

Abstract
We measured the proportion of mutant mtDNA (mutation load) in 82 primary oocytes from a woman who harbored the A3243G mtDNA mutation. The frequency distribution of mutation load indicates that random drift is the principal mechanism that determines the level of mutant mtDNA within individual oocytes.
Pathogenic mtDNA mutations are present in ∼1/8,000 members of the U.K. population (Chinnery et al. 2000a) and often cause progressive disabling neurological disease (Leonard and Schapira 2000). These mutations are often heteroplasmic, with a mixture of mutant and wild-type mtDNA present within a single individual (Larsson and Clayton 1995). For the most common point mutations, the presence of a higher percentage of mutant mtDNA (mutation load) is associated with a more severe clinical phenotype (Chinnery et al. 1997; White et al. 1999b).
For reasons that are not well understood, mtDNA deletions are rarely, if ever, transmitted from clinically affected women to their offspring. By contrast, women who harbor heteroplasmic mtDNA point mutations or mtDNA duplications transmit a variable amount of mutant mtDNA (Poulton et al. 1998). This creates a problem in the clinic, because it is difficult to predict the outcome of pregnancy. As illustrated in figure 1, the same mother may have offspring with a range of clinical features, from a severely affected fetus that dies in utero to a mildly affected child who survives well into adult life (for other examples, see Ciafaloni et al. 1992; de Vries et al. 1994; White et al. 1999b). For some mtDNA mutations there is a relationship between mutation load in the mother and the outcome of pregnancy (Chinnery et al. 1998; White et al. 1999a), but very little is known about the mechanisms of transmission of heteroplasmic mtDNA mutations in humans.
Figure 1.

Previously unpublished pedigree of a family from the northeast of England, showing the clinical variation among the offspring of a woman who transmitted the heteroplasmic A3243G mtDNA point mutation (MIM 540000). Individual I:2 had offspring that died either in utero (individuals II:1, II:2, and II:4) or <2 h after birth (individual II:3). In addition, individual I:2 had a severely affected child with short stature and bilateral sensorineural deafness who developed a recurrent encephalopathy, strokelike episodes, seizures, ataxia, diabetes mellitus, and a cardiomyopathy in early adult life (individual II:6, now 31 years old); and she had offspring who remain asymptomatic in adult life (individual II:5 [27 years old] and individual II:7 [33 years old]). TM = trimester.
Heteroplasmic female mice transmit neutral heteroplasmic mtDNA polymorphisms to their offspring, and the variation among the offspring is largely determined by random genetic drift (Jenuth et al. 1996). It is thought that a restriction in the number of mtDNA molecules early in embryogensis (the mitochondrial “genetic bottleneck”) is behind this process (Poulton et al. 1998). Although no mouse model transmits a pathogenic heteroplasmic mtDNA mutation, Inoue et al. (2000) recently generated matrilineal pedigrees of mice with high levels of a heteroplasmic mtDNA deletion. However, these mice also harbor mtDNA duplications, and it has yet to be determined whether the duplications or the deletions themselves are transmitted in the female germline (Jacobs 2000). Further studies of these mice will undoubtedly cast light on the mechanisms of inheritance of mtDNA heteroplasmy, but it remains to be established whether random drift is the principal mechanism behind the varied transmission of heteroplasmy in human pedigrees with mtDNA disease.
Attempts to study the inheritance of mtDNA heteroplasmy in humans have been hampered by a number of problems. Very few human pedigrees are large enough to permit reliable calculations of the variance between offspring. Ovarian hyperstimulation may yield many oocytes; however, because they have been artificially selected, their mutation load may not reflect the level of the whole population of primary oocytes. In addition, the variable segregation of mutant mtDNA among various tissues throughout embryonic development—and changes in the level of mutant mtDNA in various tissues throughout life—may confound any clear relationship between maternal mutation load and the level found in the offspring themselves.
To advance our understanding of the transmission of pathogenic heteroplasmic mtDNA mutations in humans, we performed the first study of mutant mtDNA within a large number of single primary oocytes from a woman who harbored the most common inherited heteroplasmic mtDNA mutation: A3243G. This mutation classically presents with mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS) (MIM 540000) (Goto et al. 1990), but it may also cause diabetes, cardiomyopathy, sensorineural deafness, and ophthalmoplegia (Leonard and Schapira 2000). If the level of mtDNA heteroplasmy in single oocytes were determined by random genetic drift, one would expect the frequency distribution of oocyte mutation load to be binomial (Poulton et al. 1998).
The patient was a 41-year-old mother of two sons who was known to have the A3243G mutation although she had no features of mitochondrial disease. (This woman is not part of the family shown in figure 1.) She underwent a total abdominal hysterectomy and bilateral salpingo-oophorectomy for endometriosis, and she gave informed consent for collection of ovarian wedge samples. Samples were frozen in liquid nitrogen immediately after resection, and 10-μm frozen sections were cut perpendicular to the serosal surface. The first section was stained with hematoxylin and eosin (H&E), to allow tissue orientation, and adjacent sections were dried in air, without staining or fixation. Primary oocytes were identified on the H&E section and then on the adjacent unstained sections, by phase-contrast microscopy. Eighty-two individual primary oocytes were dissected using a 10-μm glass micropipette. They were then lysed, and the proportion of mutant mtDNA within each oocyte was determined by last-cycle fluorescent PCR, as described elsewhere (Chinnery et al. 2000b).
The level of the A3243G mutation was 18.11% in her quadriceps and 7.24% in her leukocytes. The proportion of mutant mtDNA within the primary oocytes was 0%–45% (mean 12.64%). The shape of the frequency distribution for oocyte mutation load corresponded to a binomial distribution (fig. 2). The median level (8.18%) fell within the 95% confidence limits for the sample mean for the binomial distribution, indicating that the number of oocytes with mutation loads greater than the mean was approximately equal to the number with mutation loads less than the mean.
Figure 2.

Frequency distribution for the percentage of the A3243G mutation in 82 primary oocytes selected at random from an unstained cryostat section of freshly frozen ovarian tissue. The proportion of mutant mtDNA within the primary oocytes was 0%–45% (mean 12.64%; median 8.18%).
These findings indicate that, as in mice (Jenuth et al. 1996), the level of mutant mtDNA in human primary oocytes is largely determined by random genetic drift. The continuous distribution of mutation load shown in figure 2 contrasts with the findings by Blok et al. (1997), who showed highly skewed segregation of the T8993G mutation in oocytes harvested from a woman after ovarian follicular hyperstimulation. It is possible that the extreme levels of heteroplasmy noted by Blok et al. (1997) arose through the random sampling of only seven oocytes. This is unlikely, because they did not detect intermediate levels of heteroplasmy—each oocyte that they collected contained either 100% wild-type or >95% mutant mtDNA. It is well recognized that the rate of segregation of the T8993G/C mutations differs markedly from that of most other heteroplasmic mtDNA point mutations (Poulton and Turnbull 2000). This is reflected in the size of the pedigrees: T8993G/C families are usually small, because of the extremely rapid segregation of mutant mtDNA, leading, within one or two generations (White et al. 1999a, 1999b), to fixation or loss of the new mutation. Less-dramatic shifts in mutation load are seen in larger pedigrees, which transmit other mtDNA point mutations, including A3243G (Chinnery et al. 2000c). Because the variation in heteroplasmy between the offspring of a woman is largely generated before the primary-oocyte stage (Jenuth et al. 1996), the differences in segregation patterns seen in pedigrees is reflected by the level of mutant mtDNA measured in the harvested oocytes reported by Blok et al. (1997) and in the primary oocytes reported here.
The measurements that we made on primary oocytes can be used to estimate the size of the mitochondrial genetic bottleneck. Various authors have used the population-genetic-bottleneck model developed by Wright (1969) to study the mitochondrial genetic bottleneck (Howell et al. 1992; Jenuth et al. 1996; Poulton et al. 1998). This model assumes that a population of infinite size is instantaneously restricted to a minimum bottleneck size (N) for a number of generations (g) before the population instantaneously expands back to an infinite size. Under these circumstances, the size and duration of the bottleneck determine the variation in the genotypes among the subsequent lineages. The size of the bottleneck (measured in “segregating units”) can be calculated—provided that the duration of the bottleneck is known—according to the following equation:
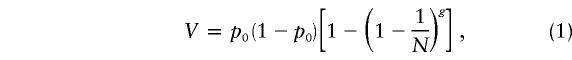 |
where V is the variation between offspring, and p0 is the initial allele frequency. In humans, g is generally taken to be the 24 synchronous cell divisions needed to produce a full quota of 8.3 million primary oocytes from a single progenitor cell (Baker 1963). An alternative approach is to assume that a single sampling event occurs once during development (Bendall et al. 1996). This greatly simplifies the calculation, which can be modeled using equation (1), where g = 1 (Poulton et al. 1998). We have applied both models to the primary-oocyte data presented here, assuming that the mean level of the A3243G mutation for the 82 oocytes corresponds to the initial allele frequency, p0, ( p0=.1264), and that V is the variance between the individual primary oocytes (V=.014321). When equation (1) with g = 24 is used, the estimated bottleneck size, as based on the primary-oocyte data, is 173 segregating units; when the single-sampling approach is used, the estimated bottleneck size is 8 segregating units. These figures closely correspond to values for human pedigrees reported elsewhere (Howell et al. 1992; Jenuth et al. 1996; Poulton et al. 1998), but they must be interpreted with caution. The size of the segregating unit is not known, and it may correspond to a single mitochondrial genome or to groups of mtDNA molecules arranged in clusters, possibly compartmentalized within nucleoids or discrete mitochondria. Although these models do not tell us about the precise biological mechanisms involved in the inheritance of mtDNA heteroplasmy, the calculations are of value in the comparison of data obtained from various sources. The mutation-load variation among primary oocytes of our patient is of the same order of magnitude as that between blood samples drawn from members of human pedigrees transmitting the same or other mutations (Chinnery et al. 2000c).
It is of interest that the level in the blood of our patient’s 15-year-old son was 11.70%, a value that fell just within the 59% confidence interval of the mutation-load distribution of the oocytes. Although the relationship between oocyte mutation load and the outcome of pregnancy remains to be determined, it is significant that 8 (9.8%) of the 82 oocytes collected from our patient contained undetectable levels of mutant mtDNA. It is well recognized that women with heteroplasmic mtDNA point mutations may have unaffected offspring (fig. 1; also see Ciafaloni et al. 1992; Hammans et al. 1993; White et al. 1999b). On the basis of our observations, it is likely that the patient whom we describe here had a 1/10 chance of not transmitting the mtDNA mutation to her offspring. This information could not be confidently determined by measuring the proportion of mutant mtDNA in tissue from any organ other than the ovary. Further studies will cast light on the relationship between the percentage of mutant mtDNA in oocytes and that in other tissues. Alternatively, under certain circumstances, an ovarian biopsy may yield information with important implications for a prospective mother.
In conclusion, although we cannot be certain that all the primary oocytes that we collected would necessarily develop into mature oocytes, we have provided evidence that the level of the A3243G mutation within human primary oocytes is determined by random genetic drift. It is possible that higher levels of the A3243G mutation may skew this distribution, particularly if they exceed the critical threshold (>85%) of mutant mtDNA needed to produce a biochemical defect in transmitochondrial cybrids (Chomyn et al. 1991). However, data from a large number of independent pedigrees suggest that this is not likely to have a major effect on the overall transmission pattern for this and other mutations (Chinnery et al. 2000c). We suspect that random genetic drift is the principal mechanism behind the transmission of all pathogenic heteroplasmic mtDNA point mutations. It is likely that differences in the segregation pattern and genetic threshold contribute to the specific inheritance patterns of various mtDNA defects.
Acknowledgments
P.F.C., D.T.B., and D.M.T. are supported by the Wellcome Trust. We are grateful for the help of M. A. Johnson.
Electronic-Database Information
The accession number and URL for data in this article are as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for MELAS [MIM 540000])
References
- Baker TG (1963) A quantitative and cytological study of germ cells in human ovaries. Proc R Soc Lond B Biol Sci 158:417–423 [DOI] [PubMed] [Google Scholar]
- Bendall KE, Macaulay VA, Baker JR, Sykes BC (1996) Heteroplasmic point mutations in the human mtDNA control region. Am J Hum Genet 59:1276–1287 [PMC free article] [PubMed] [Google Scholar]
- Blok RB, Gook DA, Thorburn DR, Dahl HH (1997) Skewed segregation of the mtDNA nt 8993 (T→G) mutation in human oocytes. Am J Hum Genet 60:1495–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery P, Howell N, Lightowlers R, Turnbull D (1997) Molecular pathology of MELAS and MERRF: the relationship between mutation load and clinical phenotype. Brain 120:1713–1721 [DOI] [PubMed] [Google Scholar]
- ——— (1998) MELAS and MERRF: the relationship between maternal mutation load and the frequency of clinically affected offspring. Brain 121:1889–1894 [DOI] [PubMed] [Google Scholar]
- Chinnery PF, Johnson MA, Wardell TM, Singh-Kler R, Hayes C, Brown DT, Taylor RW, Bindoff LA, Turnbull DM (2000a) Epidemiology of pathogenic mitochondrial DNA mutations. Ann Neurol 48:188–193 [PubMed] [Google Scholar]
- Chinnery PF, Taylor DJ, Brown DT, Manners D, Styles PJ, Lodi R (2000b) Very low levels of the mtDNA A3243G mutation associated with mitochondrial dysfunction in vivo. Ann Neurol 47:381–384 [PubMed] [Google Scholar]
- Chinnery PF, Thorburn DR, Samuels DC, White SL, Dahl HH, Turnbull DM, Lightowlers RN, Howell N (2000c) The inheritance of mtDNA heteroplasmy: random drift, selection or both? Trends Genet 16:500–505 [DOI] [PubMed] [Google Scholar]
- Chomyn A, Meola G, Bresolin N, Lai ST, Scarlato G, Attardi G (1991) In vitro genetic transfer of protein synthesis and respiration defects to mitochondrial DNA-less cells with myopathy-patient mitochondria. Mol Cell Biol 11:2236–2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciafaloni E, Ricci E, Shanske S, Moraes CT, Silvestri G, Hirano M, Simonetti S, Angelini C, Donati MA, Garcia C, Martinuzzi A, Mosewich R, Servidei S, Zammarchi E, Bonilla E, DeVivo DC, Rowland LP, Schon EA, DiMauro S (1992) MELAS: clinical features, biochemistry, and molecular genetics. Ann Neurol 31:391–398 [DOI] [PubMed] [Google Scholar]
- de Vries D, de Wijs I, Ruitenbeek W, Begeer J, Smit P, Bentlage H, van Oost B (1994) Extreme variability of clinical symptoms among sibs in a MELAS family correlated with heteroplasmy for the mitochondrial A3243G mutation. J Neurol Sci 124:77–82 [DOI] [PubMed] [Google Scholar]
- Goto Y, Nonaka I, Horai S (1990) A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 348:651–653 [DOI] [PubMed] [Google Scholar]
- Hammans SR, Sweeney MG, Brockington M, Lennox GG, Lawton NF, Kennedy CR, Morgan-Hughes JA, Harding AE (1993) The mitochondrial DNA transfer RNA(Lys) A→G(8344) mutation and the syndrome of myoclonic epilepsy with ragged red fibres (MERRF): relationship of clinical phenotype to proportion of mutant mitochondrial DNA. Brain 116:617–632 [DOI] [PubMed]
- Howell N, Halvorson S, Kubacka I, McCullough DA, Bindoff LA, Turnbull DM (1992) Mitochondrial gene segregation in mammals: is the bottleneck always narrow? Hum Genet 90:117–120 [DOI] [PubMed] [Google Scholar]
- Inoue K, Nakada K, Ogura A, Isobe K, Gota YI, Nonaka I, Hayashi JI (2000) Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nat Genet 26:176–181 [DOI] [PubMed] [Google Scholar]
- Jacobs H (2000) A mouse model of mtDNA disease. Trends Genet 16:487 [DOI] [PubMed] [Google Scholar]
- Jenuth J, Peterson AC, Fu K, Shoubridge EA (1996) Random genetic drift in the female germ line explains the rapid segregation of mammalian mitochondrial DNA. Nat Genet 14:146–151 [DOI] [PubMed] [Google Scholar]
- Larsson N-G, Clayton DA (1995) Molecular genetic aspects of human mitochondrial disorders. Annu Rev Genet 29:151–178 [DOI] [PubMed] [Google Scholar]
- Leonard JV, Schapira AVH (2000) Mitochondrial respiratory chain disorders. I. Mitochondrial DNA defects. Lancet 355:299–304 [DOI] [PubMed] [Google Scholar]
- Poulton J, Macaulay V, Marchington DR (1998) Mitochondrial genetics '98: is the bottleneck cracked? Am J Hum Genet 62:752–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulton J, Turnbull DM (2000) 74th ENMC International Workshop: Mitochondrial Diseases. Neuromuscul Disord 10:460–462 [DOI] [PubMed] [Google Scholar]
- White SL, Collins VA, Woolfe R, Cleary MA, Shanske S, DiMauro S, Dahl HM, Thorburn DR (1999a) Genetic counseling and prenatal diagnosis for the mitochondrial DNA mutations at nucleotide 8993. Am J Hum Genet 65:474–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White SL, Shanske S, McGill JJ, Mountain H, Geraghty MT, DiMauro S, Dahl H-HM, Thorburn DR (1999b) Mitochondrial DNA mutations at nucleotide 8993 show a lack of tissue of age-related variation. J Inherit Metab Dis 22:899–914 [DOI] [PubMed] [Google Scholar]
- Wright S (1969) Theory of gene frequencies. Vol 2. In: Evolution and the genetics of populations. University of Chicago Press, Chicago [Google Scholar]