

Abstract
Menkes disease and occipital horn syndrome (OHS) are allelic, X-linked recessive copper-deficiency disorders resulting from mutations in ATP7A, or MNK. Classic Menkes disease has a severe phenotype, with death in early childhood, whereas OHS has a milder phenotype, with, mainly, connective-tissue abnormalities. Data suggest that steady-state localization of ATP7A to the trans-Golgi network (TGN) is necessary for proper activity of lysyl oxidase, which is the predominant cuproenzyme whose activity is deficient in OHS and which is essential for maintenance of connective-tissue integrity. Recently, it was reported that ATP7A-transcript levels as low as 2%–5% of normal are sufficient to result in the milder phenotype, OHS, rather than the phenotype of Menkes disease. In contrast to previously reported cases of OHS, we describe a case of OHS in which, because of a frameshift mutation, no normal ATP7A is produced. Although abundant levels of mutant transcript are present, there are substantially reduced levels of the truncated protein, which lacks the key dileucine motif L1487L1488. It has been demonstrated that the dileucine motif L1487L1488 functions as an endocytic signal for ATP7A cycling between the TGN and the plasma membrane. The present report is the first to describe an ATP7A truncation that results in OHS rather than in Menkes disease. The data from the present report support the concepts that (1) OHS results from lower levels of functional ATP7A and (2) ATP7A does not require the dileucine motif to function in copper efflux.
Menkes disease (MIM 309400) and occipital horn syndrome (OHS [MIM 304150]) are allelic, X-linked recessive copper-deficiency disorders (Levinson et al. 1993; Das et al. 1995) resulting from mutations in ATP7A, or MNK (GenBank accession number NM_000052) (Chelly et al. 1993; Mercer et al. 1993; Vulpe et al. 1993). ATP7A contains 23 exons that span a genomic region of ∼140 kb (Dierick et al. 1995; Tümer et al. 1995) and has a predominant transcription product of ∼8.5 kb (Chelly et al. 1993; Mercer et al. 1993; Vulpe et al. 1993). ATP7A is ∼170 kD in size and is a member of the P-type–ATPase family (Chelly et al. 1993; Mercer et al. 1993; Vulpe et al. 1993). P-type ATPases transport cations across cellular membranes by using energy generated by ATP hydrolysis (Pedersen et al. 1987). ATP7A functions (a) to transport copper from the gastrointestinal tract into the bloodstream, (b) to efflux excess copper from the cell, and (c) for intracellular delivery, within the secretory pathway, of copper to cuproenzymes (for reviews, see Kaler 1998; Camakaris et al. 1999; Harris 2000). At basal intracellular copper levels, the steady-state localization of ATP7A is to the trans-Golgi Network (TGN) (Petris et al. 1996; Yamaguchi et al. 1996; Dierick et al. 1997). However, in an environment with excess copper, ATP7A is transported, via secretory vesicles, to the plasma membrane, where it functions to efflux copper from the cell (Petris et al. 1996, 1999; La Fontaine et al. 1998). In response to intracellular copper levels, it is believed that ATP7A cycles between the TGN and the plasma membrane. The dileucine motif L1487L1488 functions as an endocytic signal for ATP7A cycling (Petris et al. 1998, 1999; Francis et al. 1999) and is necessary for internalization of the protein from the plasma membrane (Francis et al. 1999). However, data from Petris et al. (1998) suggest that the dileucine motif L1487L1488 is not necessary for ATP7A to function in copper efflux.
Classic Menkes disease is a rare multisystem disorder with a severe phenotype including neonatal neurological degeneration, hypopigmentation, skin and joint laxity, coarse and twisted hair (pili torti), hypothermia, seizures, failure to thrive, and death typically by the age of 3 years (Menkes et al. 1962). In patients with Menkes disease, abnormal, or nonfunctional, ATP7A results in decreased intestinal absorption of copper, which leads to decreased copper in the plasma, liver, and brain (for review, see Kaler 1998). The clinical phenotype of Menkes disease can be attributed to the reduced activity of several cuproenzymes, such as lysyl oxidase, tyrosinase, copper/zinc superoxide dismutase, dopamine β-hydroxylase, and cytochrome c oxidase (Menkes 1988). In addition, it has been demonstrated that there is an accumulation of copper in certain tissues and cell types, which indicates that ATP7A in these patients is not functioning properly to efflux copper from the cell (for review, see Kaler 1998). Thus, at the tissue level, Menkes disease can be considered a disorder of copper maldistribution.
OHS, formerly known as “X-linked cutis laxa” or “Ehlers-Danlos syndrome type 9,” has a phenotype milder than that of Menkes disease, mainly composed of connective-tissue manifestations, including skin laxity, hyperextensible joints, bladder diverticula, characteristic occipital exostoses, and, occasionally, mild neurological involvement (Lazoff et al. 1975; Byers et al. 1980; for review, see Tsukahara et al. 1994). Byers et al. (1980) demonstrated that patients with OHS have decreased activity of the cuproenzyme lysyl oxidase, which functions in the initiation of cross-linking of both collagen and elastin. It has been hypothesized that OHS results from the presence of low levels of functional ATP7A, unlike Menkes disease, in which no normal ATP7A activity exists (Das et al. 1995). This hypothesis has been supported by a recent study that demonstrates that ATP7A-transcript levels as low as 2%–5% of normal are sufficient to result in the milder phenotype, OHS, as opposed to Menkes disease (Møller et al. 2000). It has been postulated that lysyl oxidase is more sensitive than other cuproenzymes to copper deficiency, thus resulting in decreased lysyl oxidase activity in patients with OHS (Das et al. 1995; Levinson et al. 1996).
There are 35–40 known cases of OHS worldwide (Møller et al. 2000), but mutations in only 8 cases have been elucidated at the molecular level (table 1). In seven of these eight cases, the mutation leads to aberrant splicing of the ATP7A transcript (Kaler et al. 1994; Das et al. 1995; Ronce et al. 1997; Qi and Byers 1998; Møller et al. 2000; Gu et al. 2001). In four of the cases of OHS with a splice mutation, low levels of normal transcript have been detected (Levinson et al. 1993; Das et al. 1995; Møller et al. 2000; Gu et al. 2001). Missense mutations in two reported cases of OHS resulted in aberrant splicing of the transcript with an amino acid change in the normal transcript, which did not adversely affect the function of ATP7A (Kaler et al. 1994; Ronce et al. 1997). Qi and Byers (1998) did not detect normal ATP7A transcript by reverse-transcription PCR (RT-PCR) in a family with OHS with a splice mutation in intron 10. However, it is possible that the affected members of that family could produce a low level of normal ATP7A, similar to that produced in affected members of a family described by Møller et al. (2000). In addition, Levinson et al. (1996) described a patient with OHS who did not have a mutation in the coding region of ATP7A but, instead, had a 98-bp deletion in the regulatory region of ATP7A. Northern analysis indicated normal transcript levels; however, a chloramphenicol acetyltransferase (CAT) reporter assay suggested that the promoter deletion would result in decreased expression of ATP7A.
Table 1.
Summary of OHS Mutations
| Mutationa | SpliceMutation? | SpliceSite | mRNA | Source(s) |
| −403del98nt | No | … | Deletion in regulatory region, normal transcript level, reduced CAT activity | Levinson et al. (1996) |
| IVS6+2del(taag) | Yes | Donor | Exon 6 skipped→frameshift→PTC, reduced normal transcript level | Møller et al. (2000), Gu et al. (2001) |
| 2055C→T | Yes | … | Exon 8 skipped→frameshift→PTC, exons 8–10 skipped, reduced transcript level with S637L | Ronce et al. (1997) |
| IVS10+3a→t | Yes | Donor | Exon 10 skipped→in-frame deletion→deletion of transmembrane domains 3 and 4 in protein, abundant abnormal transcript level | Qi and Byers (1998) |
| 2642A→G | Yes | Donor | Exon 11 skipped→frameshift→PTC, exons 11 and 12 skipped→frame-shift→PTC, reduced transcript level with S833G | Kaler et al. (1994) |
| IVS14−4a→g | Yes | Acceptor | Exon 15 skipped→in-frame deletion→deletion of transduction domain, abundant abnormal transcript level, reduced normal transcript level | Das et al. (1995) |
| IVS17+5g→a | Yes | Donor | Exon 17 skipped→frameshift→PTC, reduced normal transcript level | Das et al. (1995), Levinson et al. (1993) |
| 4497–4499delG | No | … | Frameshift→PTC, abundant abnormal transcript level | Present study |
IVS = intervening sequence.
In the present report, we describe a frameshift mutation in a family with classic OHS. The mutation is novel—that is, it does not result in aberrant splicing of the transcript, as has been seen in seven of the eight previously reported mutations. In our case, no normal ATP7A mRNA is transcribed, owing to a frameshift mutation at codon 1451, which leads to premature truncation of the predicted protein. Because the protein was truncated prior to L14871488 in the carboxy terminus, it provides insight as to the importance, with respect to phenotype, of this signal.
The pedigree of this family with classic OHS reveals a pattern of affected individuals that is consistent with X-linked inheritance (fig. 1). The proband (III-2) sat without assistance at the age of 7 mo and was able to crawl at the age of 7.5 mo. On examination, he exhibited multiple bladder diverticula, renal calculus, vesicoureteral reflux, bilateral inguinal hernia repair, neurogenic bladder, genu valgum, and pectus excavatum; he also had mildly hyperelastic skin, especially over the abdomen, and required special education. He did not exhibit chronic diarrhea, orthostatic hypotension, or dysautonomic symptoms. A skeletal survey of this individual revealed bilateral occipital horns, mild lower-thoracic and lumbar platyspondyly, marked pectus excavatum, broad scapular necks, clavicular handlebar/hammer contour, humeral and femoral diaphyseal wavy contour, bulbous ulnar-coronoid and radial bowing of the forearms, rounded iliac-wing contour with broadening in the medial/lateral dimension, bilateral coxa valga, and minimal dextroconvex scoliosis from T4 to L4. At the age of 8 years, III-2's serum copper level was slightly low, at 60 μg/dl (normal range 70–150 μg/dl), as was the serum ceruloplasmin level, which was 18.9 mg/dl (normal range 20–42 mg/dl). At the age of 10 years 9 mo, he was given the Woodcock-Johnson Tests of Cognitive Ability, the Woodcock-Johnson Tests of Achievement, the Wechsler Individual Achievement Test, the Bender Gestalt Test, and the Human Figure Drawing Task. For the Wechsler Individual Achievement Test, he had scores that were age equivalent to 8 years 3 mo in basic reading, 8 years in math reasoning, and 8 years 6 mo in spelling. He had a standard score of 84 for the Woodcock-Johnson Tests of Cognitive Ability, which is in the low-average range. III-2's affected brother (III-3), maternal uncle (II-10), and cousin (II-5) were similarly affected but with slight variability in severity. The pattern of inheritance and the clinical findings were consistent with a diagnosis of OHS.
Figure 1.

Pedigree, consistent with X-linked inheritance, of the family with OHS. Family members included in this study were II-6, III-1, III-2, and III-3. Blackened squares indicate affected males, and unblackened squares indicate unaffected males. Circles with a dot indicate carrier females, and circles without a dot indicate either unaffected or untested females.
After obtaining informed consent, we collected skin biopsies and blood samples from III-2, III-3, III-2's unaffected brother (III-1), and their carrier mother (II-6), in accordance with the procedures of the institutional review boards of the University of Michigan Medical School. Fibroblast and Epstein-Barr virus–transformed lymphoblastoid cell lines were generated and cultured according to standard procedures.
To determine whether, in our case, the mutation involved a splice site, we performed RT-PCR. PCR products were generated using cDNA reverse transcribed from total RNA (SuperscriptII Reverse Transcriptase; GibcoBRL) and eight primer pairs that spanned the coding region of ATP7A. No novel aberrant splicing was detected by RT-PCR (not shown), suggesting that in both III-2 and III-3 the mutation was not due to a splice mutation.
To identify the mutation, we sequenced ATP7A at the genomic level. By use of the DNAeasy Tissue Kit (Qiagen), genomic DNA was isolated from lymphoblastoid cell lines from each family member studied and from an unrelated normal male control cell line. PCR products for each exon were generated from the genomic DNA of both III-2 and the control by intronic primer pairs for each of the 23 exons and the 98-bp triple repeat in the promoter region (table 2). PCR products were subcloned into TA-cloning vector pCR2.1 (Invitrogen) and were sequenced, by use of an ABI model 373XL automated sequencer, by the University of Michigan DNA Sequencing Core. II-6, III-1, III-2, and III-3 all had the 98-bp triple repeat in the promoter region, indicating that the mutation in this family was not similar to the OHS-promoter mutation described by Levinson et al. (1996). A frameshift mutation was identified in exon 23 of III-2 and, subsequently, in both III-3 and II-6. A single guanine nucleotide was deleted from a guanine triplet (fig. 2, black background) at nucleotides 4497–4499 of ATP7A, in III-2 and III-3 and in one allele of II-6; an unrelated normal control and III-1 and the other allele of II-6 all contained the guanine triplet at these nucleotide positions. Conceptual translation of III-2's ATP7A gene (fig. 2, mutant, gray background) shows a frameshift at codon 1451, resulting in a carboxy terminus with 13 novel amino acids before premature truncation of the protein. The truncated protein lacks the dileucine motif L1487L1488 (fig. 2, wt, underlined), which functions as an endocytic signal (Petris et al. 1998; Francis et al. 1999).
Table 2.
Primer Pairs for Sequencing ATP7A Exons and Promoter Region
|
Primera(5′→3′) |
|||
| Region/Exon | Forward | Reverse | Size(bp) |
| Promoter | tcaatggagggaagaggat | gaaaagagagcgactggtca | 579 |
| 1 | tgaggtgttatgagtttcttcacaagc | gaatcacagcattctttcaagaggtc | 312 |
| 2 | aggggaaaagttgaggaaag | ggcatttgacttcagtttcca | 241 |
| 3 | cttcttgaatgtggtgtgatg | gccttacaaaacaatcaacacc | 588 |
| 4 | caacagaatgttttctgaagtagccc | gcaaaagtggaattacagtgcctatg | 904 |
| 5 | aaggatgcaattgaatgatcaatactg | cattactgaaaatcatgaggccttacc | 384 |
| 6 | ttatgaagcaaggtgataggtacgtca | tttaaattccatgcttgaagagtacca | 606 |
| 7 | ttaagatcttgtgtggcaaacc | aaccagaaatgattcacgggagt | 615 |
| 8 | aaagattggaaacaaatgcaatcaag | ttgtgaccatttcatccagtataagga | 356 |
| 9 | tccttatactggatgaaatggtcacaa | tctttcttaggggttaaatggcatcta | 534 |
| 10 | gcttgtgcagcaatttgaatacct | aagcatgtatttccaatgattggc | 282 |
| 11 | gctgtcttttgggtaaatgctaaagat | gacaaaatggatagatgaagtatggca | 351 |
| 12 | cttccattggcagtaattatggagc | gccacaaagtaaatctgaggaaattg | 402 |
| 13 | aaggtttatattcctggcattggc | ttgtctgcaaactgctggatagg | 909 |
| 14 | aagaggcacaaacatcaaaggtaactt | aaaccatgtcagaaatgagactaagca | 960 |
| 15 | ctggaatctcagtatgtcccaatttc | tctgtctttctgccacaaagactagtc | 695 |
| 16 | ttgaggcactagaggcgtctctt | aacaccgagctagatttgatccg | 576 |
| 17 | cttgctaatgaatgaccttgcattatc | aatgatcagaaggcatagggtattgac | 427 |
| 18 | tgaacatcactgttggaggctatg | gcattcaagatgcttttgataaccc | 399 |
| 19 | catgccctgaacaactttacttgag | aacagaaattggtgtacagtgtcacg | 506 |
| 20 | ttgctcagttatgtttcacgtactc | ttgaagatcattttattccgaatcca | 535 |
| 21 | cttcggaagctgagtctaagttttagc | gaagacaggcttccaatatgttaggtc | 353 |
| 22 | caaacattcttatgtctgtgaaaacc | gaatggtttgggcttatcattgatc | 610 |
| 23 | atcttctttcttttctgcatgtagcg | AAGATATTTTTGCCAACAGATAAACGC | 515 |
Lowercase letters denote that primer is in the intron, and uppercase letters denote that primer is in the noncoding region of the exon.
Figure 2.

Frameshift mutation in ATP7A. The 5′ and 3′ UTRs of the ∼8.5-kb ATP7A transcript are shown atop a gray background, and the 4.5-kb coding region is shown without a background. In the lower portion of the figure, the coding sequence of wild-type (wt) ATP7A exon 23 is enlarged. The wild-type guanine triplet at positions 4497–4499 is shown atop a black background. The BslI (ccnnnnnnngg) restriction site is shown above the wild-type nucleotide sequence. Conceptual translation of wild-type (without background) and mutant (atop a gray background) ATP7A is illustrated below the nucleotide sequence. The dileucine motif L1487L1488 is underlined in wild-type ATP7A. Stop codons are indicated by asterisks (*).
The mutation was confirmed by restriction analysis, since the guanine deletion results in the loss of a BslI (ccnnnnnnngg) restriction site (fig. 2). PCR products of 515 bp (fig. 3, upper panel) were generated from genomic DNA by the exon 23 primer pair given in table 2. The PCR products were digested with BslI for 3 h at 55°C and then were resolved by electrophoresis through a 2% agarose gel (fig. 3, lower panel). Normal genomic DNA has two BslI restriction sites within this PCR product and, after digestion, generates fragment sizes of 312 bp, 119 bp, and 84 bp. These fragments were detected in the control (wt), II-6, and III-1. II-6, III-2, and III-3 had digestion products of 431 bp and 84 bp, which indicated a loss of the BslI restriction site.
Figure 3.
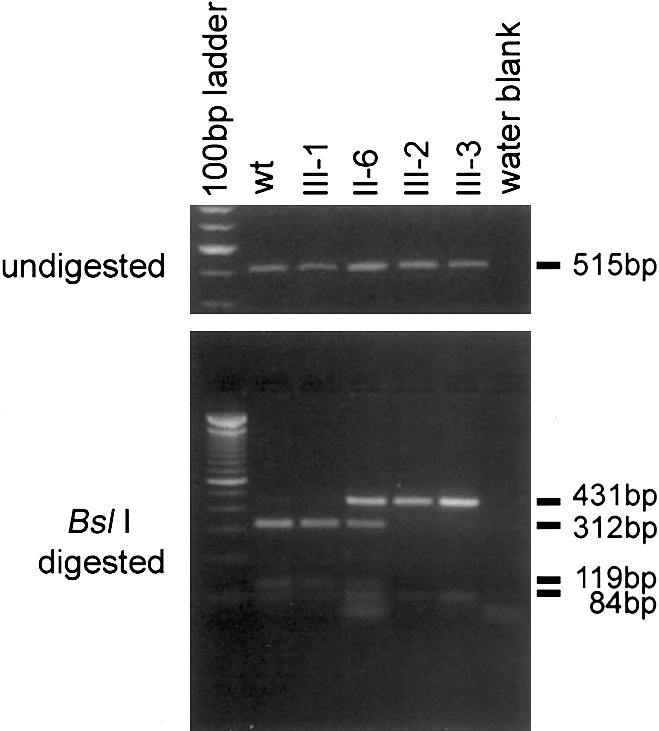
Mutation analysis of the family with OHS. To generate 515-bp PCR products (upper panel), the exon 23 primer pair (table 2) and genomic DNAs isolated from lymphoblastoid cell lines from an unrelated normal control (wt), II-6, III-1, III-2, and III-3 were used. Digests in III-2, III-3, and in one allele of II-6 demonstrate loss of 312-bp and 119-bp BslI fragments (lower panel). The presence, in all digests, of the 84-bp fragment indicates complete digestion with BslI. The smallest band in the digest for II-6 is a primer band and is also present in the water blank.
Despite the presence of the frameshift mutation, the transcript levels in III-2, III-3, and II-6 were determined, by northern blot analysis, to be either normal or somewhat increased (fig. 4). It is known that nonsense and frameshift mutations introduce premature-termination codons (PTCs) into open reading frames, which typically leads to rapid degradation, by the nonsense-mediated decay (NMD) process, of the affected mRNA (for review, see Frischmeyer and Dietz 1999). However, when a PTC occurs in the last exon of the gene, the mRNA may not be subject to NMD (Thermann et al. 1998), thus likely explaining why abundant transcript levels are present in patients with OHS in our study.
Figure 4.
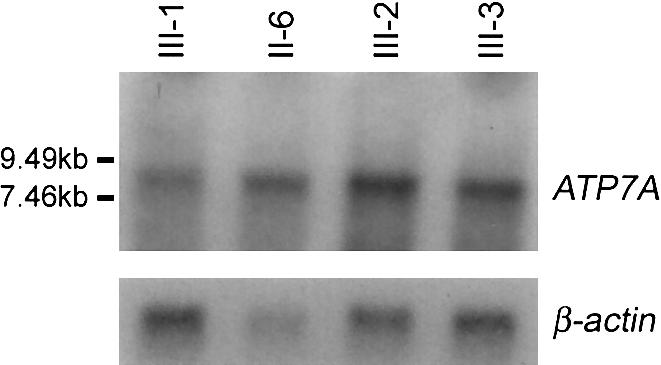
Northern blot analysis of ATP7A-transcript levels. TRIZOL (Gibco) was used to isolate total RNA from lymphoblastoid cell lines from II-6, III-1, III-2, and III-3. mRNA was obtained using the PolyATtract mRNA Isolation System IV (Promega). A northern blot containing 2 μg of poly(A)+ from each family member was hybridized with a [32P]-radiolabeled 1.2-kb cDNA probe, specific to exons 4–10 (nucleotides 1147–2335) of ATP7A. The primer sequences used to generate, via RT-PCR, the 1.2-kb product of normal lymphoblastoid cDNA were 5′- AATAGTGGCTGTATCACCGGG-3′ and 5′- GTACCAGCCTCCGAAAAACTG-3′. Subsequent hybridization with a [32P]-radiolabeled β-actin probe (Clontech) demonstrated loading consistency of poly(A)+ RNA, in each lane (lower panel). Probes were radiolabeled by the rediprimeII Random Prime Labeling System (Amersham Pharmacia Biotech).
Although abundant levels of transcript were detected by northern blots of the affected siblings' mRNA, the predicted protein is truncated and, thus, could be degraded, mislocalized, or both. Western blot analysis of proteins from lymphoblastoid cell lines from the family members, a normal control, and the female patient with Menkes disease and translocation t(X;2)(q13;q32.2) who is known as “CG” (Kapur et al. 1987; Verga et al. 1991) revealed substantially less ATP7A in III-2 and III-3 (fig. 5, upper panel). No protein was detected in CG. Blots were hybridized with tubulin-α Ab-2 antibody (Research Antibody) as a loading control (fig. 5, lower panel). Serial dilutions of normal control protein on western blots were used to estimate that III-2 and III-3 have ∼100-fold less ATP7A than is seen in normal controls. We propose that the ATP7A truncation in the carboxy terminus may lead to either misfolding or destabilization of the protein, resulting in rapid degradation. The truncated protein, with its 13 novel amino acids, may be conformationally incorrect and rapidly degraded in the endoplasmic reticulum (ER), which has a strict quality-control system for secretory and membrane proteins (Zhang et al. 1997; Lord et al. 2000). Alternatively, the carboxy terminus of ATP7A may play a role in its metabolic stability after it exits the ER. Rapid degradation has been observed in (a) protein misfolding resulting from PTC mutations in the final exon of a gene (Kainulainen et al. 1992) and (b) protein destabilization resulting from a C-terminal–tail truncation (Haardt et al. 1999). We postulate that a small percentage of the truncated protein bypasses this quality-control system. The western blot result is consistent with the findings presented by Møller et al. (2000), which suggest that as low as 2%–5% of functional ATP7A is sufficient to result in the milder phenotype, OHS, rather than the more severe phenotype, Menkes disease.
Figure 5.
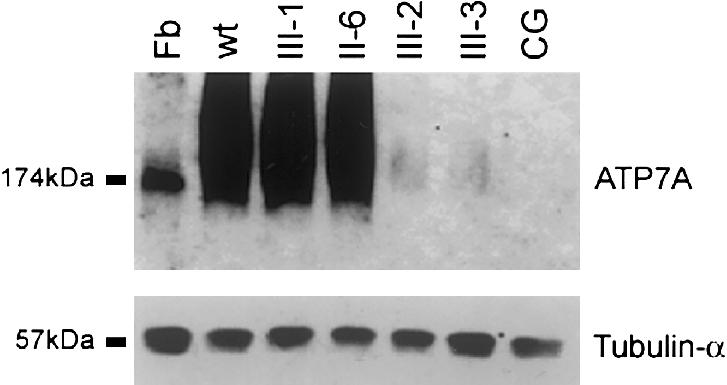
Western blot analysis of ATP7A levels. Protein from lymphoblastoid cell lines from CG, II-6, III-1, III-2, and III-3 was isolated according to a procedure described by Ambrosini and Mercer (1999). Fibroblast (Fb) protein from an unrelated normal control cell line was isolated according to a procedure described by Dierick et al. (1997). One hundred micrograms of protein per lane was fractionated through a 7.5% acrylamide gel and then was electroblotted onto a nitrocellulose membrane. This membrane was then hybridized with a 1:1,000 dilution of polyclonal α-ATP7A (upper panel), as described by Dierick et al. (1997); after several washes in Tris buffered saline solution plus Triton X-20, this membrane was incubated with a 1:2,000 dilution of horseradish peroxidase–conjugated donkey anti-rabbit antibodies. α-ATP7A hybridized to a 174-kD protein in fibroblast, consistent with ATP7A. Hybridization to a 174-kD protein in the fibroblast cell line confirms that the ATP7A antibody is hybridizing to the correct protein from lymphoblastoid cells. To demonstrate loading consistency, the blot was hybridized with a 1:10,000 dilution of tubulin-α Ab-2 antibody, which hybridized to a 57-kD protein. Ambrosini and Mercer (1999) have reported smearing of ATP7A isolated from lymphoblastoid cell lines and have suggested that it most likely was a result of protein glycosylation.
To verify these results and to attempt to localize the truncated protein, we performed immunocytochemistry on cultured fibroblasts. II-6 (not shown) and III-1 (fig. 6a) had normal ATP7A localization, to the perinuclear region, consistent with TGN localization, in cells grown in basal media. ATP7A was not detected in the TGN in III-2 (fig. 6b), III-3 (not shown), or Menkes cell line GM01981 (803del5nt; fig. 6c) (Das et al. 1994). Lack of residual ATP7A perinuclear staining indicated that ATP7A was either mislocalized or present at a level below detection via immunocytochemistry (as would be expected, on the basis of the western blot results). Colocalization experiments with α-KDEL (Grp78; BiP), which is ER specific, and with α-ATP7A demonstrated that the truncated ATP7A was not mislocalized to the ER (data not shown).
Figure 6.

Indirect immunofluorescence of ATP7A in fixed fibroblasts. Immunocytochemistry was performed according to a procedure described by Dierick et al. (1997). Fibroblasts from II-1 (a), III-2 (b), and GM01981 (c) were incubated, for 1 h, with a 1:200 dilution of polyclonal α-ATP7A (Dierick et al. 1997). To visualize the hybridization of α-ATP7A, the fibroblasts were incubated with a 1:1,000 dilution of affinity-purified fluorescein isothiocyanate (H+L) (Vector Laboratories). Cell nuclei were counterstained with 4’,6-diamidino-2-phenylindole. Results were analyzed by a Zeiss Axioscope epifluorescence microscope and were documented by a Photometrics SenSys camera and Vysis Quips imaging software (Applied Imaging).
Ambrosini and Mercer (1999) described a patient with classic Menkes disease who displayed only the OHS phenotype after early treatment with copper-histidine. Western blot analysis indicated that this patient had normal levels of ATP7A—which was truncated within transmembrane domain (TMD) 7, thus deleting all of TMD8 and the carboxy tail, including the dileucine motif L1487L1488. Immunocytochemistry of fibroblasts from this patient, using an ATP7A antibody, displayed an abundance, above background levels, of diffuse staining. Ambrosini and Mercer (1999) surmised that the protein was mislocalized—most likely to the plasma membrane—owing to the loss of the endocytic signal. Since ATP7A above background levels was not detected in patients with OHS in our study (fig. 6), the immunocytochemistry results support the western blot data and demonstrate that the truncated protein is present at very low levels.
This is the first demonstration, in OHS, of reduced ATP7A at the protein level, but not at the mRNA level. Our findings are consistent with other reports in which reduced levels of functional ATP7A appear to be sufficient to result in OHS; however, the mechanism reported here is novel. In contrast to previous reports, transcript levels are abundant, but the protein level is reduced. In addition, the protein is abnormal and is truncated prior to the dileucine motif L1487L1488, which functions as an endocytic signal. This raises the question of whether the dileucine motif is contributing to the OHS phenotype.
Recent data suggest that steady-state localization of ATP7A to the TGN is necessary for proper activity of lysyl oxidase (Ambrosini and Mercer 1999). It has been hypothesized that, in OHS, the available ATP7A is mislocalized—most likely to the plasma membrane—owing to the low protein levels in relation to the increased intracellular copper levels (Das et al. 1995), thus explaining the decreased lysyl oxidase activity. Our data are consistent with both this hypothesis and the hypothesis that the dileucine motif is not necessary for maintenance of copper efflux from the cell. Because the dileucine motif L14871488 is required for ATP7A to be recycled back to the TGN, we would expect the majority of the ATP7A present in our patients to be at the plasma membrane.
In summary, patients with OHS in our study have a deletion of a guanine in exon 23 of ATP7A, and this deletion leads to a frameshift that results in 13 novel amino acids before premature truncation of the protein, prior to the dileucine motif L1487L1488. We have demonstrated that these patients with OHS have abundant ATP7A-transcript levels but reduced levels of a truncated protein. This is the first report to show an ATP7A truncation that does not result in Menkes disease. This study supports data from Møller et al. (2000), which suggest that as low as 2%–5% of normal levels of functional ATP7A is sufficient to result in the milder phenotype, OHS, rather than the more severe phenotype, Menkes disease. Lower expression of ATP7A in these patients with OHS supports the hypothesis that lysyl oxidase is more sensitive than other cuproenzymes are to copper deficiency, owing to reduced levels of functional ATP7A (Das et al. 1995; Levinson et al. 1996). The fact that our patients have OHS rather than Menkes disease supports the hypothesis that the dileucine motif is not essential for ATP7A activity in efflux of copper from the cell.
Acknowledgments
We are grateful to Herman Dierick and Jianming Fang for their contributions and thank Martin Arlt, Mike Glynn, and Anne Loeb for their suggestions regarding the manuscript. We thank the family for their cooperation. This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, grant DK44130 (to T.W.G.).
Electronic-Database Information
The accession numbers and URLs for data in this article are as follows:
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for ATP7A [accession number NM_000052])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for Menkes disease [MIM 309400] and OHS [MIM 304150])
References
- Ambrosini L, Mercer JFB (1999) Defective copper-induced trafficking and localization of the Menkes protein in patients with mild and copper-treated classical Menkes disease. Hum Mol Genet 8:1547–1555 [DOI] [PubMed] [Google Scholar]
- Byers PH, Siegel RC, Holbrook KA, Narayanan AS, Bornstein P, Hall JG (1980) X-linked cutis laxa due to deficiency of lysyl oxidase. N Engl J Med 303:61–65 [DOI] [PubMed] [Google Scholar]
- Camakaris J, Voskoboinik I, Mercer JF (1999) Molecular mechanism of copper homeostasis. Biochem Biophys Res Commun 261:225–232 [DOI] [PubMed] [Google Scholar]
- Chelly J, Tümer Z, Tonnesen T, Petterson A, Ishikawa-Brush Y, Tommerup N, Horn N, Monaco AP (1993) Isolation of a candidate gene for Menkes disease that encodes a potential heavy metal binding protein. Nat Genet 3:14–19 [DOI] [PubMed] [Google Scholar]
- Das S, Levinson B, Vulpe C, Whitney S, Gitschier J, Packman S (1995) Similar splicing mutations of the Menkes/mottled copper-transporting ATPase gene in occipital horn syndrome and the blotchy mouse. Am J Hum Genet 56:570–576 [PMC free article] [PubMed] [Google Scholar]
- Das S, Levinson B, Whitney S, Vulpe C, Packman S, Gitschier J (1994) Diverse mutations in patients with Menkes disease often lead to exon skipping. Am J Hum Genet 55:883–889 [PMC free article] [PubMed] [Google Scholar]
- Dierick HA, Adam AN, Escara-Wilke JF, Glover TW (1997) Immunocytochemical localization of the Menkes copper transport protein (ATP7A) to the trans-Golgi network. Hum Mol Genet 6:409–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierick HA, Ambrosini L, Spencer J, Glover TW, Mercer JFB (1995) Molecular structure of the Menkes disease gene (ATP7A). Genomics 28:462–469 [DOI] [PubMed] [Google Scholar]
- Francis MJ, Jones EE, Levy ER, Martin RL, Ponnambalam S, Monaco AP (1999) Identification of a di-leucine motif within the C terminus domain of the Menkes disease protein that mediates endocytosis from the plasma membrane. J Cell Sci 112:1721–1732 [DOI] [PubMed] [Google Scholar]
- Frischmeyer PA, Dietz HC (1999) Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet 8:1893–1900 [DOI] [PubMed] [Google Scholar]
- Gu YH, Kodam H, Murata Y, Mochizuki D, Yanagawa Y, Ushijima H, Shiba T, Lee CC (2001) ATP7A gene mutations in 16 patients with Menkes disease and a patient with occipital horn syndrome. Am J Med Genet 99:217–222 [DOI] [PubMed] [Google Scholar]
- Haardt M, Benharouga M, Lechardeur D, Kartner N, Lukacs GL (1999) C-terminal truncations destabilize the cystic fibrosis transmembrane conductance regulator without impairing its biogenesis: a novel class of mutation. J Biol Chem 274:21873–21877 [DOI] [PubMed] [Google Scholar]
- Harris ED (2000) Cellular copper transport and metabolism. Annu Rev Nutr 20:291–310 [DOI] [PubMed] [Google Scholar]
- Kainulainen K, Sakai LY, Child A, Pope M, Puhakka L, Ryhänen L, Palotie A, Kaitila I, Peltonen L (1992) Two mutations in Marfan syndrome resulting in truncated fibrillin polypeptides. Proc Natl Acad Sci USA 89:5917–5921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaler SG (1998) Metabolic and molecular bases of Menkes disease and occipital horn syndrome. Pediatr Dev Pathol 1:85–98 [DOI] [PubMed] [Google Scholar]
- Kaler SG, Gallo LK, Proud VK, Percy AK, Mark Y, Segal NA, Goldstein DS, Holmes CS, Gahl WA (1994) Occipital horn syndrome and a mild Menkes phenotype associated with splice site mutations at the MNK locus. Nat Genet 8:195–202 [DOI] [PubMed] [Google Scholar]
- Kapur S, Higgins JV, Delph K, Rogers B (1987) Menkes syndrome in a girl with X-autosome translocation. Am J Med Genet 26:503–510 [DOI] [PubMed] [Google Scholar]
- La Fontaine S, Firth SD, Lockhart PJ, Brooks H, Parton RG, Camakaris J, Mercer JFB (1998) Functional analysis and intracellular localization of the human Menkes protein (MNK) stably expressed from a cDNA construct in Chinese hamster ovary cells (CHO-K1). Hum Mol Genet 7:1293–1300 [DOI] [PubMed] [Google Scholar]
- Lazoff SF, Rybak JJ, Parker BR, Luzzatti L (1975) Skeletal dysplasia, occipital horns, diarrhea and obstructive uropathy—a new hereditary syndrome. Birth Defects Orig Artic Ser 11:71–74 [PubMed] [Google Scholar]
- Levinson B, Conant R, Schnur R, Das S, Packman S, Gitschier J (1996) A repeated element in the regulatory region of the MNK gene and its deletion in a patient with occipital horn syndrome. Hum Mol Genet 5:1737–1742 [DOI] [PubMed] [Google Scholar]
- Levinson B, Gitschier J, Vulpe C, Whitney S, Yang S, Packman S (1993) Are X-linked cutis laxa and Menkes disease allelic? Nat Genet 3:6 [DOI] [PubMed] [Google Scholar]
- Lord JM, Davey J, Frigerio L, Roberts LM (2000) Endoplasmic reticulum-associated protein degradation. Semin Cell Dev Biol 11:159–164 [DOI] [PubMed] [Google Scholar]
- Menkes JH (1988) Kinky hair disease: twenty five years later. Brain Dev 10:77–79 [DOI] [PubMed] [Google Scholar]
- Menkes JHM, Alter M, Steigleder GK, Weakley DR, Sung JH (1962) A sex-linked recessive disorder with retardation of growth, peculiar hair and focal cerebral and cerebellar degeneration. Pediatrics 29:764–779 [PubMed] [Google Scholar]
- Mercer JFB, Livingston J, Hall B, Paynter JA, Begy C, Chandrasekharappa S, Lockhart P, Grimes A, Bhave M, Siemieniak D, Glover TW (1993) Isolation of a partial candidate gene for Menkes disease by positional cloning. Nat Genet 3:20–25 [DOI] [PubMed] [Google Scholar]
- Møller LB, Tümer Z, Lund C, Petersen C, Cole T, Hanusch R, Seidel J, Jensen LR, Horn N (2000) Similar splice-site mutations of the ATP7A gene lead to different phenotypes: classical Menkes disease or occipital horn syndrome. Am J Hum Genet 66:1211–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen PL, Carafoli E (1987) Ion motive ATPases. I. Ubiquity, properties, and significance to cell function. Trends Biochem Sci 12:146–150 [Google Scholar]
- Petris MJ, Camakaris J, Greenough M, LaFontaine S, Mercer JFB (1998) A C-terminal di- leucine is required for localization of the Menkes protein in the trans-Golgi network. Hum Mol Genet 7:2063–2071 [DOI] [PubMed] [Google Scholar]
- Petris MJ, Mercer JFB (1999) The Menkes protein (ATP7A; MNK) cycles via the plasma membrane both in basal and elevated extracellular copper using a C-terminal di-leucine endocytic signal. Hum Mol Genet 8:2107–2115 [DOI] [PubMed] [Google Scholar]
- Petris MJ, Mercer JF, Culvenor JG, Lockhart P, Gleeson PA, Camakaris J (1996) Ligand- regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: a novel mechanism of regulated trafficking. EMBO J 15:6084–6095 [PMC free article] [PubMed] [Google Scholar]
- Qi M, Byers PH (1998) Constitutive skipping of alternatively spliced exon 10 in the ATP7A gene abolishes Golgi localization of the Menkes protein and produces the occipital horn syndrome. Hum Mol Genet 7:465–469 [DOI] [PubMed] [Google Scholar]
- Ronce N, Moizard MP, Robb L, Toutain A, Villard L, Moraine C (1997) A C2055T transition in exon 8 of the ATP7A gene is associated with exon skipping in an occipital horn syndrome family. Am J Hum Genet 61:233–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thermann R, Neu-Yilik G, Deters A, Frede U, Wehr K, Hagemeier C, Hentze MW, Kulozik AE (1998) Binary specifications of nonsense codons by splicing and cytoplasmic translation. EMBO J 17:3484–3494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukahara M, Imaizumi K, Kawai S, Kajii T (1994) Occipital horn syndrome: report of a patient and review of the literature. Clin Genet 45:32–35 [DOI] [PubMed] [Google Scholar]
- Tümer Z, Vural B, Tonnesen T, Chelly J, Monaco AP, Horn N (1995) Characterization of the exon structure of the Menkes disease gene using vectorette PCR. Genomics 26:437–442 [DOI] [PubMed] [Google Scholar]
- Verga V, Hall BK, Wang S, Johnson S, Higgins JV, Glover TW (1991) Localization of the translocation breakpoint in a female with Menkes syndrome to Xq13.2-q13.3 proximal to PGK-1. Am J Hum Genet 48:1133–1138 [PMC free article] [PubMed] [Google Scholar]
- Vulpe C, Levinson B, Whitney S, Packman S, Gitschier J (1993) Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat Genet 3:7–13 [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Heiny ME, Suzuki M, Gitlin JD (1996) Biochemical characterization and intracellular localization of the Menkes disease protein. Proc Natl Acad Sci USA 93:14030–14035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JX, Braakman I, Matlack KES, Helenius A (1997) Quality control in the secretory pathway: the role of calreticulin, calnexin and BiP in the retention of glycoproteins and C-terminal truncations. Mol Biol Cell 8:1943–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]