

Abstract
Hereditary benign intraepithelial dyskeratosis (HBID) is an autosomal dominant disorder characterized by elevated epithelial plaques on the ocular and oral mucous membranes. It has been reported primarily, but not exclusively, in individuals of American Indian heritage in North Carolina. We have examined and obtained DNA on two large families affected by HBID. Using genetic linkage analysis we have localized the HBID gene to chromosome 4 (4q35) with a peak LOD score of 8.97. Molecular analysis of these data reveals that all individuals affected with HBID in both families demonstrate the presence of three alleles for two tightly linked markers, D4S1652 and D4S2390, which map to the telomeric region of 4q35. This suggests the presence of a duplication segregating with the disease phenotype that is most likely involved in its causation.
Von Sallmann and Paton first described autosomal dominant hereditary benign intraepithelial dyskeratosis (HBID [MIM 127600]) in 1959 (von Sallmann and Paton 1959). Several additional cases (Yanoff 1968; Shields et al. 1987) have been subsequently reported, all but one (McLean et al. 1981) trace their ancestry back to North Carolina. The onset of HBID is usually at birth or in early childhood. Lesions consist of elevated, granular, white-to-grayish epithelial plaques located within the exposed nasal and/or temporal conjunctival surface. The growths can interfere with vision (von Sallmann and Paton 1960), although this is uncommon. Occasionally, the plaques can involve the cornea or can spontaneously shed. The large plaques are frequently associated with hyperemic blood vessels producing a “red eye” appearance, which gives HBID this common colloquial name. Affected individuals report that this “bloodshot” appearance has led many to experience difficulties in job employment and social interactions, as strangers often assume their appearance is a result of alcohol or drug abuse or is secondary to infection. The oral mucosa in HBID has a white, spongy, macerated appearance and may demonstrate pinpoint elevations when stretched.
Von Sallman and Paton (1960) first reported that the disorder has an apparent seasonal interaction, becoming much worse in the spring and summer and subsiding in the cooler weather of the fall. Histologically, the epithelium in HBID is markedly thickened by the pathognomonic hyperplasia, hyperkeratosis, acanthosis, and contains individual cell dyskeratoses. Although the ocular and oral lesions associated with HBID can clinically resemble malignant neoplasms, the condition does not invade the underlying tissue and remains localized to the sites of the primary lesions.
Two families (DUK 5800 and DUK 5801) were ascertained through the Duke University Medical Center (DUMC) Department of Ophthalmology. DUK 5800 was initially described by Witkop et al. (Witkop et al. 1960) and later by Reed et al. (1979). A second family, DUK 5801, living in a neighboring county, was ascertained independently. After informed consent was obtained from all family members, individuals were examined by a board-certified ophthalmologist (R.R.A.). Clinical affection status was classified as follows: (1) “definite,” conjunctival erythema in both eyes and plaque >2 mm (greatest dimension) in each eye; (2) “probable,” conjunctival erythema in both eyes and plaque >2 mm in only a single eye; (3) “unknown,” conjunctival erythema, with or without plaque in either eye, or status not possible to determine by clinical examination; and (4) “normal,” no conjunctival abnormality.
Blood was obtained by venipuncture, and DNA was extracted using conventional methods (Ben Othmane et al. 1999). Genotyping was performed using the fluorescent allele static scanning technique (FASST) developed at the DUMC Center for Human Genetics (CHG) (Vance and Ben Othmane 1997). Radiation-hybrid scoring at the Whitehead Institute Web site was done using PCR on the GB4 hybrid panel for D4S1652 and D4S2390. Markers were run in duplicate.
All family-history, clinical, and genotyping data were maintained in the PEDIGENE database system (Haynes et al. 1995). Two-point and multipoint linkage analyses were performed using the Vitesse software package (O'Connell and Weeks 1995). The model linkage assumed autosomal dominant inheritance with a disease-allele frequency of .01. Estimation of the penetrance function was done by maximizing the log-likelihood with respect to the penetrance parameter (Speer 1998). Full penetrance was used for individuals classified as “definite” and 90% penetrance for individuals classified as “probable.” A 10% phenocopy rate was also included in the model for all susceptible individuals.
In addition, an “affecteds only” or low-penetrance model was performed, in which the disease phenotypes for at-risk family members were excluded while the marker genotypes of all family members were retained to maximize information on linkage phase. Markers were chosen from the Marshfield and Généthon genetic maps (D4S408–8cM–D4S3332–4cM–[D4S2283–0.0cM–D4S187–0.0cM–D4S2688–0.0cM–D4S2930–0.0cM–D4S1652–0.0cM–D4S2390]–4cM–D4S1523). Marker allele frequencies were generated from 38 unrelated American Indians living in the same general geographical region.
The two large families affected by HBID (DUK 5800 and 5801), according to genealogical data and self-report by family members, were not known to be related. Of 55 individuals studied, 25 were affected. Only two individuals met the classification of “probable.” Before evidence of linkage was found, 97 polymorphic markers were screened. A maximum combined LOD score of 7.32 was obtained at maximum recombination fraction (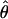 ) .00 between the disease locus and marker locus D4S1652, located on chromosome 4q35. Low-penetrance analysis confirmed these findings:
) .00 between the disease locus and marker locus D4S1652, located on chromosome 4q35. Low-penetrance analysis confirmed these findings: 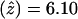 at
at 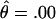 (data not shown). Subsequently, the families were genotyped with the surrounding markers D4S408, D4S3332, D4S2283, D4S18, D4S2688, D4S2930, and D4S1523. D4S1523 gave the highest score, with
(data not shown). Subsequently, the families were genotyped with the surrounding markers D4S408, D4S3332, D4S2283, D4S18, D4S2688, D4S2930, and D4S1523. D4S1523 gave the highest score, with 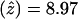 at
at 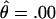 . Low-penetrance analysis of these data gave similar, although slightly less significant, LOD scores supporting the full-pedigree analysis of the data (data not shown). Multipoint analysis of the markers (D4S3332, D4S1652, D4S2390, and D4S1523) resulted in a peak LOD score of 11.58 at the D4S2390 (data not shown). Similar results were obtained when D4S1652 was substituted into the multipoint analysis for D4S2390.
. Low-penetrance analysis of these data gave similar, although slightly less significant, LOD scores supporting the full-pedigree analysis of the data (data not shown). Multipoint analysis of the markers (D4S3332, D4S1652, D4S2390, and D4S1523) resulted in a peak LOD score of 11.58 at the D4S2390 (data not shown). Similar results were obtained when D4S1652 was substituted into the multipoint analysis for D4S2390.
Examination of the marker data showed that two markers, D4S1652 and D4S2390, demonstrated three alleles in multiple affected individuals in both families. These markers are independent loci, with a GATA repeat as one marker and an ATA repeat comprising the other. The segregation of one of the markers in a portion of family DUK 5800 is shown in figure 1. Affected individuals not displaying three alleles received a copy of one of the two duplicated alleles from their unaffected parent (fig. 1). Since these individuals were homozygous for one of the two duplicated alleles, three alleles were not observed upon genotyping. Thus, these data are compatible with the segregation of a duplicated haplotype in the family. Haplotype analysis reveals that both families showed consistent segregation of the duplication and shared the same duplicated haplotype. Even though it was not possible to establish a documented link between the two families, the sharing of an identical duplicated haplotype strongly suggests that, as expected, they are derived from the same ancestral founder. The sizes, in base pairs, of the duplicated microsatellite haplotypes in both families are 150/142 for D4S1652 and 111/102 for D4S2390. For the linkage analysis, these duplicated haplotypes were run as a single, unique allele. To map the extent of the duplication, additional markers that were closely linked to D4D1652 and D4S2390 (D4S408, D4S2283, D4S187, D4S2688, D4S2930, D4S2299, and D4S1523) were also examined for evidence of duplication, but no individuals with three alleles were found for these markers. Also, three alleles for D4S2390 and D4S1652 were not found in any of the married-in or control samples. No parent-of-origin effects were seen in the segregation of the duplication. Radiation-hybrid screening demonstrated that D4S1652 and D4S2390 lie ∼6.4 cR3000 apart. This suggests a physical distance of ∼1.7 Mb between D4S2390 and D4S1652.
Figure 1.
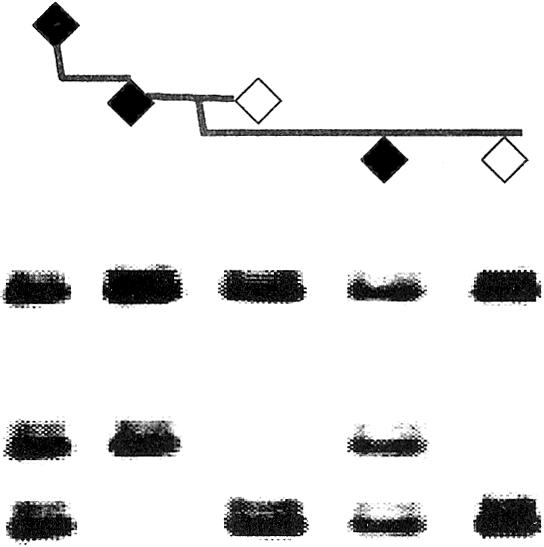
Segregation of the HBID duplication in three generations of the family DUK 5800. Affected individuals have blackened symbols.
The finding of three alleles segregating for the two markers D4S1652 and D4S2390 is very similar to the initial reports in the Charcot-Marie-Tooth disorder type 1 (CMT1A) (Couch et al. 1991; Raeymaekers et al. 1991) duplication. Whether the HBID duplication leads to similar regulatory abnormalities as seen in CMT1A (Timmerman et al. 1992) or causes a gene disruption has yet to be determined.
The most intriguing candidate gene in the region of the duplication is the human homolog of the FAT gene, an epithelial gene which can promote abnormal epithelial cell proliferation and functions as a tumor suppressor in Drosophila (Mahoney et al. 1991; Gray 1992; Dunne et al. 1995). This presents with a phenotype somewhat similar to HBID. This very large gene (15 kb) is thought to function in mammalian cell communication. This gene lies ∼7.4 cR (2.0 Mb) from D4S1652 on the Genemap 99 radiation-hybrid map.
In summary, we have localized the HBID gene to chromosome 4q35. Analysis has found a duplication segregating in the affected individuals that appears to lead to the abnormal cell proliferation in HBID patients.
Acknowledgments
We wish to thank the members of the families who participated in this study and the support personnel of the DUMC CHG. The study was supported by research grant R01-EY12012 from the National Eye Institute.
Electronic-Database Information
The accession number and URLs for data in this article are as follows:
- Center for Medical Genetics, Marshfield Medical Research Foundation, http://research.marshfieldclinic.org/genetics/ (for markers)
- Généthon, http://www.genethon.fr/genethon_en.html (for markers)
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for HBID [MIM 127600]) [PubMed]
References
- Ben Othmane K, Johnson E, Menold M, Graham FL, Barker J, Ben Hamida M, Hasewaga O, Rogala AO, Ohnishi A, Pericak-Vance MA, Hentati F, Vance JM (1999) Identification of a new locus for autosomal recessive Charcot-Marie-Tooth disease with focally folded myelin on chromosome 11p15. Genomics 62:344–349 [DOI] [PubMed] [Google Scholar]
- Couch FJ, McCarthy TV, Gregg RG, Hogan K (1991) Dinucleotide repeat polymorphism at the D17S518 locus. Nucleic Acids Res 19:5093 [PMC free article] [PubMed] [Google Scholar]
- Dunne J, Hanby AM, Poulsom R, Jones TA, Sheer D, Chin WG, Da SM Zhao Q, Beverly PCL, Owen MJ (1995) Molecular cloning and tissue expression of FAT, the human homologue of the Drosophila fat gene that is located on chromosome 4q34-q35 and encodes a putative adhesion molecule. Genomics 30:207–223 [DOI] [PubMed] [Google Scholar]
- Gray MR (1992) Detection of DNA sequence polymorphisms in human genomic DNA by using denaturing gradient gel blots. Am J Hum Genet 50:331–346 [PMC free article] [PubMed] [Google Scholar]
- Haynes C, Speer MC, Peedin M, Roses AD, Haines JL,Vance JM, Pericak-Vance MA (1995) PEDIGENE: a comprehensive data management system to facilitate efficient and rapid disease gene mapping. Am J Hum Genet Suppl 57:A193 [Google Scholar]
- Mahoney PA, Weber U, Onofrechuk P, Biessmann H, Bryant PJ, Goodman CS (1991) The fat tumor suppressor gene in Drosophila encodes a novel member of the cadherin gene superfamily. Cell 67:853–868 [DOI] [PubMed] [Google Scholar]
- McLean IW, Riddle PJ, Schruggs JH, Jones DB (1981) Hereditary benign intraepithelial dyskeratosis: a report of two cases from Texas. Ophthalmology 88:164–168 [DOI] [PubMed] [Google Scholar]
- O'Connell JR, Weeks DE (1995) The VITESSE algorithm for rapid exact multilocus linkage analysis via genotype set-recoding and fuzzy inheritance. Nat Genet 11:402–408 [DOI] [PubMed] [Google Scholar]
- Raeymaekers P, Timmerman V, Nelis E, De Jonghe P, Hoogendijk JE, Baas F, Barker DF Martin JJ, De Visser M, Bolhuis PA, Van Broeckhoven C, HMSN, Collaborative Research Group (1991) Duplication in chromosome 17p11.2 in Charcot-Marie-Tooth neuropathy type 1a (CMT1a). Neuromuscul Disord 1:93–97 [DOI] [PubMed] [Google Scholar]
- Reed JW, Cashwell F, Klintworth GK (1979) Corneal manifestations of hereditary benign intraepithelial dyskeratosis. Arch Ophthalmol 97:297–300 [DOI] [PubMed] [Google Scholar]
- Shields CL, Shields JA, Eagle RC (1987) Hereditary benign intraepithelial dyskeratosis. Arch Ophthalmol 105:422–423 [DOI] [PubMed] [Google Scholar]
- Speer MC (1998) Basic concepts in genetics. In: Pericak-Vance MA, Haines JL (eds) Approaches to gene mapping in complex human diseases. J Wiley & Sons, New York, pp 17–52 [Google Scholar]
- Timmerman V, Nelis E, Van Hul W, Nieuwenhuijsen BW, Chen KL, Wang S, Ben Othmane K, Cullen B, Leach RJ, Hanemann CO, De Jonghe P, Raeymaekers P, van Ommen GJB, Martin JJ, Muller HW, Vance JM, Fischbeck KH, Van Broeckhoven C (1992) The peripheral myelin protein gene PMP-22 is contained within the Charcot-Marie-Tooth disease type 1A duplication. Nat Genet 1:171–175 [DOI] [PubMed] [Google Scholar]
- Vance JM, Ben Othmane K (1998) Methods in genotyping. In: Pericak-Vance MA, Haines JL (eds) Approaches to gene mapping in complex human diseases. J. Wiley and Sons, New York, pp 213–228 [Google Scholar]
- von Sallmann L, Paton D (1959) Hereditary dyskeratosis of the perilimbal conjunctiva. Trans Am Ophthalmol Soc 57:53–62 [PMC free article] [PubMed] [Google Scholar]
- von Sallmann L, Paton D (1960) Hereditary benign intraepithelial dyskeratosis. I. Ocular manifestations. Arch Ophthalmol 63:421–429 [Google Scholar]
- Witkop CJ, Shankle CH, Graham JB, Murray MR, Rucknagel DL, Byerly BH (1960) Hereditary benign intraepithelial dyskeratosis. Arch Pathol 70:696–711 [PubMed] [Google Scholar]
- Yanoff M (1968) Hereditary benign intraepithelial dyskeratosis. Arch Ophthalmol 79:291–293 [DOI] [PubMed] [Google Scholar]