nidulans EST (for MCCB) (accession number n1c10 [Fungal (Aspergillus nidulans, Aspergillus parasiticus, and Neurospora crassa) Cosmid and cDNA Sequencing]). In a third TBLASTN search, this fungal amino acid sequence identified kidney (GenBank accession number AI949422 [GenBank Overview]; 0.7 kb) and uterus (GenBank accession number AI367183 [GenBank Overview]; 1.25 kb) ESTs, the sequencing of which, combined with RT-PCR using human liver mRNA, served to assemble a full-length MCCB cDNA (GenBank accession number AF310971 [GenBank Overview]).
Human MCCB encodes a 563-residue MCCβ polypeptide (fig. 1b) showing 59% sequence identity to its Aspergillus nidulans orthologue (the mccB product; see below) and 29% identity to human PCCβ. During the course of this work, the Arabidopsis thaliana MCC-B gene was reported (McKean et al. 2000). Arabidopsis MCC-B is also 59% identical to hMCCβ, which confirms the correct identification of the human sequences. Both MCCα and MCCβ contain putative signal peptides, in agreement with their predicted mitochondrial localization. A polyclonal antiserum raised against His-tagged MCCβ purified from recombinant bacteria was used to confirm the mitochondrial localization of the protein in normal human fibroblasts (fig. 1d).
Structure and Chromosomal Localization of the MCCA and MCCB Genes
The intron-exon organization of the human MCCA gene was obtained from sequence data, available from the Human Genome Project, of a bacterial artificial chromosome (BAC) (GenBank accession number AC026920 [GenBank Overview]), and from our sequencing data for a human PAC clone (RZPD RPCI-4 615H13). The human MCCA gene is split into 19 exons (fig. 1c and table 2). RH mapping with the Stanford G3 and Genebridge 4 (GB4) panels was initially used to determine the gene's chromosomal location. MCCA was linked to RH markers mapping to different chromosome 3 locations in either the G3 (3p11.2-p13) or the GB4 (3q25-27) RH panel. To solve this contradiction, we used FISH. The analysis of >25 individual metaphases from two independent experiments unambiguously mapped MCCA to the telomeric region in the long arm of chromosome 3 (fig. 2). We conclude that MCCA maps to 3q25-27 (fig. 2A).
Table 2.
Structure of Human MCCA and Human MCCB
| Exon | Size(bp) | 3′ Splice | Exon | 5′ Splice | Intron | Size(bp) |
| MCCA: | ||||||
| 1 | 222 | GCCCAAAGGTAG.......CTG CCG CCG AG | gtgggtggaggg | 1 | 4,746 | |
| 2 | 47 | Tttatgttatag | G ACA TGG GTG.........ACA GCC ACA G | gtactgacagac | 2 | 2,014 |
| 3 | 137 | Tgttttaaacag | GA AGA AAC ATT......CAT GTA GAT ATG | gtatgtgttaaa | 3 | >1,867 |
| 4 | 96 | Tttcttaaacag | GCA GAT GAA GCA.....TCT GCT GCA CAG | gtgagggttgtg | 4 | >14,033 |
| 5 | 122 | Tctcatttctag | GCT ATC CAT CCA......GGT ATA AAG AG | gtatgagtttat | 5 | 1,008 |
| 6 | 148 | Cccactctaaag | C ACA TCC AAA.......GGA GGA GGA AAA | gtaagattgtgc | 6 | 89 |
| 7 | 122 | Tttctctcttag | GGA ATG AGG ATT......GAC ACA CCG AG | gtgggttggcct | 7 | >5,793 |
| 8 | 112 | Gcttttatatag | G CAT GTA GAA...GAG GCC CCA GCG | gtaaggaccttg | 8 | 5,000 |
| 9 | 82 | Tttttttcatag | CCT GGT ATT AAA.......GTT GGA GCA G | gtaggttaagat | 9 | >6,509 |
| 10 | 128 | Tttaccttttag | GG ACT GTG GAG......TGG CAG CTT AGA | gtgagaatattt | 10 | 3,662 |
| 11 | 184 | Ttctattgccag | ATT GCA GCA GGA.......GTA CGG CAA G | gtaaagtgaaag | 11 | 2,431 |
| 12 | 110 | Tttcaatttcag | GA GAC GAA GTT......CGT CAG TAC AAT | gtgagtgatctg | 12 | 1,591 |
| 13 | 21 | Ttctcttcctag | ATT GTT GGA CTG.......CAG GCA CAT G | gtatggaaggct | 13 | >3,054 |
| 14 | 87 | Aattttttctag | AT CAA TTC TCT........GGT AAA AAC A | gtaagtaatgtt | 14 | >11,299 |
| 15 | 50 | Ttttccataaag | AT GTA GCC ATA......TAT AGC ATG CAG | gtaagccttact | 15 | 3,200 |
| 16 | 138 | Catttttttcag | ATT GAA GAT AAA.....CTA TTT TCC AAG | gtaatgtctctt | 16 | 2,179 |
| 17 | 108 | Ttctgtttctag | GAA GGA AGT ATT.....ACC ATT GAA AAG | gtaatcacatga | 17 | 2,792 |
| 18 | 72 | Ctgaatctcaag | GTG TTT GTC AAA.....ATG AAG ATG GAG | gtaaggaaatca | 18 | 1,699 |
| 19 | 347 | Gtgtctctgcag | CAT ACC ATA AAG....TTTTTATTGGAAGCC | |||
| MCCB: | ||||||
| 1 | 237 | GGGGCTGCAAGG......GCC CTC TAC CAG | gtaggctgagcg | 1 | 5,368 | |
| 2 | 67 | Tttctgttttag | GAG AAC TAC.........ATA AAA CTA G | gtaaacacagca | 2 | 3,285 |
| 3 | 85 | Tttctatcatag | GA GGT GGT.........ATA GAC CCA GG | gtgcgtacatag | 3 | 3,290 |
| 4 | 102 | Tcttcgctttag | G TCT CCA TTT......AGA GTA TCA GG | gtgagtatttac | 4 | 2,746 |
| 5 | 128 | Tctgcctttcag | A GTA GAA TGC.......ATC TAC TTA G | gcaagtcaccag | 5 | 1,720 |
| 6 | 113 | Tcttgtcttcag | TT GAT TCG GGA....AAT ATT GCA CAG | gtaatttttcat | 6 | 22,183 |
| 7 | 114 | Cttggattccag | ATC GCA GTG.......GGA CCC CCC TTG | gtaagaacataa | 7 | 5,367 |
| 8 | 65 | Tccttgttgtag | GTT AAA GCG GCA....CTT CAT TGC AG | gtgaaacagaaa | 8 | 2,767 |
| 9 | 100 | Cttattctgtag | A AAG TCT GGA.....AAG AAA TTG GAT | gtgagtacgata | 9 | 108 |
| 10 | 96 | Gtatttctgcag | GTC ACC ATT GAA...GAT GTC CGA GAG | gtatgtgaaagt | 10 | 5,753 |
| 11 | 73 | Ttttcctcttag | GTC ATT GCT AGA.....TTA GTT ACA G | gtataaaggtga | 11 | 2,743 |
| 12 | 77 | Ctttcttttgag | GA TTT GCT CGA....TCT GCA AAA AAG | gcaagtactgtt | 12 | 2,320 |
| 13 | 67 | Atttgcttatag | GGT ACT CAC TTT.....AAC ATT ACT G | gtaagaaaatag | 13 | 2,818 |
| 14 | 157 | Tctttctctcag | GA TTT ATG GTT.....AGA GCG TAT AG | gtaggtgtcatg | 14 | 815 |
| 15 | 115 | Ttcttcccccag | C CCA AGA TTT.....GAA GGA AAG CAG | gtcggtgtcgtt | 15 | 2,485 |
| 16 | 86 | Cttttcctttag | TTC TCC AGT GCT....TCC AGC GCA AG | gtgggggccaga | 16 | 3,987 |
| 17 | 475 | Ttcttttgtcag | G GTA TGG GAT.....ATTTTCACTGAAATG |
Figure 2.

Chromosomal localization of MCCA (A) and MCCB (B). RH-mapping results with the GB4 panel (for MCCA) and with the G3 and GB4 panels (for MCCB) are shown. The nearly telomeric chromosomal localization of MCCA was confirmed by FISH (see insets of individual chromosomes, in panel A).
We used three Human Genome Sequence clones (GenBank accession numbers AC010279, AC026775, and AC025958 [GenBank Overview]) to determine the structure of the 17-exon human MCCB gene (fig. 1c and table 2). The MCCB “AA465612” EST sequence, included within these genomic clones, belongs to a UniGene cluster (H. 167531; STS accession number SGC34463 or SHGC-34463 [GeneMap'99]) mapping either to 5q12-13.1 (reference interval D5S637–D5S1977) in the GB4 map or to 5q12 (reference interval D5S647–D5S637) in the G3 map. In agreement, our RH experiments using primers that generate an amplicon in the 3′ UTR of exon 17 (table 1) consistently linked the human MCCB gene to markers mapping to chromosome 5q12-q13.2, in both the G3 panel and the GB4 panel.
Transcription Pattern of the MCCA and MCCB Genes: Agreement with the Predicted Physiological Role
Unlike most amino acids absorbed across the intestine, the branched-chain amino acids leucine, valine, and isoleucine are not taken up to any great extent by the liver, which leads to a large increment in their arterial concentration after a protein-rich meal (Wahren et al. 1976). Muscle is the primary site for branched-chain–amino acid transamination (Goldberg and Chang 1978; Chuang and Shih 1995). The resulting branched-chain ketoacids can be either further metabolized there (Goldberg and Chang 1978) or transported to liver, kidney, and heart, where they are oxidized (Chuang and Shih 1995). In the liver, α-isocaproic acid, the product of leucine transamination, is further metabolized, via 3-methycrotonyl-CoA and MCC, to acetyl-CoA (fig. 3a), which can serve as a precursor for ketone-body and isoprenoid biosynthesis. In the muscle, leucine can be a source of energy, via full oxidation to CO2 (Goldberg and Chang 1978). In agreement with both the predicted involvement of MCC in the oxidation of leucine in these tissues and the heteromeric nature of MCC, northern blot hybridization showed that the MCCA and MCCB transcripts are coordinately expressed at high levels in liver, kidney, heart, and skeletal muscle (fig. 3b). MCCA is transcribed as a single 2.6-kb message; MCCB gives rise to a minor 2.4-kb transcript and a major 4-kb transcript. This long transcript almost certainly results from further downstream polyadenylation of exon 17, which would lead to a long (∼1.5 kb) 3′ UTR. The presence of this long 3′ UTR within the human transcriptome was confirmed by both RT-PCR (table 1) and the identification of EST clones (GenBank accession numbers AW439494 and AI869038 [GenBank Overview]) containing this region.
Figure 3.

A, Leucine-degradation pathway. The absence of MCC leads to the accumulation of the upstream compounds isovaleryl-CoA and methylcrotonyl-CoA, which lead to 3-hydroxyisovaleric (3-HIVA) and 3-methylcrotonylglycine (3-MC-Gly), which are biochemical landmarks of the isolated, biotin-insensitive MCC deficiency. B, Multitissue northern blot hybridization, showing coordinated, high-level expression of MCCA and MCCB in liver, kidney, heart, and skeletal muscle. β-Actin was used as a loading control. MCCA and MCCB probes were ESTs “AA605162” and “AI949422,” respectively. B = brain; H = heart; SM = skeletal muscle; C = colon; T = thymus; S = spleen; K = kidney; Li = liver; Si = small intestine; P = placenta; Lu = lung; PBL = peripheral blood lymphocytes. The human multiple-tissue northern filter was purchased from Clontech.
Mutations in MCCA or MCCB, in Patients with MCG
To confirm the involvement of MCCA and MCCB in MCG, we screened for mutations in the cloned genes in seven probands. These patients (one from Argentina, one from Spain, and five from the United States) were unrelated, although two of them (Gibson et al. 1998) belong to the Amish/Mennonite population in Lancaster County, Pennsylvania. These patients either showed elevated excretion of the diagnostic compounds 3-methylcrotonylglycine or 3-hydroxyisovaleric acid or came to clinical attention because of their abnormally increased blood levels of C5-OH. All of them had been shown to be deficient for MCC but were normal for at least one of the other biotin-dependent carboxylases (Gibson et al. 2000) and showed normal [3H]-biotin labeling of PC and PCCα (see below). These data rule out a multiple-carboxylase deficiency (i.e., a deficiency of all four biotin-dependent carboxylases) resulting from defects either in the insertion of the biotin cofactor into the apoenzymes or in the recycling of the cofactor (Wolf 1995).
Polyethylene glycol–mediated heterokaryosis between fibroblast cell lines from five of these patients (fibroblasts were not available for the remaining two patients) was used to classify them into two complementation groups (table 3). By analogy to the deficiency of the dicistronic enzyme PCC (Gravel et al. 1977; Ugarte et al. 1999), each of these two complementation groups would be defined by recessive loss-of-function mutations in one of the two genes encoding the MCC subunits. Fibroblasts from the Argentine patient were arbitrarily used as the reference. Two complementing patients were included within complementation group A (CGA), and two noncomplementing patients were included, along with the reference patient, within complementation group B (CGB) (table 3). In addition, we used [3H]-biotin labeling of mitochondrial biotin carboxylases, followed by SDS-PAGE and autoradiography, to resolve PC, MCCα, and PCCα (Lamhonvah et al. 1983). Fibroblasts of all of these five patients showed normal PC and PCCα labeling (data not shown). However, whereas the three CGB patients showed normal MCCα labeling, one of the two CGA patients did not (fig. 4), strongly indicating that CGA is defined by mutations in MCCA.
Table 3.
MCCA and MCCB Mutations in Patients with MCG
|
Mutation inb |
||||||
| Patient (Origin) | Groupa | MCCα [3H]-Biotin Labeling? | MCCA | MCCB | Nucleotide Changec | Predicted Consequence(s) |
| 15468 (Spain) | CGB | Yes | C167R | MCCB c. 499T→C | Cys167Arg substitution in MCCβ | |
| 15469 (Argentina) | CGB | Yes | A218T | MCCB c. 652G→A | Ala218Thr substitution in MCCβ | |
| 15626 (United States) | … | Yes | D172fs | MCCB c. 517insT | MCCβ sequence frameshifted after residue 172 | |
| 15628 (United States) | … | Yes | D172fs | MCCB c. 517insT | MCCβ sequence frameshifted after residue 172 | |
| 15767 (United States) | CGB | Yes | IVS3+5→T | MCCB c. 281+5G→T | MCCB exon 3 skipping, frameshift after residue 66 | |
| 15765 (United States) | CGA | No | M325R | MCCA c.974T→G | Met325Arg substitution in MCCα | |
| 15766 (United States) | CGA | Yes | R385S | MCCA c.1155A→C | Arg385Ser substitution in MCCα | |
An ellipsis (…) denotes that the individual was not classified.
Found in apparent homozygosis (or in a hemizygosis-like situation; see the text). Missense mutations, which are underlined, were absent from 100 normal chromosomes.
Nucleotide numbering referred to the A in the initiation ATG codons of the cDNA sequences in figure 1.
Figure 4.
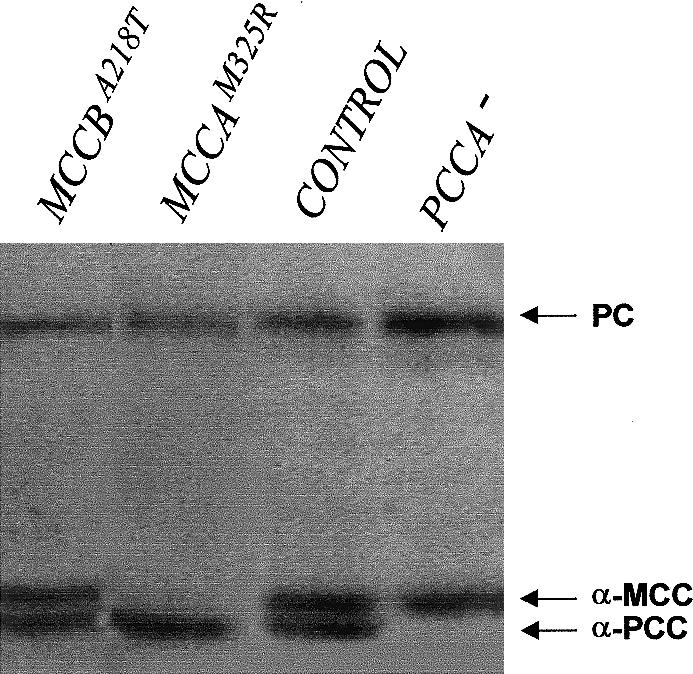
Results of in vivo [3H]-biotin labeling of mitochondrial biotin-containing polypeptides PC, PCCα, and MCCα. Cultured fibroblasts (relevant genotypes are shown at the top of each lane) were incubated in the presence of the labeled vitamin and were lysed, and the protein extracts were loaded onto a 7.5% SDS-polyacrylamide gel. Labeled proteins were revealed by fluorography.
We detected two missense mutations in MCCA in the two CGA patients (fig. 5 and table 3). These mutations were absent in a sample of 100 control chromosomes. M325R involves a nonconservative substitution leading to absence of [3H]-biotin MCCα labeling (fig. 4), which would indicate that it affects either protein stability or biotinylation. R385S involves a substitution of an arginine residue that is strictly conserved among humans, plants, and fungi. Human MCCα Arg385 corresponds to Arg338 in the biotin-carboxylating component of E. coli ACC, which appears to form part of a positively charged pocket for bicarbonate binding (Thoden et al. 2000). This mutation could lead to a kinetic defect without MCCα destabilization, as indicated by the normal [3H]-biotin labeling of mutant MCCα in fibroblasts of our patient (table 3). In addition to these mutations, we detected two frequent polymorphisms in MCCA—the synonymous point mutation c.396T→C (L132L) and the missense mutation c.1391A→C (H464P). Proline rather than histidine is present in the corresponding position in Arabidopsis MCCα (Song et al. 1994).
Figure 5.

Mutations in MCCA and MCCB. A, Mutations in MCCB: results of direct sequencing of PCR-amplified genomic DNA from controls and from CGB patients who have MCG are shown. Arrows indicate the mutated positions. B, Results of direct sequencing of PCR-amplified genomic DNA from controls and from CGA patients who have MCG. Arrows indicate the mutated positions. C, IVS3+5G→T MCCB mutation, which results in skipping of exon 3. The gel shows the results of agarose-gel electrophoresis and direct sequence analysis of MCCB cDNA from the region surrounding the exon2/exon3 junction, amplified by RT-PCR with fibroblast mRNA as template, from a control (lane C) and from the patient (lane P) carrying this mutation. Lane M, molecular-wieght marker.
We detected three mutations in the MCCB gene in the three CGB patients (fig. 4 and table 3). An additional MCCB mutation was found in the two patients who were not included in complementation studies (table 3 and fig. 5). No mutation was detected in the MCCA gene of the latter two patients, who, like the three CGB patients, showed normal MCCα labeling (table 3). Two of these four MCCB mutations almost certainly represent null alleles: D172fs (c.516insT) truncates MCCβ after residue 172, and IVS3+5G→T is a splicing mutation leading to skipping of exon 3, frameshift, and early truncation (fig. 5). The remaining two mutations (C167R and A218T) are missense mutations involving nonconservative substitutions of MCCβ residues that are strictly conserved among humans, fungi, and plants. Both mutations are absent from a sample of 100 normal chromosomes.
In Vivo Involvement of MCC in Leucine Catabolism
The haploid fungus Aspergillus nidulans is able to grow on a number of amino acids, as the sole carbon source, using catabolic pathways that are very similar to those found in humans (Fernández-Cañón and Peñalva 1995a). In previous studies, we have exploited this metabolic versatility and the relatively facile genetic manipulation of this lower eukaryote in order to characterize the human genes of phenylalanine metabolism (Fernández-Cañón and Peñalva 1995a, 1995b, 1996, 1998). Aspergillus nidulans grows on leucine and isoleucine—and, to a lesser extent, on valine—as the sole carbon source. dbEST searches allowed us to identify Aspergillus nidulans mccA and mccB cDNAs, which encode fungal MCCα and MCCβ, respectively. Their genomic characterization showed that, together with the isovaleryl-CoA dehydrogenase gene, these genes are closely linked within a 14-kb region, in a likely leucine-degradation cluster (J.M.R. and M.A.P, unpublished data). We constructed, by homologous recombination, a null ΔmccA strain. In contrast to the parental strain, this ΔmccA strain is unable to utilize leucine but grows normally on threonine, isoleucine, or valine (which are catabolized via propionyl-CoA and PCC) (fig. 6). These data establish in vivo the specific involvement of MCCα (and, by extension, MCC) in leucine catabolism.
Figure 6.

Knockout of the gene for MCCα in Aspergillus nidulans. An Aspergillus nidulans ΔmccA strain carrying a complete deletion of the gene encoding fungal MCCα is unable to utilize leucine as the sole carbon source but grows on other branched-chain amino acids. The parental strain and three independent transformants (denoted “#1”–“#3”) deleted for the mccA gene were grown on synthetic minimal medium with ammonium chloride as the nitrogen source and with 30 mM of the indicated amino acids as the sole carbon source. Plates were incubated for 5 d at 37°C. Fungi can synthesize carbohydrates from acetyl-CoA, via the glyoxylate cycle—thereby bypassing the Krebs cycle, which leads to complete oxidation of acetyl-CoA to CO2 and water.
Discussion
We have characterized, at both the genomic level and the cDNA level, the human MCCA and MCCB genes encoding the subunits of MCC, which is an enzyme of leucine catabolism and which, when deficient, leads to MCG; and we have detected six MCCA or MCCB mutations in patients diagnosed with the disease. All these mutations are most likely in homozygosis. However, since no relatives of the patients were available for segregation analysis, we cannot formally rule out the possibility that, among the probands, there are compound heterozygotes carrying a partial or complete deletion of either the MCCA gene or the MCCB gene.
The six mutations reported here fulfill the criteria for pathogenic mutations (Cotton and Horaitis 2000): (i) each of these mutations was detected in a particular proband after direct sequencing of the complete coding region of either MCCA or MCCB; (ii) they were confirmed in two independent RT-PCR experiments; (iii) they were confirmed by direct sequencing of genomic DNA; (iv) they were confirmed by RFLP or SSCP analysis of the corresponding amplified exons; (v) all six were absent from a sample of 100 normal chromosomes; and (vi) the M325R MCCA missense mutation leads to absence of biotinylated MCCα, which is a formal proof that this mutation is pathogenic, since the biotin cofactor is essential for enzyme function (see Wolf 1995); (vii) Arg385, the MCCα residue affected by the second MCCA missense mutation (R385S), is predicted to be involved in the binding of one of the enzyme substrates (see the Results section); (viii) D172fs and IVS3+5G→T, two of the four mutations found in MCCB in homozygosis (or in a hemizygosis-like situation), result in early protein truncation (table 3) and almost certainly lead to null alleles; and (ix) the other two mutations found in MCCB involve nonconservative substitutions of residues that are strictly conserved among the three MCCβ sequences available (i.e., human, Arabidopsis, and Aspergillus). We therefore conclude that MCG results from loss-of-function mutations in either MCCA or MCCB, the genes encoding the α and β subunits, respectively, of MCC.
The MCCA M325R mutation leading to absence of biotinylated MCCα deserves particular attention. In human PCCα, a polypeptide comprising the carboxyl-terminal 67 amino acids that include the biotin-attachment sequence Ala-Met-Lys-Met functions as an independent domain that can be biotinylated in E. coli (León del Río and Gravel 1994). Met325, affected by this MCCA mutation, lies considerably upstream of the corresponding region in hMCCα (see fig. 1), and therefore the absence of biotinylated mutant MCCα almost certainly reflects the absence of the protein, rather than an impairment of biotinylation. With regard to the efficiency of RT-PCR amplification, we noticed no differences between the MCCA transcript from M325R fibroblasts and that from normal fibroblasts, which would suggest both that this mutation does not result in mRNA destabilization and that the absence of MCCα labeling possibly results from decreased protein stability.
Only two of the probands in the present study carry the same mutation (D172fs). These individuals are female adults shown to be deficient for MCC but clinically asymptomatic at the moment of diagnosis (Gibson et al. 1998). They belong to the Amish/Mennonite population and came to clinical attention because their children, later shown to be normal obligate carriers (Gibson et al. 1998; present study), showed increased quantities of 5C-OH in blood, possibly as a result of the accumulation of maternal 5C-OH in the red cells of these children during pregnancy (Ranieri et al. 2000b). The absence of symptoms in these Mennonite female adults is an extreme example of the variable clinical phenotype of MCG (Sweetman and Williams 1995), in which the symptoms range from mild muscular weakness to severe episodes of metabolic decompensation. It is noteworthy that these two clinically asymptomatic female adults carry an MCCβ null mutation, apparently in homozygosis, and show highly elevated levels of 5C-OH and 3-methylcrotonylglycine, in blood and urine, respectively (Gibson et al. 1998), which is consistent with their genotype. This shows the significant influence that environmental factors (such as minor intercurrent infections or adoption of a protein-rich diet, which frequently trigger metabolic crises) and/or genetic makeup may have in the clinical outcome of this potentially severe disease. Therefore, in MCG, as in other metabolic monogenic traits, the enzyme-structural gene(s) is the major determinant of the biochemical phenotype, but the associated clinical features are complex, somehow resembling the so-called complex traits (Scriver and Waters 1999).
Although MCG previously had been thought to be a rare inborn error of metabolism, the introduction of massive neonatal screening by MS/MS of urine or blood spots in neonates has revealed that MCG is unexpectedly frequent, with an incidence, for example, of ∼1/27,000 in South Australia (Ranieri et al. 2000a), ∼1/52,000 in North Carolina (Smith et al. 2000), ∼1/110,000 in New South Wales (Wilcken et al. 2000), and ∼1/30,000 in Bavaria, where MCG appears to be the most frequent of the organic acidurias (Roscher et al. 2000). Our work provides the molecular tools to analyze the molecular basis of this emergent disease, paves the way for future studies on phenotype-genotype correlations, and should facilitate the detection of heterozygote carriers and false positives of neonatal screening. Finally, this work completes the characterization of all four biotin-dependent carboxylases of human intermediary metabolism.
Acknowledgments
We thank Herbert N. Arst, for critical reading of the manuscript; R. Navarrete, E. Reoyo, and staff in the sequencing facility at the Centro de Investigaciones Biologícas, for technical assistance; the Fundación Ramón Areces, for an institutional grant to the Centro de Biología Molecular CSIC-UAM; and the Dirección General de Investigacion Cientifica y Técnica, for support through grant 2FD97-1292.
Note added in proof.
—We have recently learned that the molecular basis of MCG has also been identified by Baumgartner et al. (in press).
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- BLAST, http://www.ncbi.nlm.nih.gov/BLAST/
- Expressed Sequence Tags database, http://www.ncbi.nlm.nih.gov/dbEST/index.html
- Fungal (Aspergillus nidulans, Aspergillus parasiticus, and Neurospora crassa) Cosmid and cDNA Sequencing, http://www.genome.ou.edu/fungal.html (for Aspergillus nidulans [accession number n1c10])
- Fungal Genetics Stock Center, http://www.fgsc.net/ (for fungal EST clones)
- GenBank Overview, http://www.ncbi.nlm.nih.gov/Genbank/GenbankOverview.html (for human MCCA ESTs, [accession numbers AA134548, AA605162, and AW410916], human MCCB ESTs [accession numbers AA465612, AI949422, AI367183, AW439494, and AI869038], full-length human MCCA cDNA [accession number AF310972], full-length MCCB cDNA [accession number AF310971], human MCCA BAC [accession number AC026920], and human MCCB BACs [accession numbers AC010279, AC026775, and AC025958])
- GeneMap'99, http://www.ncbi.nlm.nih.gov/genemap99/ (for overview of human chromosome maps, for Unigene MCCB cluster H. 167531 [STS accession number SGC34463 or SHGC-34463])
- Incyte Genomics http://www.incyte.com/ (for human EST clones)
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for MCG [MIM 210200])
- Radiation Hybrid Mapping, http://carbon.wi.mit.edu:8000/cgi-bin/contig/rhmapper.pl (for Genebridge 4 panel)
- Resource Center/Primary Database RZPD, http://www.rzpd.de/ (for human PAC clones) (for MCCA, RPCI-4 615H13)
- Stanford Human Genome Center, http://shgc-www.stanford.edu/RH/index.html (for G3 panel)
References
- Baumgartner MR, Almashanu S, Suormala T, Obie C, Cole RN, Morton DH, Packman S, Baumgartner ER, Valle D. The molecular basis of human 3-methylcrotonyl CoA carboxylase deficiency. J Clin Invest (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrán-Valero de Bernabé D, Granadino B, Chiarelli I, Porfirio B, Mayatepek E, Aquaron R, Moore MM, Festen JJ, Sanmartí R, Peñalva MA, Rodríguez de Córdoba S (1998) Mutation and polymorphism analysis of the human homogentisate 1, 2- dioxygenase gene in alkaptonuria patients. Am J Hum Genet 62:776–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang DT, Shih VE (1995) Disorders of branched chain amino acid and ketoacid metabolism. In: Scriver C, Beaudet A, Sly W, Valle D (eds) The metabolic basis of inherited disease, 7th ed. McGraw-Hill, New York, pp 1239–1276 [Google Scholar]
- Cotton RGH, Horaitis O (2000) Quality control in the discovery, reporting and recording of genomic variation. Hum Mutat 15:16–21 [DOI] [PubMed] [Google Scholar]
- Fernández-Cañón JM, Granadino B, Beltrán-Valero de Bernabé D, Renedo M, Fernández-Ruiz E, Peñalva MA, Rodríguez de Córdoba S (1996) The molecular basis of alkaptonuria. Nat Genet 14:19–24 [DOI] [PubMed] [Google Scholar]
- Fernández-Cañón JM, Peñalva MA (1995a) Fungal metabolic model for human type I hereditary tyrosinemia. Proc Natl Acad Sci USA 92:9132–9136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— (1995b) Molecular characterization of a gene encoding a homogentisate dioxygenase from Aspergillus nidulans and identification of its human and plant homologues. J Biol Chem 270:21199–21205 [DOI] [PubMed] [Google Scholar]
- ——— (1998) Characterization of a fungal maleylacetoacetate isomerase gene and identification of its human homologue. J Biol Chem 273:329–337 [DOI] [PubMed] [Google Scholar]
- Gibson KM, Bennet MJ, Naylor EW, Morton DH (1998) 3-Methylcrotonyl-coenzyme A carboxylase deficiency in Amish/Mennonite adults identified by detection of increased acylcarnitines in blood spots of their children. J Pediatr 132:519–523 [DOI] [PubMed] [Google Scholar]
- Gibson KM, Ugarte M, Fukao T, Mitchell GA (2000) Molecular and enzymatic methods for detection of genetic defects in distal pathways of branched-chain amino acid metabolism. Methods Enzymol 324:432–453 [DOI] [PubMed] [Google Scholar]
- Goldberg AL, Chang TW (1978) Regulation and significance of amino acid metabolism in skeletal muscle. Fed Proc 37:2301–2307 [PubMed] [Google Scholar]
- Gravel RA, Lam KF, Scully KJ, Hia Y (1977) Genetic complementation of propionyl-CoA carboxylase deficiency in cultured human fibroblasts. Am J Hum Genet 29:378–388 [PMC free article] [PubMed] [Google Scholar]
- Gyapay G, Schmitt K, Fizames C, Jones H, Vega-Czarny N, Spillett D, Muselet D, Prud'Homme JF, Dib C, Auffray C, Morissette J, Weissenbach J, Goodfellow PN (1996) A radiation hybrid map of the human genome. Hum Mol Genet 5:339–346 [DOI] [PubMed] [Google Scholar]
- Lamhonwah AM, Barankiewicz TJ, Willard HF, Mahuran DJ, Quan F, Gravel RA (1986) Isolation of cDNA clones coding for the alpha and beta chains of human propionyl-CoA carboxylase: chromosomal assignments and DNA polymorphisms associated with PCCA and PCCB genes. Proc Natl Acad Sci USA 83:4864–4868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamhonwah AM, Lam KF, Tsui F, Robinson B, Saunders ME, Gravel RA (1983) Assignment of the a and b chains of human propionyl-CoA carboxylase to genetic complementation groups. Am J Hum Genet 35:889–899 [PMC free article] [PubMed] [Google Scholar]
- Lau EP, Cochran BC, Fall RR (1980) Isolation of 3-methylcrotonyl-coenzyme A carboxylase from bovine kidney. Arch Biochem Biophys 205:352–359 [DOI] [PubMed] [Google Scholar]
- León del Río A, Gravel RA (1994) Sequence requirements for the biotinylation of carboxyl-terminal fragments of human propionyl-CoA carboxylase alpha subunit expressed in Escherichia coli. J Biol Chem 269:22964–22968 [PubMed] [Google Scholar]
- McKean AL, Ke J, Song J, Che P, Achenbach S, Nikolau BJ, Wurtele ES (2000) Molecular characterization of the non-biotin-containing subunit of 3-methylcrotonyl-CoA carboxylase. J Biol Chem 275:5582–5590 [DOI] [PubMed] [Google Scholar]
- Moorhead PS, Nowell PC, Mellman WJ, Battips DM, Hungerford DA (1960) Chromosome preparation of leukocytes cultured from human peripheral blood. Exp Cell Res 20:613–616 [DOI] [PubMed] [Google Scholar]
- Naylor EW, Chace DH (1999) Automated tandem mass spectrometry for mass newborn screening for disorders in fatty acid, organic acid, and amino acid metabolism. J Child Neurol 14 Suppl 1:S4–S8 [DOI] [PubMed] [Google Scholar]
- Ranieri E, Gerace R, Barlett B, Barnard K, Fletcher JM (2000a) The introduction of tandem mass spectrometry into the South Australian neonatal screening program: benefits and costs. J Inherit Metab Dis 23 Suppl 1:006 (poster) [Google Scholar]
- Ranieri E, Gerace R, Metz M, Harrison JR, Nelson PV, Fietz M, Fletcher JM (2000b) Localisation of the 3-hydroxyvalerylcarnitine within the cellular fraction of blood in a case of 3-methylcrotonyl coenzyme A carboxylase deficiency. J Inherit Metab Dis 23 Suppl 1:177 (oral presentation) [Google Scholar]
- Roscher A, Liebl B, Fingerhut R, Olgemöller B (2000) Prospective study of MS-MS newborn screening in Bavaria, Germany: interim results. J Inherit Metab Dis 23 Suppl 1:008 (poster) [Google Scholar]
- Scriver CR, Waters PJ (1999) Monogenic traits are not simple: lessons from phenylketonuria. Trends Genet 15:267–272 [DOI] [PubMed] [Google Scholar]
- Smith WE, Muenzer J, Frazier D, Millington DS Kishnani PS, McDonald M, Koeberl DD (2000) Evaluation of elevated hydroxyisovalerylcarnitine in the newborn screen by tandem mass spectrometry. Am J Hum Genet 67 Suppl 2:292 [Google Scholar]
- Song J, Wurtele ES, Nikolau BJ (1994) Molecular cloning and characterization of the cDNA coding for the biotin-containing subunit of 3-methylcrotonoyl-CoA carboxylase: identification of the biotin carboxylase and biotin-carrier domains. Proc Natl Acad Sci USA 91:5779–5783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart EA, McKusick KB, Aggarwal A, Bajorek E, Brady S, Chu A, Fang N, et al (1997) An STS-based radiation hybrid map of the human genome. Genome Res 7:422–433 [DOI] [PubMed] [Google Scholar]
- Sweetman L, Williams JC (1995) Branched chain organic acidurias. In: Scriver C, Beaudet A, Sly W, Valle D (eds) The metabolic basis of inherited disease, 7th ed. McGraw-Hill, New York, pp 1387–1422 [Google Scholar]
- Thoden JB, Blanchard CZ, Holden HM, Waldrop GL (2000) Movement of the biotin carboxylase B-domain as a result of ATP binding. J Biol Chem 275:16183–16190 [DOI] [PubMed] [Google Scholar]
- Ugarte M, Pérez-Cerdá C, Rodríguez-Pombo P, Desviat LR, Pérez B, Richard E, Muro S, Campeau E, Ohura T, Gravel RA (1999) Overview of mutations in the PCCA and PCCB genes causing propionic acidemia. Hum Mutat 14:275–282 [DOI] [PubMed] [Google Scholar]
- van Hove JL, Rutledge SL, Nada MA, Kahler SG, Millington DS (1995) 3-Hydroxyisovalerylcarnitine in 3-methylcrotonyl-CoA carboxylase deficiency. J Inherit Metab Dis 18:592–601 [DOI] [PubMed] [Google Scholar]
- Wahren J, Felig P, Hagenfeldt L (1976) Effect of protein ingestion on splanchic and leg metabolism in normal man and in patients with diabetes mellitus. J Clin Invest 57:987–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldrop GL, Rayment I, Holden HM (1994) Three-dimensional structure of the biotin carboxylase subunit of acetyl-CoA carboxylase. Biochemistry 33:10249–10256 [DOI] [PubMed] [Google Scholar]
- Wilcken B, Wiley V, Carpenter K (2000) Two years of routine newborn screening by tandem mass spectrometry (MSMS) in New South Wales, Australia. J Inherit Metab Dis 23 Suppl 1:007 (poster) [Google Scholar]
- Wolf B (1995) Disorders of biotin metabolism. In: Scriver C, Beaudet A, Sly W, Valle D (eds) The metabolic basis of inherited disease, 7th ed. McGraw-Hill, New York, pp 3151–3177 [Google Scholar]