

Abstract
Cancer occurrence in 164 families with breast/ovarian cancer and germline BRCA2 mutations was studied to evaluate the evidence for genotype-phenotype correlations. Mutations in a central portion of the gene (the “ovarian cancer cluster region” [OCCR]) were associated with a significantly higher ratio of cases of ovarian:breast cancer in female carriers than were mutations 5′ or 3′ of this region (P<.0001), extending previous observations. The optimal definition of the OCCR, as judged on the basis of deviance statistics, was bounded by nucleotides 3059–4075 and 6503–6629. The relative and absolute risks of breast and ovarian cancer associated with OCCR and non-OCCR mutations were estimated by a conditional likelihood approach, conditioning on the set of mutations observed in the families. OCCR mutations were associated both with a highly significantly lower risk of breast cancer (relative risk [RR] 0.63; 95% confidence interval (95% CI) 0.46–0.84; P=.0012) and with a significantly higher risk of ovarian cancer (RR = 1.88; 95% CI = 1.08–3.33; P=.026). No other differences in breast or ovarian cancer risk, by mutation position, were apparent. There was some evidence for a lower risk of prostate cancer in carriers of an OCCR mutation (RR = 0.52; 95% CI = 0.24–1.00; P=.05), but there was no evidence of a difference in breast cancer risk in males. By age 80 years, the cumulative risk of breast cancer in male carriers of a BRCA2 mutation was estimated as 6.92% (95% CI = 1.20%–38.57%). Possible mechanisms for the variation in cancer risk are suggested by the coincidence of the OCCR with the RAD51-binding domain.
Introduction
Germline mutations in BRCA2 [MIM 600185] strongly predispose to breast and ovarian cancer (Wooster et al. 1994, 1995; Tavtigian et al. 1996). BRCA2 mutations also predispose to breast cancer in men (Wooster et al. 1994). There is evidence for an increased risk of several other cancers—including prostate cancer, pancreatic cancer, gall-bladder and bile-duct cancer, stomach cancer, and malignant melanoma—in carriers of BRCA2 (Breast Cancer Linkage Consortium 1999). Estimates of the cumulative risk of breast cancer by age 70 years in BRCA2 carriers vary from 37%, in a study based on carriers of the 999del5 mutation in Iceland (Thorlacius et al. 1998), to 84%, based on multiple-case families collected through the Breast Cancer Linkage Consortium (BCLC) (Ford et al. 1998). The ovarian cancer risk by age 70 year has been estimated to be 20%–27% (Ford et al. 1998; Breast Cancer Linkage Consortium 1999).
From the viewpoint of genetic counseling, it is important to determine whether these variations in cancer risk are associated with different mutations. In a previous study, based on 25 families with germline BRCA2 mutations, Gayther et al. (1997) reported that families with a high proportion of ovarian cancers, relative to the frequency of breast cancer, tended to have mutations located within a 3.3-kb region in exon 11. They called this region of BRCA2, bounded by nucleotides 3035 and 6629, the “ovarian cancer cluster region” (OCCR). In an attempt to confirm and extend this observation, we have analyzed a much larger data set, of 164 BRCA2 families. We have also attempted to estimate absolute breast and ovarian cancer risks associated with mutations in different regions of the gene, and we have additionally been able to evaluate variations in the risks of prostate cancer and of breast cancer in males.
Families and Methods
Families
The data set used in this study was based on the same set of families previously used to evaluate risks, in BRCA2 carriers, of cancers other than breast and ovarian cancer (Breast Cancer Linkage Consortium 1999). These families were collected by members of the BCLC from 20 centers in western Europe, the United States, and Canada (68 families from North America and 96 from Europe). Twenty-three of the families were ascertained through systematic, population-based studies of patients with either breast cancer or ovarian cancer. The remainder of the families were ascertained on the basis of the presence of at least two relatives with either breast cancer diagnosed at age <60 years or ovarian cancer at any age, with more restrictive criteria being used by some centers. Families were eligible for inclusion in this study if at least one family member tested positive for a pathological BRCA2 mutation. Four families that, on the basis of linkage evidence alone, had been included in the previous analyses of this data set were excluded from these analyses. We also excluded five families with putative missense mutations. These mutations would not necessarily be expected, a priori, to have the same phenotypic effect as would be produced by protein-truncating mutations at the same position in the gene, and there were too few to allow them to be analyzed as a separate group. With these exclusions, a total of 164 families with 92 distinct mutations were included in this analysis. The 164 families include 25 used in the study by Gayther et al. (1997), and herein are referred to as the “CRC families”; details of these families have been updated since that study.
Statistical Methods
To evaluate the evidence for a difference, in the breast:ovarian cancer ratio, between OCCR and non-OCCR mutations, we first computed the numbers of females with breast cancer and the number of females with ovarian cancer, over families with OCCR mutations and families with non-OCCR mutations, ignoring the second cancer in an individual with bilateral breast cancer. The resulting 2×2 contingency table was used to calculate the deviance statistic, D = 2Σ(observed)ln(observed/expected), asymptotically equivalent to Pearson’s χ2 statistic. Counts were for cancers at any age, with only tested noncarriers and known sporadic cases excluded. Thirteen families were collected by the Montreal center on the basis of the fact that they carry the Ashkenazi 6174delT mutation, but data on these families did not contribute to this analysis. To overcome the problem that the occurrence of breast/ovarian cancer in different members of the same family are not independent, the significance of the test statistic was estimated by simulation. Within each center, families were randomly permuted between the three types of mutation 10,000 times, and the resulting deviance statistics, calculated under the null hypothesis of no genotype-phenotype relation, were compared with that observed. The significance level was calculated as the proportion of the random deviance statistics that were as large as or larger than that observed.
The definition of the OCCR was chosen by Gayther et al. (1997) because of its good fit to the 25 CRC families. With the BCLC set of 164 families, including the original CRC families, we were able to consider whether this definition could be improved. For simplicity, we restricted our candidate OCCRs to be single contiguous regions of BRCA2. The deviance statistic for the 2×2 contingency table of number of breast cancers and number of ovarian cancers inside and outside the candidate OCCR was computed, as described above, and was maximized over the set of all possible candidate OCCRs. This analysis was performed to find the optimal OCCR definition for the CRC families, for the set of all non-CRC families, and for all 164 families together.
Risks of breast and ovarian cancer associated with mutations inside and outside the OCCR were estimated by a maximum-likelihood approach. This approach allows the disease information from untyped individuals to be incorporated appropriately into the analysis. A full likelihood would take the form
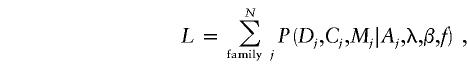 |
where Dj is the vector of disease status of all females in family j, Cj is the vector of carrier status of all individuals, Mj is the mutation in the tested proband of family j, and Aj is the ascertainment events for family j. Ascertainment was assumed to depend on the first breast/ovarian cancer in each woman and on the fact that the proband must, by definition, be a carrier. Hence, the information contained in vector Aj is a subset of the information contained in Dj and Cj. λ is the vector of age-specific incidence rates of breast cancer and of ovarian cancer in carriers of non-OCCR mutations, and β is the log-RR (log value of the relative risk [RR]) to carriers of OCCR mutations relative to carriers of non-OCCR mutations, which is assumed to be constant with age. f is a vector of the population frequencies of the mutations. In practice, the population frequencies of each specific mutation are unknown. We therefore based inference on the likelihood conditional on the set of mutations observed in each center, which is of the form
 |
The sum in the denominator is over all permutations π of mutations found in center i. Analyses were stratified by center to allow for the possibility that ascertainment criteria and mutation frequencies might vary between centers. This will also take into account any mutation-detection–sensitivity differences between centers. For this purpose, the Montreal series of 13 Ashkenazi Jewish families with the 6174delT mutation were considered to be a single center. The parameters of interest related to the comparison of cancer risks associated with mutations inside and outside the OCCR; hence, the specific mutation Mj was replaced by an indicator variable taking values 1 and 0 for mutations inside and outside the OCCR, respectively.
Breast cancer follow-up was censored at the date of either prophylactic mastectomy or oophorectomy and at the date of ovarian cancer diagnosis, because of potential hormonal effects on breast cancer risk. Ovarian cancer follow-up was censored at the date of prophylactic oophorectomy, and both sets of follow-up were censored at the earliest of date of death, 70th birthday, or 1 January 1996; that is, ovarian cancer follow-up subsequent to either diagnosis of a breast cancer or mastectomy was included in the analysis. The penetrance function was constructed by use of a Poisson model for the number of each type of cancer (0 or 1) diagnosed in each woman, with the mean being dependent on carrier status. Among carriers, the mutation-specific mean was parameterized by use of the Cox model. The cancer rates assumed in noncarriers were given separately for seven calendar periods during 1960–1996 and for 17 5-year age groups. The rates were obtained from the series of publications titled Cancer Incidence in Five Continents (Waterhouse et al. 1976, 1982; Muir et al. 1987; Parkin et al. 1992, 1997) and from information provided by the International Agency for Research on Cancer. The likelihood was maximized over the risk and log-RR parameters by use of the program MENDEL (Lange et al. 1988). For untested individuals, MENDEL estimates their carrier probability on the basis of both their phenotype and the phenotype and carrier status of each of their relatives; their contribution to the likelihood is weighted accordingly. Maximum likelihoods were also found for models with one or both of the log-RRs fixed at 0. The maximum-likelihood estimates (MLEs) of the parameters were used to compute the cumulative risks, by age, of breast cancer and ovarian cancer, that are associated with OCCR and non-OCCR mutations.
Among BRCA2 mutation carriers, the proportion of cases of breast cancer at age t that have OCCR mutations was estimated by use of Bayes’s theorem, as follows:
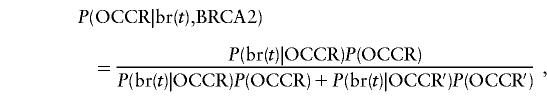 |
where br(t) is the event of breast cancer diagnosed at age t and P(br(t)|OCCR) and P(br(t)|OCCR′) are given by the MLEs of the incidence rates. Equivalent calculations were made for ovarian cancer.
In addition to the elevated risks of breast/ovarian cancer in female carriers, BRCA2 mutations are also associated with increased risks of breast cancer and prostate cancer in males. (We have previously reported that BRCA2 mutations are also associated with increased risks of pancreatic cancer, gallbladder and bile-duct cancer, stomach cancer, and malignant melanoma (Breast Cancer Linkage Consortium 1999), but the small numbers of cases prohibits an extension of the genotype-phenotype analysis to these cancer sites). The RRs of male breast cancer and prostate cancer for carriers of OCCR mutations relative to carriers of other BRCA2 mutations were estimated simultaneously, by use of the conditional maximum-likelihood technique described above. The likelihood maximized took a form similar to that used in the estimation of the RR of breast/ovarian cancer (eq. [1]). Disease information from females was included, as for the likelihood of breast/ovarian cancer, with parameters fixed at their MLEs. Males were censored at age 80 years, because of the higher average age at diagnosis of these cancers. Ascertainment was deemed to be independent of prostate cancer occurrence but was deemed to depend on breast cancer occurrence in males. MLEs of the risks and RRs were obtained, and the cumulative risks were computed, as for the female carriers.
Results
Figure 1 shows the relative proportions of breast cancer and ovarian cancer for each group of mutations, with the original CRC families excluded. Consistent with the results of Gayther et al. (1997), the OCCR mutations are associated with the highest proportion of ovarian cancers (23.8%, with the CRC families excluded), with the regions 5′ and 3′ of the OCCR each having similar proportions (8.8% and 9.6%, respectively). The deviance statistic for testing the independence of mutation position and cancer site is significant (D=6.50; P=.027). The significance of this effect is considerably strengthened when the original 25 CRC families are included (D=37.13; P<.0001).
Figure 1.

Counts of breast cancer and ovarian cancer, by mutation location within BRCA2. Counts exclude tested noncarriers and other known sporadic cases.
When we maximized the deviance statistic over all possible OCCR positions, the optimal OCCR definition was nucleotides [3059–4075,6503–6629] inclusive (the boundaries cannot be stated more precisely than this because no mutations fall within these intervals). The 5′ boundary differed slightly from the original OCCR, [3035,6629], but the change in deviance was small (39.39 vs. 37.13). The change in definition would affect only three families, and, since the difference in deviance was slight, we chose to use the original definition in the main analysis, rather than a new post hoc definition.
The odds ratios (ORs) for the region-by-region comparisons are shown in table 1. The most significant OR is for the comparison of the OCCR with the non-OCCR (OR = 3.86; P<.0001), suggesting either that ovarian cancers are more common in families with OCCR mutations or that breast cancers are more common in families with non-OCCR mutations, or a combination of the two effects. The effect is less strong when the CRC families are excluded (the OR for the comparison of OCCR vs. non-OCCR when only the 25 CRC families are used is 108.7). The OR for the comparison of the 5′ and 3′ regions is not significantly different from unity.
Table 1.
OR for Breast:Ovarian Cancer, for Comparison of Mutations in Different Regions of BRCA2
| Regions Compareda | OR | P |
| All families: | ||
| 2 vs. 1 | 3.70 | .0003 |
| 1 vs. 3 | 1.09 | .54 |
| 2 vs. 3 | 4.02 | .0001 |
| OCCR vs. non-OCCR | 3.86 | <.0001 |
| Families without CRC: | ||
| 2 vs. 1 | 2.06 | .023 |
| 1 vs. 3 | .91 | .40 |
| 2 vs. 3 | 1.88 | .082 |
| OCCR vs. non-OCCR | 1.98 | .023 |
Region 1 is 5′ of OCCR, region 2 is the OCCR, and region 3 is 3′ of OCCR.
Cancer risks for carriers of different mutations were evaluated by maximization of the conditional likelihood over the parameters (table 2). For the complete set of 164 families, the OCCR is associated with a significantly reduced risk of breast cancer (RR = 0.63 [95% CI = 0.46–0.84]; P=.001) and with an increased risk of ovarian cancer, although this effect is less statistically significant (RR = 1.88 [95% CI = 1.08–3.33]; P=.026). Repeating the analysis with the CRC families excluded weakens the breast cancer effect (RR = 0.68 [95% CI = 0.49–0.94]; P=.011), whereas the RR of ovarian cancer ceases to be significant and actually changes direction (RR = 0.85 [95% CI = 0.39–1.85]; P=.2). As predicted, replacing the original OCCR boundaries with those given by maximizing the deviance over all possible boundaries enhances both effects only slightly (when all families are included, RR of breast cancer = 0.59 [95% CI = 0.42–0.82] and RR of ovarian cancer = 1.95 [95% CI = 1.05–3.63]).
Table 2.
MLEs of Age-Specific Incidence of Breast Cancer and of Ovarian Cancer, for Non-OCCR Mutations, and MLEs of OCCR:Non-OCCR RRs
| Parameter | MLE [95% CI] |
| Breast cancer: | |
| <40 years | .0027 [.001–.004] |
| 40–49 years | .0169 [.007–.026] |
| 50–59 years | .0196 [.006–.033] |
| 60–69 years | .0148 [.001–.028] |
| Ovarian cancer: | |
| <40 years | .0001 [.0–0.0004] |
| 40–49 years | .0010 [.0–.003] |
| 50–59 years | .0050 [.001–.009] |
| 60–69 years | .0051 [.0–.010] |
| RR: | |
| Breast cancer | .63 [.46–.84] |
| Ovarian cancer | 1.88 [1.08–3.33 ] |
Figure 2A shows the cumulative risks of breast cancer, by age, for carriers of BRCA2 mutations inside and outside the OCCR, and figure 2B shows the corresponding cumulative risks of ovarian cancer. The cumulative risk of breast cancer by age 70 years is 46.2% (95% CI = 34.1%–60.3%) for non-OCCR mutations, compared with 32.6% (95% CI = 23.2%–44.5%) for mutations within the OCCR. The corresponding cumulative risks of ovarian cancer by age 70 years are 10.9% (95% CI = 5.5%–20.9%) for non-OCCR mutations and 19.5% (95% CI = 11.6%–31.7%) for OCCR mutations. When it is assumed that there is no relationship between cancer risk and mutation location, the cumulative risks by age 70 years are 40.6% (95% CI = 30.1%–53.3%) for breast cancer and 13.8% (95% CI = 8.6%–21.8%) for ovarian cancer.
Figure 2.

Cumulative incidence, in BRCA2-mutation carriers, of breast cancer (A) and ovarian cancer (B), by mutation location
The corresponding estimates for males are shown in table 3. The data set included 59 cases of breast cancer in males and 72 cases of prostate cancer. Preliminary estimations revealed that the OCCR:non-OCCR RR of male breast cancer appeared to vary with age, whereas the prostate RR remains approximately constant. Therefore, the likelihood was modeled by use of a single parameter for the log-RR of prostate cancer and two separate parameters for the male breast cancer log-RR, one at age <60 years and the other at age ⩾60 years. Overall, there was no evidence of a difference, by mutation position, in male breast cancer risk: the OCCR RR of male breast cancer was >1 at age <60 years and <1 at age ⩾60; however, both 95% CIs include 1. The RR of prostate cancer, 0.52, achieved borderline significance (95% CI = 0.24–1.00; P=.05). The cumulative risk of prostate cancer by age 80 years was estimated as 33.6% for non-OCCR mutations (95% CI = 25.1%–44.1%) and 19.2% for OCCR mutations (95% CI = 10.7%–33.1%). The estimated cumulative risk of male breast cancer, estimated under the model of no OCCR effect, was 2.8% (95% CI = 0.6%–13.0%) by age 70 years, rising to 6.9% by age 80 years (95% CI = 1.2%–38.6%).
Table 3.
MLEs of Age-Specific Rates of Prostate Cancer and of Male Cases of Breast Cancer, for Non-OCCR Mutations, and MLEs of OCCR:Non-OCCR RRs
| Parameter | MLE [95% CI] |
| Breast: | |
| 40-49 years | .0003 [.0–.001] |
| 50-59 years | .0008 [.0–.003] |
| 60-69 years | .0016 [.0–.007] |
| 70-79 years | .0066 [.0–.036] |
| Prostate: | |
| 40-49 years | .0003 [.0–.0001] |
| 50-59 years | .0043 [.002–.007] |
| 60-69 years | .0099 [.005–.015] |
| 70-79 years | .0265 [.014–.039] |
| RR: | |
| Breast: | |
| <60 years | 1.73 [.52–5.78] |
| ⩾60 years | .59 [.20–1.74] |
| Prostate | .52 [.24–1.00] |
Discussion
This report has described the evidence for genotype-phenotype correlations in 164 families with BRCA2 mutations. After excluding the families originally reported by Gayther et al. (1997), we found significant evidence of a reduced breast:ovarian cancer–risk ratio in families with mutations in the OCCR, thus confirming their observation. The limited number of families available to Gayther et al. (1997) meant that they were unable to separate the effects of mutation position on risk of breast cancer versus its effect on risk of ovarian cancer. We performed maximum-likelihood estimation of the RR parameters for mutations in the OCCR relative to those outside it , finding that, for mutations inside the OCCR, the risk of breast cancer was 37% lower than that for other BRCA2 mutations. We also found evidence of a higher risk of ovarian cancer associated with OCCR mutations, but this effect was less significant, and it was not significant when the CRC families were excluded. The stronger effect in the CRC families might be a reflection of their different pattern of ascertainment, since they contain a high proportion of families that have multiple cases of ovarian cancer but comparatively few cases of breast cancer.
We also found a lower prostate cancer risk in the OCCR, with the OCCR risk being 48% lower than the non-OCCR risk. Perhaps surprisingly, we found no evidence of variation in risk of male breast cancer, although the comparatively small number of cases of this disease gives the lowest power to detect any such difference. It is interesting that eight of nine families with more than one case of male breast cancer have mutations outside the OCCR.
The OCCR includes the 6174delT mutation, present in ∼1% of the Ashkenazi Jewish population (Oddoux et al. 1996) and seen in 23 families in our data set. Thirteen of these families were ascertained in Montreal on the basis of the fact that they carry the 6174delT mutation, and, throughout the analysis, these families were considered as a separate center. To assess whether the observed effects could be explained entirely by this mutation, the maximum-likelihood estimation was repeated without these 23 families. When this restricted set is used, the OCCR-associated reduction in breast cancer risk is weakened very slightly (RR = 0.67 [95% CI = 0.48–0.93]; P=.008), whereas the increase in ovarian risk is identical in size but is less significant (RR = 1.88 [95% CI = 1.05–3.40]; P=.034). Hence, the OCCR effect does not appear to be simply a consequence of the Ashkenazi founder mutation. Recent studies of mutation frequencies in unselected Ashkenazi patients with either breast cancer or ovarian cancer suggest that the 6174delT mutation is nearly four times as frequent in patients with ovarian cancer as it is in patients with breast cancer (Warner et al. 1999; Moslehi et al. 2000). This is in contrast to the Icelandic mutation (999del5, 5′ of the OCCR), the frequency of which is similar in patients with breast cancer and patients with ovarian cancer, a finding that again is consistent with the OCCR effect (Johannesdottir et al. 1996). In other, nonfounder populations, our results suggest that approximately half of BRCA2 mutations in patients with ovarian cancer would fall within the OCCR but that only one quarter of those in patients with breast cancer do so.
The distinctive phenotype associated with OCCR mutations relative to other BRCA2 mutations appears to be predominantly a reduced risk of breast cancer rather than an increased risk of ovarian cancer. In view of these data, the original term—“ovarian cancer cluster region”—appears to be something of a misnomer. However, most high-risk families are selected on the basis of a certain number of cases of breast/ovarian cancer; hence, the two cancer risks are somewhat hard to disentangle (the correlation between the RR estimates was .26). It may be that the ovarian cancer effect is actually stronger and that the breast cancer effect weaker, or vice versa. A further potential complication is that the risk of breast cancer and the risk of ovarian cancer are likely to be correlated in families because of other shared genetic or environmental factors. Although this does not affect the evidence in favor of mutation-specific effects, the precise estimates may need adjustment to incorporate other familial factors.
At present, we can only speculate about possible biological mechanisms that might be responsible for the breast/ovarian cancer–risk variation that we have observed. The function of BRCA2 remains unclear, partly because its predicted amino acid sequence bears little resemblance to proteins of known function (Wooster et al. 1995; Tavtigian et al. 1996). The most significant data suggest that BRCA2 is a nuclear protein that is involved in DNA repair, via its association with the double-strand DNA break-repair protein RAD51 (Bertwistle et al. 1997; Wong et al. 1997; Venkitaraman 2000). In vitro studies have shown that an interaction between BRCA2 and RAD51 is mediated via a series of eight amino acid repeats (i.e., BRC repeats) located in a central region of the gene, as shown in figure 1 (Bork et al. 1996). The observation that at least six of these RAD51-binding motifs lie within the boundaries of the OCCR defined in this study suggests that they may play a role in the variation in cancer risk we observed. Furthermore, mice with homozygous truncating BRCA2 mutations 5′ of the BRC repeats are embryonic lethal, whereas mice in which at least three of the BRC repeats are retained have only a partially lethal phenotype; some mice survive until birth but have stunted growth and skeletal defects (e.g., see Connor et al. 1997; Sharan et al. 1997; reviewed in Gayther and Ponder 1998). Although a C-terminus RAD51-binding site has been reported in murine BRCA2, the most recent data suggest that, in humans, the RAD51-BRCA2 interaction takes place principally via the BRC repeats (Sharan et al. 1997; Wong et al. 1997; Aihara et al. 1999; A. R. Venkitaraman, personal communication).
Biological mechanisms that may explain these genotype-phenotype variations rely on the assumption that protein products of BRCA2 truncating mutations retain partial or complete stability, although this has not been demonstrated formally. One possibility is that mutations occurring before the OCCR have no function and are effectively null. Mutations occurring within the OCCR would be predicted to retain one or more of the BRC motifs and, presumably, some ability to bind with RAD51 and the complex with which it is associated. BRCA2 truncated by these mutations may have a partial function or could behave in a dominant-negative manner to negate the function of the RAD51-associated complex. Truncated proteins occurring as a result of mutations 3′ of the OCCR would be predicted to retain all of the BRC repeats and may have a residual function instead of behaving in a dominant-negative fashion. The opposing effects that OCCR mutations have on the risk of breast cancer and on the risk of ovarian cancer could be explained if the DNA-repair pathways through which BRCA2 acts differ between the two types of epithelial tissue. However, this theory is complicated by a recent report that two nuclear-localization signals (NLSs) located at the 3′ end of BRCA2 are essential for its cellular localization (Spain et al. 1999). All mutations in the present study are predicted to truncate BRCA2 5′ to the NLS, rendering it cytoplasmic and ruling out any interaction with the RAD51 complex unless it is transported into the nucleus by an alternative means, such as homodimerization with wild-type BRCA2.
Another possible biological mechanism assumes the existence of an in-frame alternative splicing of the BRCA2 protein, a splicing that skips the OCCR, resulting in a stable RNA isoform that retains some degree of BRCA2 functionality; OCCR mutations would still truncate the full-length protein but would have no effect on the isoform, having been spliced out. If this isoform were more frequent in breast epithelial tissue than in ovarian epithelial tissue, then the partial rescue—and, hence, the reduced penetrance—associated with OCCR mutations would be evident only in breast cancer. At least two stable RNA isoforms of BRCA1, each skipping almost all of exon 11, have been found in breast epithelial cells (Lu et al. 1996; Thakur et al. 1997; Wilson et al. 1997). However, although an in-frame alternative BRCA2 splicing that removes exon 12 has been discovered, no isoform lacking exon 11 has been reported (Bièche and Lidereau 1999). More-detailed functional studies will be required to support or refute these hypotheses.
Although the main aim of this study has been to assess the variation, due to different mutations, in the risk of breast cancer and in the risk of ovarian cancer, we were also able to provide estimates of the absolute risks for mutations within and outside the OCCR (33% and 46%, respectively, for breast cancer by age 70 years; and 20% and 11%, respectively for ovarian cancer by age 70 years). Estimation of the risk of breast cancer employed only information about first breast cancers, in contrast to the previous penetrance estimation performed on essentially the same data set, which made use only of information from second, contralateral breast cancers (Breast Cancer Linkage Consortium 1999). The contralateral method gave higher breast cancer–risk estimates (52.3% [95% CI = 41.7%–61.0%] by age 70 years), as would be expected. Both estimates are lower than the 84% (95% CI = 43%–95%) reported by Ford et al. (1998), who used a smaller number of families with BRCA2 linkage information. Some of this difference is likely to be due to chance, given the wide confidence limits; however, selection of families may also be a factor. The set of families in the present study has been less strongly selected for family history than were the linkage families reported in earlier studies. It is possible that this would lead to a lower estimate of absolute risk, as a result of other familial factors being weaker. Also, Ford et al. (1998) estimated the risk of ovarian cancer by age 70 years to be 27% (95% CI = 0%–47%); this is considerably higher than the 14% derived in the present study—although not inconsistent with it, given the very wide 95% confidence interval for the earlier estimate.
Although many studies have established male breast cancer as a characteristic element of the BRCA2 phenotype (e.g., see Wooster et al. 1994; Tonin et al. 1995), to date only one attempt has been made to estimate the risk of breast cancer in male carriers of BRCA2 (Easton et al. 1997). That study estimated that the cumulative risk of breast cancer by age 70 years is 6.3% (95% CI = 1.4%–25.6%), but this was based on just four cases observed in two large BRCA2 pedigrees. Our estimated cumulative risk of breast cancer by age 70 years, based on 59 cases, is somewhat smaller (2.8%) and has a narrower 95% CI (0.6%–13.0%). Our estimated risk of breast cancer by age 80 years is 6.9% (95% CI = 1.2%–38.6%), equivalent to an RR of ∼80. If it is assumed that the population frequency of BRCA2 mutations is .0006 (Ford et al. 1995), this would imply that ∼10% of male cases of breast cancer would be expected to be due to BRCA2 mutations. The published studies of BRCA2 prevalence in male cases of breast cancer are consistent with an estimate of this order of magnitude. In five studies of non-Icelandic populations, the average prevalence is 13%, based on a total of 181 cases (Couch et al. 1996; Friedman et al. 1997; Mavraki et al. 1997; Haraldsson et al. 1998; Csokay et al. 1999). The higher prevalence, in Iceland, of BRCA2 mutations among male patients with breast cancer (38% of 34 cases [Thorlacius et al. 1998]) is consistent with the high frequency (∼1/200) of carriers of the 999del5 mutation in the Icelandic population (Johannesdottir et al. 1996; Thorlacius et al. 1997).
In conclusion, the present study confirms that mutations in the OCCR of BRCA2 confer different risks of breast, ovarian, and, possibly, prostate cancer than are conferred by other mutations. At this stage, it seems unlikely that the observed differences in risks are sufficiently large to justify that clinical management should differ according to the position of the mutation in a family. Nevertheless, these differences should be borne in mind when risk information is provided to mutation carriers.
Appendix: Members of the Breast Cancer Linkage Consortium
The following are the contributing centers and the names of the principal investigators (numbers in square brackets are the number of families contributed by each center):
| CRC Genetic Epidemiology Unit (Cambridge) (coordinating center): D. Easton, D. Thompson, and L. McGuffog |
| University of Aberdeen: N. Haites and A. Schofield [1] |
| Humangenetik, Kantonsspital (Basel): R. J. Scott [2] (present address : University of Newcastle [Australia]) |
| Departments of Medicine and Genetics, University of Washington (Seattle): M.-C. King and E. Schubert [6] |
| Centre Jean Perrin, Clermont-Ferrand: Y. Bignon [1] |
| Institute of Cancer Research: M. Stratton, D. Ford, J. Peto, and R. Eeles [19] |
| CRC Human Cancer Genetics Research Group: B. Ponder and S. Gayther [11] |
| Deutsches Krebsforschungszentrum (Heidelberg) and University of Würzburg: J. Chang-Claude, B. H. F. Weber, and U. Hamann [5] |
| Centro Nacional de Investigaciones Oncológicas (Madrid): J. Benítez and A. Osorio [4] |
| University Central Hospital, Departments of Oncology and Obstetrics and Gynaecology (Helsinki): H. Eerola and H. Nevanlinna [11] |
| Creighton University (Omaha) and International Agency for Research on Cancer (Lyon): H. T. Lynch, S. Narod, D. Goldgar, and G. Lenoir [8] |
| Institut Curie (Paris): D. Stoppa-Lyonnet and S. Gad [9] |
| University Hospital of Iceland (Reykjavik): A. Arason, R. B. Barkardottir, and V. Egilsson [5] |
| Icelandic Cancer Society (Reykjavik): J. Eyfjord and H. Tulinius [5] |
| Imperial Cancer Research Fund (Leeds): D. T. Bishop [3] |
| University of Lund: A. Borg, N. Loman, O. Johannsson, and H. Olsson [20] |
| McGill University (Montreal): P. Tonin and W. Foulkes [11] |
| University of Montreal: P. Ghadirian, A. M. Mes-Masson, and D. Provencher [8] |
| University of Pennsylvania: B. Weber [11] |
| University of Leiden and Foundation for the Detection of Hereditary Tumours (Leiden) and Erasmus Medical Center and Daniel den Hoed Cancer Center (Rotterdam) P. Devilee, H. Vasen, C. J. Cornelisse, H. Meijers-Heijboer, and J. G. M. Klijn [8] |
| University of Toronto: S. Narod, J.-S. Brunet, and R. Moslehi [19] |
| University of Utah: S. L. Neuhausen and L. Cannon-Albright [6] |
Electronic-Database Information
The accession number and URL for data in this article are as follows:
- Online Mendelian Inheritance in Man (OMIN), http://www.ncbi.nlm.nih.gov/Omim (for inherited breast cancer type 2 [MIM 600185])
References
- Aihara H, Ito Y, Kurumizaka H, Yokoyama S, Shibata T (1999) The N-terminal domain of the human Rad51 binding surface as revealed by NMR. J Mol Biol 290:495–504 [DOI] [PubMed] [Google Scholar]
- Bertwistle D, Swift S, Marston NJ, Jackson LE, Crossland S, Crompton MR, Marshall CJ, Ashworth A (1997) Nuclear location and cell cycle regulation of the BRCA2 protein. Cancer Res 57:5485–5488 [PubMed] [Google Scholar]
- Bièche I, Lidereau R (1999) Increased level of exon 12 alternatively spliced BRCA2 transcripts in tumour breast tissue compared with normal tissue. Cancer Res 59:2546–2550 [PubMed] [Google Scholar]
- Bork P, Blomberg N, Niges M (1996) Internal repeats in the BRCA2 protein sequence. Nat Genet 13:22–23 [DOI] [PubMed] [Google Scholar]
- Breast Cancer Linkage Consortium (1999) Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst 91:1310–1316 [DOI] [PubMed] [Google Scholar]
- Connor F, Bertwistle D, Mee PJ, Ross GM, Swift S, Grigorieva E, Tybulewicz VLJ, Ashworth A (1997) Tumorigenesis and a DNA repair defect in mice with a truncating BRCA2 mutation. Nat Genet 17:423–430 [DOI] [PubMed] [Google Scholar]
- Couch FJ, Farid LM, DeShano ML, Tavtigian SV, Calzone K, Campeau L, Peng Y, Bogden B, Chen Q, Neuhausen S, Shattuck-Eidens D, Godwin AK, Daly M, Radford DM, Sedlack S, Rommens J, Simard J, Garber J, Merajver S, Weber B (1996) BRCA2 germline mutations in male breast cancer cases and breast cancer families. Nat Genet 13:123–125 [DOI] [PubMed] [Google Scholar]
- Csokay B, Udvarhelyi N, Sulyok Z, Besznyak I, Ramus S, Ponder B, Olah E (1999) High frequency of germ-line BRCA2 mutations among Hungarian male breast cancer patients without family history. Cancer Res 59:995–998 [PubMed] [Google Scholar]
- Easton DF, Steele L, Fields P, Ormiston W, Averill D, Daly PA, McManus R, Neuhausen SL, Ford D, Wooster R, Cannon-Albright LA, Stratton MR, Goldgar DE (1997) Cancer risks in two large breast cancer families linked to BRCA2 on chromosome 13q12-13. Am J Hum Genet 61:120–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford D, Easton DF, Peto J (1995) Estimates of the gene frequency of BRCA1 and its contribution to breast and ovarian cancer incidence. Am J Hum Genet 57:1457–1462 [PMC free article] [PubMed] [Google Scholar]
- Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, Bishop DT, et al (1998) Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. Am J Hum Genet 62:676–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman LS, Gayther SA, Kurosaki T, Gordon D, Noble B, Casey G, Ponder BAJ, Anton-Culver H (1997) Mutation analysis of BRCA1 and BRCA2 in a male breast cancer population. Am J Hum Genet 60:313–319 [PMC free article] [PubMed] [Google Scholar]
- Gayther SA, Mangion J, Russell PA, Seal S, Basford R, Ponder BAJ, Stratton M, Easton D (1997) Variation in risks of breast an ovarian cancer associated with different germline mutations of the BRCA2 gene. Nat Genet 15:103–105 [DOI] [PubMed] [Google Scholar]
- Gayther SA, Ponder BAJ (1998) Clues to the function of the tumour susceptibility gene BRCA2. Dis Markers 14:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraldsson K, Loman N, Zhang Q-X, Johannsson O, Olsson H, Borg A (1998) BRCA2 germ-line mutations are frequent in male breast cancer patients without a family history of the disease. Cancer Res 58:1367–1371 [PubMed] [Google Scholar]
- Johannesdottir G, Gudmundsson J, Bergthorsson JT, Arason A, Agnarsson BA, Eiriksdottir G, Johannsson OT, Borg A, Ingvarsson S, Easton DF, Egilsson V, Barkardottir RB (1996) High prevalence of the 999del5 mutation in Icelandic breast and ovarian cancer patients. Cancer Res 56:3663–3665 [PubMed] [Google Scholar]
- Lange K, Weeks D, Boehnke M (1988) Programs for pedigree analysis: MENDEL, FISHER and dGENE. Genet Epidemiol 5:471–472 [DOI] [PubMed] [Google Scholar]
- Lu M, Conzen S, Cole C, Arrick A (1996) Characterization of functional messenger RNA splice variants of BRCA1 expressed in nonmalignant and tumor-derived breast cells. Cancer Res 56:4578–4581 [PubMed] [Google Scholar]
- Mavraki E, Gray IC, Bishop DT, Spurr NK (1997) Germline BRCA2 mutations in men with breast cancer. Br J Cancer 76:1428–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moslehi R, Chu W, Karlan B, Fishman D, Risch H, Fields A, Smotkin D, Ben-David Y, Rosenblatt J, Russo D, Schwartz P, Tung N, Warner E, Rosen B, Friedman J, Brunet J-S, Narod SA (2000) BRCA1 and BRCA2 mutation analysis of 208 Ashkenazi Jewish women with ovarian cancer. Am J Hum Genet 66:1259–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir C, Waterhouse J, Mack T, Powell J, Whelan S (eds) (1987) Vol 5, IARC sci publ 88, in: Cancer Incidence in Five Continents. International Agency for Research on Cancer, Lyon [Google Scholar]
- Oddoux C, Streuwing JP, Clayton CM, Neuhausen S, Brody LC, Kaback M, Haas B, Norton L, Borgen P, Jhanwar S, Goldgar D, Ostrer H, Offit K (1996) The carrier frequency of the BRCA2 6174delT mutation among Ashkenazi Jewish individuals is approximately 1%. Nat Genet 14:188–190 [DOI] [PubMed] [Google Scholar]
- Parkin DM, Muir CS, Whelan SL, Gao YT, Ferlay J, Powell J (eds) (1992) Vol 6, IARC sci publ 120, in: Cancer incidence in five continents. International Agency for Research on Cancer, Lyon [Google Scholar]
- Parkin DM, Whelan SL, Ferlay J, Raymond L, Young J (eds) (1997) Vol 7, IARC sci publ 143, in: Cancer incidence in five continents. International Agency for Research on Cancer, Lyon [Google Scholar]
- Sharan SK, Morimatsu M, Albrecht U, Lim DS, Regel E, Dinh C, Sands A, Eichele G, Hasty P, Bradley A (1997) Embryonic lethality and radiation hypersensitivity mediated by RAD51in mice lacking BRCA2. Nature 386:804–810 [DOI] [PubMed] [Google Scholar]
- Spain BH, Larson CJ, Shihabuddin LS, Gage FH, Verma IM (1999) Truncated BRCA2 is cytoplasmic: implications for cancer-linked mutations. Proc Natl Acad Sci USA 96:13920–13925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian SV, Simard J, Rommens J, Couch F, Shattuck-Eidens D, Neuhausen S, Merajver S, et al (1996) The complete BRCA2 gene in chromosome 13q-linked kindreds. Nat Genet 12:333–337 [DOI] [PubMed] [Google Scholar]
- Thakur S, Zhang HB, Peng Y, Le H, Carroll B, Ward T, Yao J, Farid LM, Couch FJ, Wilson RB, Weber BL (1997) Localization of BRCA1 and a splice site variant identifies the nuclear localization signal. Mol Cell Biol 17:444–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorlacius S, Sigurdsson S, Bjarnadottir H, Olafsdottir G, Jonasson JG, Tryggvadottir L, Tulinius H, Eyfjörd JE (1997) Study of a single BRCA2 mutation with high carrier frequency in a small population. Am J Hum Genet 60:1079–1084 [PMC free article] [PubMed] [Google Scholar]
- Thorlacius S, Struewing JP, Hartge P, Olafsdottir GH, Sigvaldason H, Tryggvadottir L, Wacholder S, Tulinius H, Eyfjörd JE (1998) Population-based study of risk of breast cancer in carriers of BRCA2 mutation. Lancet 352:1337–1339 [DOI] [PubMed] [Google Scholar]
- Tonin P, Ghadirian P, Phelan C, Lenoir GM, Lynch HT, Letendre F, Belanger D, Monté M, Narod SA (1995) A large multisite cancer familiy is linked to BRCA2. J Med Genet 32:982–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkitaraman AR (2000) Multiplying functions for BRCA1 and BRCA2? meeting report, The Breakthrough Breast Cancer Second International Workshop on the Function of BRCA1 and BRCA2, Cambridge, UK, 9–10 September 1999. Biochem Biophys Acta 1470:41–47 [DOI] [PubMed] [Google Scholar]
- Warner E, Foulkes W, Goodwin P, Meschino W, Blondal J, Paterson C, Ozcelik H, Goss P, Allingham-Hawkins D, Hamel N, Di Prospero L, Contiga V, Serruya C, Klein M, Moslehi R, Honeyford J, Liede A, Glendon G, Brunet J-S, Narod S (1999) Prevalence and penetrance of BRCA1 and BRCA2 gene mutations in unselected Ashkenazi Jewish women with breast cancer. J Natl Cancer Inst 91:1241–1247 [DOI] [PubMed] [Google Scholar]
- Waterhouse JAH, Muir C, Correa P, Powell J (eds) (1976) Vol 3, IARC sci publ 15, in: Cancer incidence in five continents. International Agency for Research on Cancer, Lyon [Google Scholar]
- Waterhouse J , Muir C, Shanmugaratnam K, Powell J (eds) (1982) Vol 4, IARC sci publ 42, in: Cancer incidence in five continents. International Agency for Research on Cancer, Lyon [Google Scholar]
- Wilson CA, Payton MN, Elliott GS, Buaas FW, Cajulis EE, Grosshans D, Ramos L, Reese DM, Slamon DJ, Calzone FJ (1997) Differential subcellular localization, expression and biological toxicity of BRCA1 and the splice variant BRCA1-delta11b. Oncogene 14:1–16 [DOI] [PubMed] [Google Scholar]
- Wong AKC, Pero R, Ormonde PA, Tavtigian SV, Bartel PL (1997) RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene BRCA2. J Biol Chem 272:31941–31944 [DOI] [PubMed] [Google Scholar]
- Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, et al (1995) Identification of the breast cancer susceptibility gene BRCA2. Nature 378:789–792 [DOI] [PubMed] [Google Scholar]
- Wooster R, Neuhausen SL, Mangion J, Quirk Y, Ford D, Collins N, Nguyen K, et al (1994) Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science 265:2088–2090 [DOI] [PubMed] [Google Scholar]