

Abstract
CoIII complexes have recently become an important focus in photophysics and photoredox catalysis due to metal-centered excited states with strong oxidizing properties. Optimizing chelate ligand bite angles is a widely used strategy to strengthen metal–ligand interactions in coordination complexes, with the resulting enhanced ligand fields often contributing to extended excited-state lifetimes that are advantageous for photochemical applications. We demonstrate that bite-angle optimization exerts the opposite effect on CoIII polypyridines compared to previously studied transition metal complexes, as polypyridine ligands function as π-donors to CoIII rather than π-acceptors. Our findings reveal two counterintuitive paradigms: while bite-angle optimization weakens the ligand field in CoIII complexes, the resulting lower-energy metal-centered excited states can be accompanied by extended excited-state lifetimes, driven by increased rigidification through intramolecular π–π interactions. These insights, along with additional experiments investigating the possibility of photoreactions from higher excited states, advance the current understanding of the photophysics and photochemistry of first-row transition metal complexes and highlight key distinctions from the more extensively studied photoactive complexes of second- and third-row transition metals.

Introduction
Transition metal complexes with a d6 valence electron configuration, such as RuII (4d6), IrIII and OsII (5d6), have become essential in applications including solar energy conversion, − photocatalysis, − light-emitting devices, − bioimaging, − photodynamic therapy, − molecular sensors, and probes. − This prominence is attributed to the metal-to-ligand charge transfer (MLCT) excited states of the respective noble metal complexes, which exhibit favorable properties such as high energy storage capacity, redox activity, prolonged excited-state lifetimes, and often excellent luminescence properties. In the pursuit of more cost-effective and earth-abundant alternatives to precious compounds, first-row 3d6 transition metals such as CoIII, − FeII, − MnI, , and Cr0, , have received substantial attention over the past decade. However, fine-tuning the photophysical properties of 3d metal complexes remains challenging due to the primogenic effect, which leads to a more contracted nature of the 3d metal orbitals compared to the 4d and 5d orbitals, reducing metal–ligand orbital overlap and thus decreasing ligand field strength (10 Dq). , This limited orbital overlap leads in consequence to the stabilization of highly distorted metal-centered (MC) states, which further complicates the utilization of charge-transfer (CT) states due to their rapid nonradiative deactivation into the ground state through those MC states. ,− Recent studies show that in strong ligand fields, the lowest MC excited states of CoIII complexes, although strongly distorted, can become relatively long-lived and photochemically useful. ,,
A common design strategy for photoactive metal complexes involves using π-acceptor ligands and carefully tuning coordination bite angles to enhance metal–ligand orbital overlap and thereby maximize the ligand field strength. ,,,, This approach has proven particularly effective in CrIII polypyridine complexes with a d3 electron configuration (Figure ), − as it increases the energy of the highly distorted 4T2 excited state and thereby enables “spin-flip” emission with prolonged lifetimes and enhanced luminescence quantum yields from largely undistorted and energetically lower-lying 2E/2T1 states.
1.

Effect of bite-angle optimization on the lifetimes (τ) of the doublet metal-centered (2E) excited state and emission quantum yields (Φ) of pseudo-octahedral CrIII polypyridine complexes in solution at room temperature. − Selected cis- and trans-N–Cr–N angles are marked in the molecular drawings to highlight the important geometrical differences between the individual molecular structures. The geometry distortion parameter Σ was calculated as 12Σi=1|90 – φi|, where φi represents cis-N–Cr–N angles. The Σ parameter quantifies the deviation from the ideal octahedral geometry. ,,
Thus, in [Cr(dqp)2]3+ (dqp = 2,6-di(quinolin-8-yl)pyridine) the increased σ-donation effect associated with its comparatively highly octahedral coordination environment is further combined with the π-acceptor character of the pyridine and quinoline moieties, leading to a large ligand field splitting and consequently an excited-state lifetime (τ(2E)) of 1.2 ms and a photoluminescence quantum yield of 5.2% in solution at room temperature (Figure c). The performance of [Cr(tpy)2]3+ (tpy = 2,2′:6′,2″-terpyridine) and [Cr(bpy)3]3+ (bpy = 2,2′-bipyridine) is exceeded by orders of magnitude (Figure a,b). − This is primarily due to the stronger CrIII–N interactions in [Cr(dqp)2]3+, which arise from improved metal–ligand orbital overlap enabled by a coordination geometry that more closely approximates an ideal octahedron compared to [Cr(tpy)2]3+ and [Cr(bpy)3]3+. The deviation from the ideal geometry can be quantified by the octahedral distortion parameter Σ, which is defined as the sum of deviations from 90° for all 12 cis-N–Cr–N angles (12Σi=1|90 – φi|). The Σ parameter decreases from 104° to 67° and then to 29° across the series of complexes shown in Figure , resulting in significant enhancements in τ(2E) and Φ as described above.
Beyond the d3 complexes of CrIII and MnIV, − strong ligand fields generated by π-acceptor ligands and chelates with optimized bite angles have been most effective in complexes of d6 metals. In Cr0 and MnI complexes, ,,− the combined σ-donating and π-accepting electronic properties of isocyanide chelate ligands, paired with nearly ideal octahedral geometries, make it possible to achieve emissive 3MLCT excited states with lifetimes in the nanosecond range at room temperature in solution. ,,−
Among photoactive 3d6 systems, FeII complexes have received by far the most attention, and efforts to modify their photophysical properties have also focused on creating strong ligand fields and idealized coordination geometries. ,,− In pursuit to extend the 3MLCT excited state lifetimes of FeII complexes, several research groups have developed ligands based on N-heterocyclic carbene (NHC) and mesoionic carbene (MIC) moieties, which can act as strong σ-donors and π-acceptors, increasing the ligand field strength. ,,,,,,, Ultimately, this molecular design extended the 3MLCT lifetime up to ∼500 ps in the 6-fold MIC coordination environment of [Fe(btz)3]2+ (btz = 3,3′-dimethyl-1,1′-bis(p-tolyl)-4,4′-bis(1,2,3-triazol-5-ylidene)), about 5000 times longer than in the benchmark [Fe(bpy)3]2+ complex.
The higher oxidation state of CoIII relative to FeII inherently generates a stronger ligand field, providing a priori an advantageous electronic environment. , Recent work demonstrated how to control 3MC excited state deactivation in CoIII polypyridine complexes, leading to promising application potential of these states in photoredox catalysis. ,,, In [Co(4,4′-R2bpy)3]3+ complexes the relationship between ligand field strength and the lifetimes of 3MC excited states was rationalized in terms of Marcus inverted region behavior, analogous to the common behavior of 3CT excited states in RuII or OsII complexes. In this regime, an increase in the excited state energies leads to a decrease in the (nonradiative) deactivation rate of the excited state. This finding is essential for designing CoIII systems possessing long-lived 3MC excited states with high energy capacity, which are crucial for photocatalytic applications. Furthermore, this behavior contrasts with that commonly seen for isoelectronic FeII complexes. Previous observations on [Co(PhB(MeIm)3)2]+ (PhB(MeIm)3 – = tris(3-methylimidazolin-2-ylidene)phenyl borate) and [Co(CN)6]3– complexes align well with the introduced concept of Marcus inverted behavior. In these complexes, the very strong ligand field splitting due to the 6-fold NHC or cyanido environments caused the 3MC states to become emissive, exhibiting lifetimes of 1 μs and 2.6 ns, respectively, owing to the sufficiently large energy gap between these states and the ground state (∼1.7–1.8 eV). − However, the impact of coordination bite angles on the photophysical properties of CoIII polypyridine complexes remains unexplored.
In the present work, we focus on investigating how the properties of 3MC excited states in CoIII complexes are influenced by modification of the ligand bite angles, expansion of the aromatic π-system of the ligand framework, and cooperative rigidity due to π–π and steric interactions between individual ligands coordinated to the same metal center. While it is well established for CrIII that σ-donation and π-backbonding play key roles in the ligand design based on polypyridine scaffolds (Figure a,b), comparatively little is known about those effects in CoIII complexes. ,
2.

Anticipated main electronic effects upon bite-angle optimization shown in a simplified orbital scheme: a. σ-donation in CrIII and CoIII polypyridine complexes, b. π-backbonding in CrIII polypyridine complexes, c. π-donation in CoIII polypyridine complexes. In a–c molecular orbital diagrams show relevant bonding interactions of metal 3d orbitals and (symmetry-adapted) ligand group orbitals (LGOs). The magnitude of the illustrated electronic effects increases as the coordination geometry approaches a more ideal octahedral arrangement.
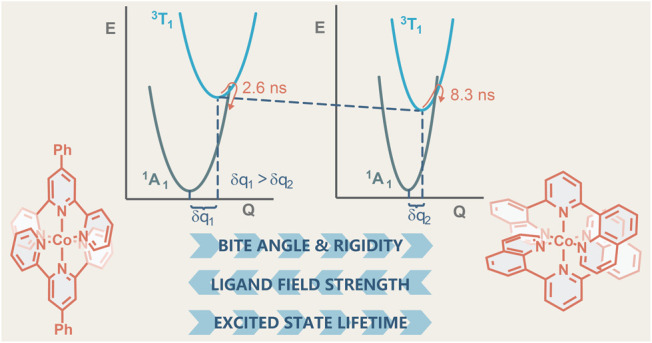
We hypothesize that improving the metal–ligand orbital overlap via enhancing the ligand bite angles for an octahedral geometry can enhance σ-donation, similar to the case for CrIII complexes (Figures and a). However, literature reports indicate that polypyridine ligands behave as π-donors rather than π-acceptors toward CoIII, which would be expected to stabilize MC states (Figure c) and could entail shorter excited-state lifetimes. , To experimentally test this hypothesis, we compare the photophysical properties of compounds of known structure[Co(phtpy)2]3+ (phtpy = 4′-phenyl-2,2′:6′,2″-terpyridine) and [Co(dqp)2]3+ (Figure a), − where the differences between coordination geometries are similarly pronounced to those observed in the CrIII complexes shown in Figure a,c. Our hypothesis is confirmed to the extent that π-donation, rather than π-acceptor behavior, predominates in the CoIII complexes; however, the resulting stabilization of the 3MC excited state does not lead to the anticipated shortening of its lifetime but rather to an unexpected extension. This finding suggests enhanced cooperative rigidity arising from the extended π-conjugated framework of the dqp ligand, which enables intramolecular π–π interactions that rigidify the overall coordination complex and reduce the efficiency of nonradiative 3MC relaxation. − The observed simultaneous lowering of excited-state energy and prolongation of its lifetime contradicts the typical photophysical behavior seen in most metal complexes and organic compounds but can be attributed to a rationalizable interplay between ligand field strength, excited-state distortions, and force constants.
3.

(a) X-ray crystal structures of [Co(phtpy)2]3+, and [Co(dqp)2]3+ (Supporting Information); counterions and solvent molecules are omitted for clarity, and thermal ellipsoids are shown at 50% probability; π–π interactions are indicated with distance (Å) between the quinoline units. (b) 1H NMR spectra of the free dqp ligand (top) and [Co(dqp)2]3+ in CD3CN (bottom), signals corresponding to the protons of the quinoline moieties, involved in π–π interactions, are marked in brown, while the protons of the pyridine unit are marked in blue.
Results and Discussion
Two Sister Complexes with Different Coordination Geometries
The 4′-phenyl-2,2′:6′,2″-terpyridine ligand (phtpy) is synthetically more accessible than unsubstituted tpy, so we used phtpy instead of tpy for our study but expect the primary coordination sphere to be largely unaffected by the additional phenyl ring in the backbone. The phtpy ligand and the compound [Co(phtpy)2](PF6)3 were synthesized following procedures adapted from the literature (Supporting Information). , [Co(dqp)2](PF6)3 was synthesized in a manner similar to [Co(phtpy)2](PF6)3. , In the first step, the dqp ligand (2.05 equiv) reacted with CoCl2·6H2O (1.0 equiv) in CH3CN, followed by CoII/III oxidation with Br2, and finally, the anion was exchanged with KPF6 in aqueous solution. After purification, the complex was characterized by standard analytical techniquesNMR spectroscopy, high-resolution mass spectrometry, and combustion analysis (Supporting Information).
Orange crystals of [Co(dqp)2](PF6)3·2CH3CN, suitable for X-ray diffraction (XRD) measurements, were obtained via vapor diffusion of Et2O into a saturated CH3CN solution. Analysis of the crystal data revealed the Co–N bond lengths to be in the range of 1.952(2)–1.972(2) Å, in line with the values for previously reported tridentate polypyridine complexes. Compared to [Co(phtpy)2]3+, upon extension of the aromatic system in [Co(dqp)2]3+, bite angles increase from 164.7(3)° to 179.42(10)°–179.21(10)°, approaching the ideal octahedral 180°, similar to what has been reported previously for [Ru(dqp)2]2+, − and for CrIII complexes with bite-angle optimized chelate ligands. ,,,, The analysis of the octahedral distortion parameter Σ reveals a highly octahedral coordination environment with a Σ = 13° in [Co(dqp)2]3+ as opposed to a Σ = 65° in [Co(phtpy)2]3+. Interestingly, the Σ value in [Co(dqp)2]3+ is lower than in [Cr(dqp)2]3+ (Σ = 29°), presumably due to a smaller ionic radius of CoIII compared to CrIII.
These structural properties as obtained in the solid state are in good agreement with the fully relaxed, calculated singlet ground-state structure of [Co(dqp)2]3+ in acetonitrile. Density functional theory (DFT) predicts Co–N bond lengths ranging from 1.961 to 1.978 Å, while a bite angle of 178.7° is obtained. All calculated equilibrium structures (as well as high-resolution images) are available via the free online repository Zenodo. In the crystal structure of [Co(dqp)2](PF6)3 π-stacking effects of the adjacent ligand “side-arms,” namely the quinoline units, are evident (Figure a), which could contribute to a favorable rigidity-enhancing effect. π–π interactions are indicated by roughly 3.4–3.5 Å distances between the relevant quinoline centroids (Figure a), matching the values reported for [Cr(dqp)2]3+. Notably, the structure exhibits helical twisting, compatible with P- and M-chirality according to Cahn–Ingold–Prelog nomenclature, and pairs of PP- and MM-enantiomers are present in the crystal unit cell, reminiscent of what has been reported for [Cr(dqp)2]3+ and related CrIII complexes. ,,
Evidence for the intramolecular π-stacking effects between individual dqp ligands is further provided by comparison of the 1H NMR spectra of [Co(dqp)2]3+ and the free dqp ligand (Figure b). Upon coordination, the quinoline protons (marked in brown in Figure b) of the ligand experience a drastic upfield shift, while the pyridine protons (marked in blue in Figure b) undergo a downfield shift. Typically, without any structural constraints, a downfield shift is anticipated for the ligand protons due to an electron density displacement from the ligand to the metal upon coordination. However, the observed upfield shift of the quinoline protons indicates that those units are likely located in close proximity to one another. These data are also consistent with π–π interactions observed in the 1H NMR spectrum of [Ru(dqp)2]2+.
Cyclic voltammetry of [Co(dqp)2]3+ in dry and deaerated CH3CN with 0.1 M (n-Bu4N)PF6 revealed two reversible reduction events (Figure S1). The first wave at +0.33 V vs SCE (E red, see below) can be assigned to the CoII/III redox pair and the second wave at −0.99 V vs SCE can be attributed to a ligand-based reduction process. The assignment of the metal-centered reduction is supported by the quantum chemical simulations, which reveal the character (or in an atomic orbital picture) of the lowest unoccupied molecular orbital (LUMO) of the [Co(dqp)2]3+ complex, while the LUMO-1associated with the second reductionis of π*dqp nature (Figure S23). When comparing the electrochemical behavior of [Co(dqp)2]3+ to that of other CoIII polypyridine complexes, the metal reduction event was observed at very similar potentials of +0.32 V vs SCE and +0.26 V vs SCE in [Co(4,4′-Br2bpy)3]3+ and [Co(phtpy)2]3+, respectively. ,,
Bite-Angle Optimization Weakens the Ligand Field
In the UV–vis absorption spectra of [Co(phtpy)2]3+ and [Co(dqp)2]3+, the strongest bands are observed below 400 nm (Figure a,b). In the case of [Co(phtpy)2]3+ and in agreement with the experimental observations our TD-DFT simulations associate the main absorption feature at 340 nm with a strongly dipole-allowed intraligand charge transfer (ILCT, ph → tpy) into S4 (at 3.27 eV, 378 nm, Figure c and Table S4). Similarly, [Co(dqp)2]3+ shows an absorption band with mixed LC/LMCT character (LC = π–π* ligand-centered, LMCT = ligand-to-metal charge transfer) at slightly lower energy, appearing at 360 nm (ε = 26,000 M–1 cm–1), while the main electronic transition, predicted at 369 nm (into S12, 3.36 eV, Figure d and Table S8), is related to a local excitation of the π-system of the coordinated dqp ligands with only very minor LMCT character. The bathochromic shift of the absorption band at 360 nm in [Co(dqp)2]3+ compared to that at 340 nm in [Co(phtpy)2]3+ is likely the result of the increased aromatic system on the dqp ligand.
4.

Top row: Experimental UV–vis absorption spectra in CH3CN (filled); experimental UV–vis transient absorption spectra in deaerated CH3CN at room temperature following excitation at 355 nm with picosecond pulses (∼6 mJ per pulse) time-integrated over 20 ns (solid traces); kinetic transient absorption traces at the indicated wavelengths, and their monoexponential fitting results (insets). The individual data sets correspond to a. [Co(phtpy)2]3+ and b. [Co(dqp)2]3+; TD-DFT calculated state energies (S0 – singlet ground state geomerty, T1 – first excited triplet state geometry, corresponding to the 3T1 excited state) with their character assignment (see the insets for the color coding), and simulated ground-state absorption spectra for the respective complexes: c. [Co(phtpy)2]3+; d. [Co(dqp)2]3+.
Additionally, weak absorption bands observed in both spectra in the visible region beyond 400 nm, with a molar absorptivity below 500 M–1 cm–1, can be attributed to the spin-forbidden MC transitions. Gaussian deconvolution analysis of those weak bands in both [Co(phtpy)2]3+ and [Co(dqp)2]3+ (Figure ), allowed to determine the 1MC (1T1) and 3MC (3T1) transition energies, required for the ligand field parameter analysis. The mixed 1MC states S1–S3 in [Co(phtpy)2]3+ are predicted by the calculations at 490–432 nm (2.53–2.87 eV) with slight LMCT contributions, similarly in [Co(dqp)2]3+ the 1MC states appear at 506–480 nm (S1–S3, 2.45–2.58 eV), while 1LMCT states are predicted at 442 and 410 nm (S4 and S5, 2.81 and 3.02 eV), see Tables S4 and S8 for details.
5.

Low-energy parts of the experimental UV–vis absorption spectra of a. [Co(phtpy)2]3+ and b. [Co(dqp)2]3+ in CH3CN (brown) with Gaussian deconvolution and assignment of the 1MC (1T1) and 3MC (3T1 and 3T2) electronic transitions. The cumulative fitted curve is shown with dashed black lines. MC transition energies: a. ΔE (1T1–1A1) = 22,600 cm–1, ΔE (3T2–1A1) = 16,600 cm–1, ΔE (3T1–1A1) = 14,500 cm–1; b. ΔE (1T1–1A1) = 21,700 cm–1, ΔE (3T1–1A1) = 14,300 cm–1.
We could not confidently determine the energies of all of the MC transitions required for a precise estimation of the 10 Dq and Racah parameters in [Co(dqp)2]3+ and [Co(phtpy)2]3+ due to the tailing of intense CT transitions in the same spectral range. However, the observed electronic transitions from the singlet ground state (1A1) to the 1T1 and 3T1 excited states allowed an estimation of 10 Dq and the Racah parameter C for both CoIII complexes (see Figure and the footnote of Table for details). The relevant parameters for the [Co(bpy)3]3+ complex have been reported previously, enabling a comparison of 10 Dq values across the series [Co(phtpy)2]3+, [Co(bpy)3]3+, and [Co(dqp)2]3+. Within this series, the symmetry around the CoIII center becomes progressively more octahedral while the Σ parameter decreases (Table ), mirroring the trend observed for the CrIII complexes shown in Figure . For the 10 Dq values, however, opposite trends are observed for CoIII and CrIII. Whereas 10 Dq increases for CrIII across the ligand series tpy < bpy < dqp (as discussed in the Introduction), it decreases for CoIII along the series phtpy > bpy > dqp (Table ). This fundamentally different behavior of CrIII and CoIII complexes can be rationalized by taking into account the effective nuclear charges. Due to its smaller ionic radius, CoIII has a higher effective nuclear charge, resulting in lower energies of its t2g orbitals and a closer energy alignment with the ligand π-orbitals than in the case of CrIII. Consequently, while the tpy/phtpy, bpy, and dqp ligands act as π-acceptors to CrIII, they behave as π-donors toward CoIII. Whereas π-acceptors enhance the ligand field strength, π-donors lead to weaker ligand fields (Figure b,c), consistent with the trends observed in Table .
1. Ligand Field Parameters, Racah Parameters, and Octahedral Distortion Parameters in CoIII and CrIII Complexes along with the Lifetimes of Their Lowest Electronically Excited States ,,,,,
| Complex | 10 Dq†/cm–1 | C†/cm–1 | τ(3T1)/ns | Σ/° | Complex | 10 Dq/cm–1 | τ(2E)/μs | Σ/° |
|---|---|---|---|---|---|---|---|---|
| [Co(phtpy)2]3+ | 26,650 | 4050 | 2.6 | 65 | [Cr(tpy)2]3+ | 20,390 | 30 | 104 |
| [Co(bpy)3]3+ | 25,900 | 3730 | 5.0 | 47 | [Cr(bpy)3]3+ | 23,300 | 63 | 67 |
| [Co(dqp)2]3+ | 25,400 | 3700 | 8.3 | 13 | [Cr(dqp)2]3+ | 24,900 | 1200 | 29 |
10 Dq and Racah parameter C values for [Co(phtpy)2]3+ and [Co(dqp)2]3+ were approximated using the Tanabe–Sugano formalism: ΔE(3T1–1A1) = 10 Dq – 3C, ΔE (1T1–1A1) = 10 Dq – C (experimental values were extracted from the data in Figure ).
Acetonitrile solution, room temperature.
Aqueous solution, room temperature.
This is in line with recent work demonstrating that bidentate and tridentate polypyridine ligands in CoIII complexes primarily act as π-donors. This behavior contrasts the π-acceptor character of polypyridines in second- and third-row transition metal complexes, and supports the previously observed trend that [Co(tpy)2]3+, with its more distorted bite angles, exhibits a stronger ligand field splitting than [Co(bpy)3]3+ (Table ). McCusker and colleagues proposed that this difference between tpy and bpy complexes arises due to a weaker π-donation by the tpy ligand owing to less favorable metal–ligand orbital overlap (Figure b), leading to a weaker destabilization of the t2g orbitals and a (slightly) stronger ligand field in the tpy complex compared to the bpy complex (Figure c). Following that logic in our case, the π-donation is strongest with the bite-angle optimized dqp ligand, leading to the weakest ligand field. The calculated triplet energy of [Co(dqp)2]3+ lies at 1.26 eV (see T1 in Figure d), reflecting an approximately 0.1 eV stabilization of the lowest 3T1 state compared to 1.35 eV in [Co(phtpy)2]3+ (see T1 Figure c). The calculated decrease in the 3T1 energy between these two bis(tridentate) coordination environments thus aligns with the experimentally observed decrease of 10 Dq, at least in terms of the overall trend.
Prolonged Excited-State Lifetimes Despite Lower Energies
The excited-state dynamics of the CoIII complexes were investigated via picosecond UV–vis transient absorption spectroscopy in deaerated acetonitrile at room temperature and in combination with computational modeling. Following the excitation of [Co(phtpy)2]3+ at 355 nm, multiple excited-state absorption (ESA) bands could be observed throughout the visible region (Figure a), decaying with a lifetime of 2.6 ns. In this case, TD-DFT simulations enabled the assignment of these ESA spectral signatures to electronic transitions from the lowest 3MC state to higher-lying excited states. In particular, the ESA in [Co(phtpy)2]3+ was associated with a low-lying 3LMCT transition (into T9 at 647 nm) as well as with 3ILCT excitation into T36 at 379 nm (Figure S20 and Table S6).
Shifting our focus to the complex with an optimized bite angle [Co(dqp)2]3+, its TA spectrum is also compatible with the lowest excited state of 3MC character (Figure S22), with a lifetime of 8.3 ns. ESA bands of the 3MC state are observed at 410, 465, and 650 nm (Figure b), along with a ground-state bleach (GSB) at 390 nm, corresponding to the LC/LMCT band in the ground-state absorption spectrum. The computational analysis assigns these ESA bands mostly to an LMCT absorption at 627 nm (T11), several highly mixed and weakly allowed transitions (e.g., T31, T33 and T35 between 432 and 402 nm) as well as with a strongly allowed LC transition (T54, 364 nm; Figure S22 and Table S10).
Across the series of CoIII complexes reported in Table , the lifetime of the lowest 3T1 excited state increases as the ligand field strength decreases (i.e., as 10 Dq decreases) and the energy of the 3T1 state becomes lower. Specifically, the 3T1 lifetime increases from 2.6 ns in [Co(phtpy)2]3+ to 5.0 ns in [Co(bpy)3]3+ and to 8.3 ns in [Co(dqp)2]3+ (Table , Figure a,b). This contrasts the Marcus inverted region behavior recently unraveled for polypyridine complexes of CoIII, where an increase of the 3MC energy led to an increase of the 3MC lifetime. , It seems plausible that the discrepant behavior observed here for [Co(phtpy)2]3+ and [Co(dqp)2]3+ could be due to different extents of structural rearrangements in the 3MC excited state relative to the electronic ground state. According to its X-ray crystal structure and the DFT simulations in a solvent environment, [Co(phtpy)2]3+ (Figure a) has a strong geometric distortion in the electronic ground state (Σ = 65°, Figure a and Table ) due to the strained bite angle (164(10)°) and axial compression, with the two axial Co–N bonds of 1.862(7) Å being considerably shorter than the four equatorial Co–N bonds (1.952(5) Å).
By contrast, the ground state of [Co(dqp)2]3+ shows minor deviations from the ideal octahedral geometry (Σ = 13°, Figure a and Table ) and reveals significant π-interactions between individual dqp ligands coordinated to the same metal (Figure S23, MOs 165–178), which could impart structural rigidity and greater force constants associated with molecular distortions both in the ground state and in the 3MC excited state. This in turn could lead to more nested 3T1/ground state potentials (Figure b), making the 3T1 lifetime more than a factor of 2 longer in [Co(dqp)2]3+ than in [Co(phtpy)2]3+, despite a decrease in triplet energy of about 0.1 eV (see above). The ca. 40% increase in the 3T1 lifetime from [Co(phtpy)2]3+ to [Co(bpy)3]3+ is comparatively modest and more difficult to rationalize within a simple qualitative framework. This is especially true given the difference in coordination environments (bis(tridentate) versus tris(bidentate)), which complicates direct comparisons. Therefore, it seems more appropriate to focus the following discussion on the tridentate systems, specifically comparing phtpy/tpy-based and dqp-based complexes.
6.

Main electronic and cooperative rigidity effects in CrIII and CoIII complexes upon bite-angle optimization and extension of the π-conjugation. Anticipated changes in the energies, nuclear coordinates, and force constants of the electronic ground and relevant excited states upon optimization of bite angles, π-conjugation, and cooperative rigidity in simplified illustrations with harmonic potential well diagrams. Q = nuclear coordinate, δq = nuclear coordinate displacement/distortion in the excited state relative to the ground state (GS), and E a = activation energy for relaxation from 3T1 to 1A1.
This leads us to the proposed picture in Figure b, where the 3T1 energy decreases from [Co(phtpy)2]3+ to [Co(dqp)2]3+ due to enhanced π-donation. However, the barrier (E a) for nonradiative relaxation from 3T1 to the singlet ground state (1A1) increases, as a result of larger force constants associated with the relevant potential energy surfaces, thereby extending the lifetime of the 3T1 state. In the case of the structurally comparable CrIII complexes, the increased σ-donation and π-acceptance from [Cr(tpy)2]3+ to [Cr(dqp)2]3+ shifts the ligand-field dependent 4T2 state to higher energies (Figure a). This reduces the likelihood of reverse intersystem crossing from the luminescent 2E state to the 4T2 state, thereby prolonging the 2E lifetime. ,, It seems plausible that intramolecular π–π-interactions in [Cr(dqp)2]3+ could induce a similar rigidifying effect as proposed for [Co(dqp)2]3+, contributing to the exceptionally slow deactivation rate of the luminescent 2E state reported previously.
To conclude this section, the picture for CrIII in Figure a has become a widely accepted model in the field and has been found applicable to several other d-block metal species, ,,,,, including FeII, ,, RuII, − ,, MnI, and Cr0. ,,, Optimization of the bite angles enhances the ligand field strength and prolongs excited-state lifetimes. The model for CoIII shown in Figure is unconventional, demonstrating that optimization of bite angles with π-donor ligands decreases the ligand field strength. Nonetheless, prolonged excited-state lifetimes can still be achieved through the simultaneous optimization of rigidity.
Energy Losses between Excitation and the Photoactive Excited State
CoIII polypyridines have emerged as exceptionally strong oxidizing agents in their photoactive 3T1 excited states, surpassing the oxidizing power of several benchmark IrIII photocatalysts. This is particularly remarkable given the significant energy loss that occurs between photoexcitation and the photochemical reaction in this class of compounds. CoIII polypyridines typically absorb strongly only in the blue and UV regions of the spectrum, yet their 3T1 state retains just ∼1.35 eV of energy, indicating that more than 50% of the energy from a 400 nm excitation photon is not retained. In the following, we aim to gain a deeper insight into the initial energy dissipation processes within the [Co(dqp)2]3+ complex.
Specifically, we performed femtosecond UV–vis TA spectroscopy in acetonitrile at room temperature and used spectroelectrochemistry to aid the assignment of the observed spectral features, recognizing the limitations of this approach. , The TA spectra of [Co(dqp)2]3+ (Figure , top) were analyzed in the spectral region from 390 to 490 nm using global fitting, revealing three species-associated spectra (SAS) (Figure S3). The SAS2 corresponds to the long-lived 3T1 state (8.3 ns); its time component was fixed as a constant for the global fit. The initially populated state (SAS0) is tentatively assigned to a 1ILCT state because a distinct ESA band at 460 nm matches with a broad absorption band in the differential absorption spectrum following ligand reduction in this complex (Figures S2a and S4). Unfortunately, the spectral features of the oxidized ligand are outside the spectral region accessible by the femtosecond TA experiment (Figures S2c and S3). We propose that this 1ILCT state undergoes vibrational cooling and an intersystem crossing to the triplet manifold (SAS1) within <1 ps. The spectral features of SAS1 are reminiscent of the lower-lying 3T1 state (SAS2), differing only in the somewhat more intense ESA band at 410 nm. A lifetime of 23 ps can be attributed to the internal conversion (IC) and vibrational cooling (VC) processes between the respective higher- and lower-lying 3MC excited states. This relatively slow process is phenomenologically reminiscent of CuI complexes, , where prolonged ISC and IC lifetimes can result from a geometrical rearrangement in the excited-state landscape.
7.
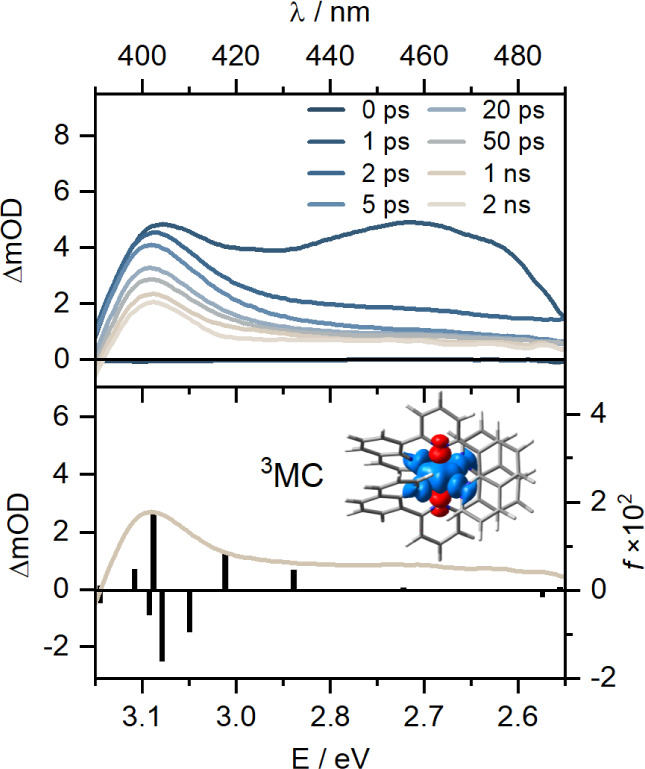
Top: UV–vis transient absorption spectra of [Co(dqp)2]3+ in acetonitrile at 293 K, recorded at different delay times following excitation at 360 nm with pulses of ca. 190 fs duration; bottom: experimental TA spectrum, obtained at a delay time of 1 ns (solid line) and simulated TA spectrum of [Co(dqp)2]3+ (black bars) at the TD-DFT level of theory with the spin density of the triplet state displayed.
While nanosecond excited-state lifetimes are typically required for diffusion-controlled bimolecular reactions, productive photoreactions can occur on the picosecond time scale when the photocatalyst and substrate molecules are preaggregated. ,, A lifetime of 23 ps would, in principle, be sufficient for such processes. It is therefore conceivable that in appropriately preorganized systems such as pairs with anionic reaction partners, − the 23 ps excited-state lifetime permits bimolecular (anti-Kasha) reactivity. This would represent an unconventional strategy for minimizing energy losses between excitation and the ensuing photochemical reaction.
To explore whether such a reaction can be observed, we attempted a photoinduced bimolecular triplet–triplet energy transfer (TTET) from the [Co(dqp)2]3+ complex to perylene. Perylene has a triplet energy of 1.53 eV, significantly higher than the energy accessible from the relaxed 3T1 state of [Co(dqp)2]3+ (1.26 eV) (Figure S9b). Thus, we were speculating that TTET could perhaps occur prior to internal conversion to the 3T1 state and its vibrational cooling (Figures S9b and d), accessed via preaggregation of the CoIII complex and perylene. The NMR titration experiment suggests the presence of the anticipated complex-substrate aggregates in the ground state (Figures S6–S8). The corresponding Benesi–Hildebrand plot yielded an association constant of K a = 190 ± 50 M–1 in acetonitrile at room temperature.
Polypyridine CoIII complexes are known to form highly oxidizing species in their lowest triplet excited state (3T1), , and [Co(dqp)2]3+ is no exception, exhibiting an excited-state reduction potential of E red* = +1.59 V vs SCE. This excited-state redox potential is sufficient to oxidize perylene (E ox = +0.85 V vs SCE) via a highly exergonic (ΔG ET 0 = −0.74 eV) photoinduced bimolecular single-electron transfer (SET) process. Therefore, upon photoexcitation of [Co(dqp)2]3+ in the presence of perylene, we anticipated to observe, using TA spectroscopy, the formation of both the perylene radical cation (perylene+·) via the SET mechanism from the fully relaxed 3T1 state of [Co(dqp)2]3+ and the triplet perylene (3perylene) via the TTET mechanism potentially involving anti-Kasha behavior. ,, Contrary to our initial expectations, we could only observe the formation of perylene+· (Figure S9a) with its characteristic ESA signal at 540 nm and no evidence for an energy transfer from the higher triplet excited states of [Co(dqp)2]3+.
In an attempt to shut down ordinary diffusion-controlled SET reactivity from the relaxed 3T1 excited state of [Co(dqp)2]3+, as observed in the case of perylene discussed above, we aimed to explore the possibility of SET to more strongly oxidizing triplet excited states (with excited-state potentials exceeding 1.85 V vs SCE), accessible only through anti-Kasha reactivity of [Co(dqp)2]3+. To this end, we investigated the possibility of photoinduced SET between [Co(dqp)2]3+ and redox-challenging aromatic compounds such as durene (E ox = +1.75 V vs SCE), 3,3′-dimethyl biphenyl (E ox = +1.84 V vs SCE) and biphenyl (E ox = +1.95 V vs SCE). , However, in all cases, no evidence of radical cation formation was observed, suggesting that both thermodynamic and kinetic electron transfer remained unfavorable. Direct laser spectroscopic evidence for photoreactivity from higher excited states of [Co(dqp)2]3+ is therefore difficult to provide at present, but could perhaps be found more indirectly in long-term light irradiation experiments, such as those carried out in classical synthetic photoredox catalysis.
The inherent photostability of [Co(dqp)2]3+ in deaerated acetonitrile at room temperature is promising for potential photocatalytic applications, as the photodegradation quantum yield under these conditions is remarkably low, only 0.003% (Figure S5). To put this value into perspective, the benchmark complex [Ru(bpy)3]2+ has been reported to exhibit a significantly higher photodegradation quantum yield under comparable conditions. Furthermore, among the CoIII complexes, [Co(ppy)3] and [Co(CN)6]3– were previously shown to undergo photodissociation from the 3T1 excited state. , The [Co(ppy)3] complex undergoes significant Jahn–Teller distortion in an electronically excited state, leading to its ∼9 ps deactivation via Co–Cph bond rupture. The photoaquation reaction of [Co(CN)6]3– occurs in a similar fashion.
Conclusions
Our work reveals two counterintuitive insights into the molecular design principles and photophysical behavior of d-metal complexes. First, the widely accepted paradigm that optimizing the coordination bite angle strengthens the ligand field is not supported in this study. ,,,,,,,, Instead, we find that such optimization can actually weaken the ligand field when the ligands function primarily as π-donors. Second, the well-known “energy gap law,” which states that nonradiative excited-state relaxation accelerates as the excited-state energy decreases, does not hold in the case investigated here. This is because intramolecular rigidification exerts a counteracting influence that can significantly prolong excited-state lifetimes. Both of these key findings are, in principle, predictable based on ligand field theory (Figure ) and simple considerations of the relevant potential well diagrams (Figure ). Although π-donor ligands in photoactive metal complexes have been receiving growing attention, ,,,,,− and the concept of cooperative rigidity among ligands coordinated to the same metal center is becoming increasingly well established, ,− ,, the direct comparison between [Co(phtpy)2]3+ and [Co(dqp)2]3+ presented in this work provides rare and fundamental insight into both of these aspects.
Our study further suggests the potential for conducting bimolecular photochemistry from higher electronically excited states. This approach could help minimize energy losses between light absorption and photochemical energy storage, especially in CoIII polypyridines, which absorb in the blue spectral range but have low-lying excited states that store significantly less energy than those of traditional RuII polypyridines or cyclometalated IrIII complexes.
The specific insights gained from our study can guide the future designs of photoactive complexes based on both abundant first-row and noble or non-noble second- and third-row transition metals, as metal-centered excited states and their deactivation pathways universally play a critical role in determining the photophysical and photochemical properties. The broader concept that a ligand may act as a π-acceptor with one metal but as a π-donor with another could become increasingly relevant as unconventional metal oxidation states are more widely explored, and novel ligand frameworks continue to emerge. Similar effects to those observed in this study may therefore be anticipated in yet-to-be-discovered metal complexes, where a delicate interplay between metal and ligand orbital energies, orbital overlap, and intramolecular π–π interactions shapes the excited-state energy landscape that ultimately governs luminescence properties and photochemical behavior.
Supplementary Material
Acknowledgments
Funding from the Swiss National Science Foundation through grant number 200020_207329, and by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) within the TRR 234 CataLight (project A4; project no. 364549901) is acknowledged.
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.5c09616.
Experimental procedures, materials and methods; synthesis and characterization of complexes, single crystal X-ray data, NMR and HR-ESI mass spectra, photophysical and electrochemical data as well as computational details and in-depth quantum chemical results (PDF)
Swiss National Science Foundation (grant number 200020_207329) and Deutsche Forschungsgemeinschaft (DFG) within the TRR 234 CataLight (project A4; project no. 364549901).
The authors declare no competing financial interest.
References
- Ardo S., Meyer G. J.. Photodriven heterogeneous charge transfer with transition-metal compounds anchored to TiO2 semiconductor surfaces. Chem. Soc. Rev. 2009;38:115–164. doi: 10.1039/B804321N. [DOI] [PubMed] [Google Scholar]
- Mills I. N., Porras J. A., Bernhard S.. Judicious Design of Cationic, Cyclometalated Ir(III) Complexes for Photochemical Energy Conversion and Optoelectronics. Acc. Chem. Res. 2018;51:352–364. doi: 10.1021/acs.accounts.7b00375. [DOI] [PubMed] [Google Scholar]
- Gray H. B., Maverick A. W.. Solar Chemistry of Metal Complexes. Science. 1981;214:1201–1205. doi: 10.1126/science.214.4526.1201. [DOI] [PubMed] [Google Scholar]
- Balzani V., Credi A., Venturi M.. Photochemical Conversion of Solar Energy. ChemSuschem. 2008;1:26–58. doi: 10.1002/cssc.200700087. [DOI] [PubMed] [Google Scholar]
- Yoon T. P., Ischay M. A., Du J.. Visible light photocatalysis as a greener approach to photochemical synthesis. Nat. Chem. 2010;2:527–532. doi: 10.1038/nchem.687. [DOI] [PubMed] [Google Scholar]
- Campagna, S. ; Puntoriero, F. ; Nastasi, F. ; Bergamini, G. ; Balzani, V. . Photochemistry and Photophysics of Coordination Compounds: Ruthenium. In Photochemistry and Photophysics of Coordination Compounds I; Springer: Berlin, Heidelberg, 2007. Vol. 280; pp. 117–214. [Google Scholar]
- Singh-Rachford T. N., Castellano F. N.. Photon upconversion based on sensitized triplet–triplet annihilation. Coord. Chem. Rev. 2010;254:2560–2573. doi: 10.1016/j.ccr.2010.01.003. [DOI] [Google Scholar]
- Kim D., Teets T. S.. Strategies for accessing photosensitizers with extreme redox potentials. Chem. Phys. Rev. 2022;3(2):021302. doi: 10.1063/5.0084554. [DOI] [Google Scholar]
- Shon J.-H., Kim D., Rathnayake M. D., Sittel S., Weaver J., Teets T. S.. Photoredox catalysis on unactivated substrates with strongly reducing iridium photosensitizers. Chem. Sci. 2021;12:4069–4078. doi: 10.1039/D0SC06306A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevernaegie R., Wehlin S. A. M., Elias B., Troian-Gautier L.. A Roadmap Towards Visible Light Mediated Electron Transfer Chemistry with Iridium(III) Complexes. ChemPhotochem. 2021;5:217–234. doi: 10.1002/cptc.202000255. [DOI] [Google Scholar]
- Bizzarri C., Spuling E., Knoll D. M., Volz D., Bräse S.. Sustainable metal complexes for organic light-emitting diodes (OLEDs) Coord. Chem. Rev. 2018;373:49–82. doi: 10.1016/j.ccr.2017.09.011. [DOI] [Google Scholar]
- Yersin H., Rausch A. F., Czerwieniec R., Hofbeck T., Fischer T.. The triplet state of organo-transition metal compounds. Triplet harvesting and singlet harvesting for efficient OLEDs. Coord. Chem. Rev. 2011;255:2622–2652. doi: 10.1016/j.ccr.2011.01.042. [DOI] [Google Scholar]
- Costa R. D., Ortí E., Bolink H. J., Monti F., Accorsi G., Armaroli N.. Luminescent Ionic Transition-Metal Complexes for Light-Emitting Electrochemical Cells. Angew. Chem., Int. Ed. 2012;51:8178–8211. doi: 10.1002/anie.201201471. [DOI] [PubMed] [Google Scholar]
- Henwood A. F., Zysman-Colman E.. Luminescent Iridium Complexes Used in Light-Emitting Electrochemical Cells (LEECs) Top. Curr. Chem. 2016;374(4):36. doi: 10.1007/s41061-016-0036-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D., He H., Leung K., Chan D. S., Leung C.. Bioactive Luminescent Transition-Metal Complexes for Biomedical Applications. Angew. Chem., Int. Ed. 2013;52:7666–7682. doi: 10.1002/anie.201208414. [DOI] [PubMed] [Google Scholar]
- Gill M. R., Thomas J. A.. Ruthenium(II) polypyridyl complexes and DNAfrom structural probes to cellular imaging and therapeutics. Chem. Soc. Rev. 2012;41:3179–3192. doi: 10.1039/c2cs15299a. [DOI] [PubMed] [Google Scholar]
- Yuan L., Wang L., Agrawalla B. K., Park S.-J., Zhu H., Sivaraman B., Peng J., Xu Q.-H., Chang Y.-T.. Development of Targetable Two-Photon Fluorescent Probes to Image Hypochlorous Acid in Mitochondria and Lysosome in Live Cell and Inflamed Mouse Model. J. Am. Chem. Soc. 2015;137:5930–5938. doi: 10.1021/jacs.5b00042. [DOI] [PubMed] [Google Scholar]
- Heinemann F., Karges J., Gasser G.. Critical Overview of the Use of Ru(II) Polypyridyl Complexes as Photosensitizers in One-Photon and Two-Photon Photodynamic Therapy. Acc. Chem. Res. 2017;50:2727–2736. doi: 10.1021/acs.accounts.7b00180. [DOI] [PubMed] [Google Scholar]
- Pham T. C., Nguyen V.-N., Choi Y., Lee S., Yoon J.. Recent Strategies to Develop Innovative Photosensitizers for Enhanced Photodynamic Therapy. Chem. Rev. 2021;121:13454–13619. doi: 10.1021/acs.chemrev.1c00381. [DOI] [PubMed] [Google Scholar]
- McKenzie L. K., Bryant H. E., Weinstein J. A.. Transition metal complexes as photosensitisers in one- and two-photon photodynamic therapy. Coord. Chem. Rev. 2019;379:2–29. doi: 10.1016/j.ccr.2018.03.020. [DOI] [Google Scholar]
- Pages B. J., Ang D. L., Wright E. P., Aldrich-Wright J. R.. Metal complex interactions with DNA. Dalton Trans. 2015;44:3505–3526. doi: 10.1039/C4DT02700K. [DOI] [PubMed] [Google Scholar]
- Rogers C. W., Wolf M. O.. Luminescent molecular sensors based on analyte coordination to transition-metal complexes. Coord. Chem. Rev. 2002;233-234:341–350. doi: 10.1016/S0010-8545(02)00023-1. [DOI] [Google Scholar]
- Poynton F. E., Bright S. A., Blasco S., Williams D. C., Kelly J. M., Gunnlaugsson T.. The development of ruthenium(II) polypyridyl complexes and conjugates for in vitro cellular and in vivo applications. Chem. Soc. Rev. 2017;46:7706–7756. doi: 10.1039/C7CS00680B. [DOI] [PubMed] [Google Scholar]
- Balzani V., Bergamini G., Marchioni F., Ceroni P.. Ru(II)-bipyridine complexes in supramolecular systems, devices and machines. Coord. Chem. Rev. 2006;250:1254–1266. doi: 10.1016/j.ccr.2005.11.013. [DOI] [Google Scholar]
- Krämer J., Kang R., Grimm L. M., De Cola L., Picchetti P., Biedermann F.. Molecular Probes, Chemosensors, and Nanosensors for Optical Detection of Biorelevant Molecules and Ions in Aqueous Media and Biofluids. Chem. Rev. 2022;122:3459–3636. doi: 10.1021/acs.chemrev.1c00746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha N., Wenger O. S.. Photoactive Metal-to-Ligand Charge Transfer Excited States in 3d6 Complexes with Cr0, MnI, FeII, and CoIII . J. Am. Chem. Soc. 2023;145(9):4903–4920. doi: 10.1021/jacs.2c13432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockin B. M., Li C., Robertson N., Zysman-Colman E.. Photoredox catalysts based on earth-abundant metal complexes. Catal. Sci. Technol. 2019;9:889–915. doi: 10.1039/C8CY02336K. [DOI] [Google Scholar]
- Sinha N., Yaltseva P., Wenger O. S.. The Nephelauxetic Effect Becomes an Important Design Factor for Photoactive First-Row Transition Metal Complexes. Angew. Chem., Int. Ed. 2023;135(30):e202303864. doi: 10.1002/ange.202303864. [DOI] [PubMed] [Google Scholar]
- Chan A. Y., Ghosh A., Yarranton J. T., Twilton J., Jin J., Arias-Rotondo D. M., Sakai H. A., McCusker J. K., MacMillan D. W. C.. Exploiting the Marcus inverted region for first-row transition metal–based photoredox catalysis. Science. 2023;382:191–197. doi: 10.1126/science.adj0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A., Yarranton J. T., McCusker J. K.. Establishing the origin of Marcus-inverted-region behaviour in the excited-state dynamics of cobalt(III) polypyridyl complexes. Nat. Chem. 2024;16:1665–1672. doi: 10.1038/s41557-024-01564-3. [DOI] [PubMed] [Google Scholar]
- Burton S. T., Lee G., Moore C. E., Sevov C. S., Turro C.. Cyclometallated Co(III) Complexes with Lowest-Energy Charge Transfer Excited States Accessible with Visible Light. J. Am. Chem. Soc. 2025;147:13315–13327. doi: 10.1021/jacs.4c18299. [DOI] [PubMed] [Google Scholar]
- Liu Y., Persson P., Sundström V., Wärnmark K.. Fe N -Heterocyclic Carbene Complexes as Promising Photosensitizers. Acc. Chem. Res. 2016;49:1477–1485. doi: 10.1021/acs.accounts.6b00186. [DOI] [PubMed] [Google Scholar]
- de Groot L. H. M., Ilic A., Schwarz J., Wärnmark K.. Iron Photoredox Catalysis–Past, Present, and Future. J. Am. Chem. Soc. 2023;145:9369–9388. doi: 10.1021/jacs.3c01000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierks P., Vukadinovic Y., Bauer M.. Photoactive iron complexes: more sustainable, but still a challenge. Inorg. Chem. Front. 2022;9:206–220. doi: 10.1039/D1QI01112J. [DOI] [Google Scholar]
- Jiang T., Bai Y., Zhang P., Han Q., Mitzi D. B., Therien M. J.. Electronic structure and photophysics of a supermolecular iron complex having a long MLCT-state lifetime and panchromatic absorption. Proc. Natl. Acad. Sci. U. S. A. 2020;117:20430–20437. doi: 10.1073/pnas.2009996117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus B. C., Nielsen K. C., Tichnell C. R., Carey M. C., McCusker J. K.. A Modular Approach to Light Capture and Synthetic Tuning of the Excited-State Properties of Fe(II)-Based Chromophores. J. Am. Chem. Soc. 2021;143:8086–8098. doi: 10.1021/jacs.1c02451. [DOI] [PubMed] [Google Scholar]
- Witas K., Nair S. S., Maisuradze T., Zedler L., Schmidt H., Garcia-Porta P., Rein A. S. J., Bolter T., Rau S., Kupfer S., Dietzek-Ivanšić B., Sorsche D. U.. Beyond the First Coordination Sphere–Manipulating the Excited-State Landscape in Iron(II) Chromophores with Protons. J. Am. Chem. Soc. 2024;146:19710–19719. doi: 10.1021/jacs.4c00552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herr P., Kerzig C., Larsen C. B., Häussinger D., Wenger O. S.. Manganese(I) complexes with metal-to-ligand charge transfer luminescence and photoreactivity. Nat. Chem. 2021;13:956–962. doi: 10.1038/s41557-021-00744-9. [DOI] [PubMed] [Google Scholar]
- Wegeberg C., Wenger O. S.. Luminescent First-Row Transition Metal Complexes. JACS Au. 2021;1:1860–1876. doi: 10.1021/jacsau.1c00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegeberg C., Sinha N., Häussinger D., Prescimone A., Wenger O. S.. Photoredox-active Cr(0) luminophores featuring photophysical properties competitive with Ru(II) and Os(II) complexes. Nat. Chem. 2023;15:1730–1736. doi: 10.1038/s41557-023-01297-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker J. K.. Electronic structure in the transition metal block and its implications for light harvesting. Science. 2019;363:484–488. doi: 10.1126/science.aav9104. [DOI] [PubMed] [Google Scholar]
- Dorn, M. ; East, N. R. ; Förster, C. ; Kitzmann, W. ; Moll, J. ; Reichenauer, F. ; Reuter, T. ; Stein, L. ; Heinze, K. . d-d and charge transfer photochemistry of 3d metal complexes. In Comprehensive Inorganic Chemistry III; Elsevier, 2023; pp. 707–788. [Google Scholar]
- Wenger O. S.. Is Iron the New Ruthenium? Chem. Eur. J. 2019;25(24):6043–6052. doi: 10.1002/chem.201806148. [DOI] [PubMed] [Google Scholar]
- Wenger O. S.. Photoactive Complexes with Earth-Abundant Metals. J. Am. Chem. Soc. 2018;140:13522–13533. doi: 10.1021/jacs.8b08822. [DOI] [PubMed] [Google Scholar]
- Arias-Rotondo D. M., McCusker J. K.. The photophysics of photoredox catalysis: a roadmap for catalyst design. Chem. Soc. Rev. 2016;45:5803–5820. doi: 10.1039/C6CS00526H. [DOI] [PubMed] [Google Scholar]
- Alowakennu M. M., Ghosh A., McCusker J. K.. Direct Evidence for Excited Ligand Field State-based Oxidative Photoredox Chemistry of a Cobalt(III) Polypyridyl Photosensitizer. J. Am. Chem. Soc. 2023;145:20786–20791. doi: 10.1021/jacs.3c09374. [DOI] [PubMed] [Google Scholar]
- Kaufhold S., Wärnmark K.. Design and Synthesis of Photoactive Iron N-Heterocyclic Carbene Complexes. Catalysts. 2020;10:132. doi: 10.3390/catal10010132. [DOI] [Google Scholar]
- Otto S., Grabolle M., Förster C., Kreitner C., Resch-Genger U., Heinze K.. [Cr(ddpd)2]3+: A Molecular, Water-Soluble, Highly NIR-Emissive Ruby Analogue. Angew. Chem., Int. Ed. 2015;54:11572–11576. doi: 10.1002/anie.201504894. [DOI] [PubMed] [Google Scholar]
- Reichenauer F., Wang C., Förster C., Boden P., Ugur N., Báez-Cruz R., Kalmbach J., Carrella L. M., Rentschler E., Ramanan C., Niedner-Schatteburg G., Gerhards M., Seitz M., Resch-Genger U., Heinze K.. Strongly Red-Emissive Molecular Ruby [Cr(bpmp)2]3+ Surpasses [Ru(bpy)3]2+ . J. Am. Chem. Soc. 2021;143:11843–11855. doi: 10.1021/jacs.1c05971. [DOI] [PubMed] [Google Scholar]
- Jiménez J., Poncet M., Míguez-Lago S., Grass S., Lacour J., Besnard C., Cuerva J. M., Campaña A. G., Piguet C.. Bright Long-Lived Circularly Polarized Luminescence in Chiral Chromium(III) Complexes. Angew. Chem., Int. Ed. 2021;60:10095–10102. doi: 10.1002/anie.202101158. [DOI] [PubMed] [Google Scholar]
- Kitzmann W. R., Heinze K.. Charge-Transfer and Spin-Flip States: Thriving as Complements. Angew. Chem., Int. Ed. 2023;62(15):e202213207. doi: 10.1002/anie.202213207. [DOI] [PubMed] [Google Scholar]
- Jones R. W., Auty A. J., Wu G., Persson P., Appleby M. V., Chekulaev D., Rice C. R., Weinstein J. A., Elliott P. I. P., Scattergood P. A.. Direct Determination of the Rate of Intersystem Crossing in a Near-IR Luminescent Cr(III) Triazolyl Complex. J. Am. Chem. Soc. 2023;145:12081–12092. doi: 10.1021/jacs.3c01543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha N., Jiménez J., Pfund B., Prescimone A., Piguet C., Wenger O. S.. A Near-Infrared-II Emissive Chromium(III) Complex. Angew. Chem., Int. Ed. 2021;60:23722–23728. doi: 10.1002/anie.202106398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicka N., Craze C. J., Horton P. N., Coles S. J., Richards E., Pope S. J. A.. Long-lived, near-IR emission from Cr(III) under ambient conditions. Chem. Commun. 2022;58:5733–5736. doi: 10.1039/D2CC01434C. [DOI] [PubMed] [Google Scholar]
- Cheng Y., Yang Q., He J., Zou W., Liao K., Chang X., Zou C., Lu W.. The energy gap law for NIR-phosphorescent Cr(III) complexes. Dalton Trans. 2023;52:2561–2565. doi: 10.1039/D2DT02872G. [DOI] [PubMed] [Google Scholar]
- Serpone N., Jamieson M. A., Henry M. S., Hoffman M. Z., Bolletta F., Maestri M.. Excited-State Behavior of Polypyridyl Complexes of Chromium(III) J. Am. Chem. Soc. 1979;101:2907–2916. doi: 10.1021/ja00505a019. [DOI] [Google Scholar]
- Jiménez J.-R., Doistau B., Cruz C. M., Besnard C., Cuerva J. M., Campaña A. G., Piguet C.. Chiral Molecular Ruby [Cr(dqp)2]3+ with Long-Lived Circularly Polarized Luminescence. J. Am. Chem. Soc. 2019;141:13244–13252. doi: 10.1021/jacs.9b06524. [DOI] [PubMed] [Google Scholar]
- Büldt L. A., Wenger O. S.. Chromium Complexes for Luminescence, Solar Cells, Photoredox Catalysis, Upconversion, and Phototriggered NO Release. Chem. Sci. 2017;8:7359–7367. doi: 10.1039/C7SC03372A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramasinghe W. A., Bird P. H., Serpone N.. Interligand Pockets in Polypyridyl Complexes. Crystal and Molecular Structure of the Bis(Terpyridyl)Chromium(III) Cation. Inorg. Chem. 1982;21:2694–2698. doi: 10.1021/ic00137a032. [DOI] [Google Scholar]
- Goodwin K. V., Pennington W. T., Petersen J. D.. A Redetermination of the Crystal and Molecular Structure of Tris(Bipyridine)Chromium(III) Hexafluorophosphate. Inorg. Chem. 1989;28:2016–2018. doi: 10.1021/ic00309a051. [DOI] [Google Scholar]
- Ryu C. K., Endicott J. F.. Synthesis, spectroscopy, and photophysical behavior of mixed-ligand mono- and bis(polypyridyl)chromium(III) complexes. Examples of efficient, thermally activated excited-state relaxation without back intersystem crossing. Inorg. Chem. 1988;27:2203–2214. doi: 10.1021/ic00286a002. [DOI] [Google Scholar]
- Guionneau, P. ; Marchivie, M. ; Bravic, G. ; Létard, J.-F. ; Chasseau, D. . Structural Aspects of Spin Crossover. Example of the [FeIILn(NCS)2] Complexes. In Spin Crossover in Transition Metal Compounds II; Springer: Berlin, Heidelberg, 2004; Vol. 234, pp. 97–128. [Google Scholar]
- Harris J. P., Reber C., Colmer H. E., Jackson T. A., Forshaw A. P., Smith J. M., Kinney R. A., Telser J.. Near-infrared 2Eg → 4A2g and visible LMCT luminescence from a molecular bis-(tris(carbene)borate) manganese(IV) complex. Can. J. Chem. 2017;95:547–552. doi: 10.1139/cjc-2016-0607. [DOI] [Google Scholar]
- Huang T., Du P., Cheng X., Lin Y.-M.. Manganese Complexes with Consecutive Mn(IV) → Mn(III) Excitation for Versatile Photoredox Catalysis. J. Am. Chem. Soc. 2024;146:24515–24525. doi: 10.1021/jacs.4c07084. [DOI] [PubMed] [Google Scholar]
- Kaul N., Asempa E., Valdez-Moreira J. A., Smith J. M., Jakubikova E., Hammarström L.. Enter MnIV – NHC: A Dark Photooxidant with a Long-Lived Charge-Transfer Excited State. J. Am. Chem. Soc. 2024;146:24619–24629. doi: 10.1021/jacs.4c08588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegeberg C., Häussinger D., Kupfer S., Wenger O. S.. Controlling the Photophysical Properties of a Series of Isostructural d6 Complexes Based on Cr0, MnI, and FeII . J. Am. Chem. Soc. 2024;146:4605–4619. doi: 10.1021/jacs.3c11580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegeberg C., Wenger O. S.. Luminescent chromium(0) and manganese(I) complexes. Dalton Trans. 2022;51:1297–1302. doi: 10.1039/D1DT03763C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büldt L. A., Guo X., Vogel R., Prescimone A., Wenger O. S.. A Tris(diisocyanide)chromium(0) Complex Is a Luminescent Analog of Fe(2,2′-Bipyridine)3 2+ . J. Am. Chem. Soc. 2017;139:985–992. doi: 10.1021/jacs.6b11803. [DOI] [PubMed] [Google Scholar]
- Wegeberg C., Häussinger D., Wenger O. S.. Pyrene-Decoration of a Chromium(0) Tris(diisocyanide) Enhances Excited State Delocalization: A Strategy to Improve the Photoluminescence of 3d6 Metal Complexes. J. Am. Chem. Soc. 2021;143:15800–15811. doi: 10.1021/jacs.1c07345. [DOI] [PubMed] [Google Scholar]
- Carey M. C., Adelman S. L., McCusker J. K.. Insights into the excited state dynamics of Fe(II) polypyridyl complexes from variable-temperature ultrafast spectroscopy. Chem. Sci. 2019;10:134–144. doi: 10.1039/C8SC04025G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhouse M. D., McCusker J. K.. Mechanistic Origin of Photoredox Catalysis Involving Iron(II) Polypyridyl Chromophores. J. Am. Chem. Soc. 2020;142:16229–16233. doi: 10.1021/jacs.0c08389. [DOI] [PubMed] [Google Scholar]
- Marri A. R., Marekha B., Penfold T., Haacke S., Gros P. C.. Towards panchromatic Fe(II) NHC sensitizers via HOMO inversion. Inorg. Chem. Front. 2022;10:118–126. doi: 10.1039/D2QI01903E. [DOI] [Google Scholar]
- Braun J. D., Lozada I. B., Kolodziej C., Burda C., Newman K. M. E., van Lierop J., Davis R. L., Herbert D. E.. Iron(II) coordination complexes with panchromatic absorption and nanosecond charge-transfer excited state lifetimes. Nat. Chem. 2019;11:1144–1150. doi: 10.1038/s41557-019-0357-z. [DOI] [PubMed] [Google Scholar]
- Leis W., Argüello Cordero M. A., Lochbrunner S., Schubert H., Berkefeld A.. A Photoreactive Iron(II) Complex Luminophore. J. Am. Chem. Soc. 2022;144:1169–1173. doi: 10.1021/jacs.1c13083. [DOI] [PubMed] [Google Scholar]
- Lindh L., Rosemann N. W., Losada I. B., Persson S., Goriya Y., Fan H., Gordivska O., Wärnmark K., Uhlig J., Chábera P., Yartsev A., Persson P.. Side-Group switching between metal-to-ligand charge-transfer and metal-centered excited state properties in iron(II) N-heterocyclic carbene complexes. Coord. Chem. Rev. 2024;506:215709. doi: 10.1016/j.ccr.2024.215709. [DOI] [Google Scholar]
- Prakash O., Lindh L., Gupta A. K., Hoang Hai Y. T., Kaul N., Chábera P., Lindgren F., Ericsson T., Häggström L., Strand D., Yartsev A., Lomoth R., Persson P., Wärnmark K.. Tailoring the Photophysical Properties of a Homoleptic Iron(II) Tetra N-Heterocyclic Carbene Complex by Attaching an Imidazolium Group to the (C∧N∧C) Pincer Ligand A Comparative Study. Inorg. Chem. 2024;63:2909–2918. doi: 10.1021/acs.inorgchem.3c02890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter T., Kruse A., Schoch R., Lochbrunner S., Bauer M., Heinze K.. Higher MLCT lifetime of carbene iron(II) complexes by chelate ring expansion. Chem. Commun. 2021;57:7541–7544. doi: 10.1039/D1CC02173G. [DOI] [PubMed] [Google Scholar]
- Liu Y., Kjær K. S., Fredin L. A., Chábera P., Harlang T., Canton S. E., Lidin S., Zhang J., Lomoth R., Bergquist K.-E.. et al. A heteroleptic ferrous complex with mesoionic bis(1,2,3-triazol-5-ylidene) ligands: taming the MLCT excited state of iron(II) Chem. Eur. J. 2015;21:3628–3639. doi: 10.1002/chem.201405184. [DOI] [PubMed] [Google Scholar]
- Fredin L. A., Pápai M., Rozsályi E., Vankó G., Wärnmark K., Sundström V., Persson P.. Exceptional Excited-State Lifetime of an Iron(II) – N-Heterocyclic Carbene Complex Explained. J. Phys. Chem. Lett. 2014;5:2066–2071. doi: 10.1021/jz500829w. [DOI] [PubMed] [Google Scholar]
- Fatur S. M., Shepard S. G., Higgins R. F., Shores M. P., Damrauer N. H.. A Synthetically Tunable System To Control MLCT Excited-State Lifetimes and Spin States in Iron(II) Polypyridines. J. Am. Chem. Soc. 2017;139:4493–4505. doi: 10.1021/jacs.7b00700. [DOI] [PubMed] [Google Scholar]
- Zimmer P., Burkhardt L., Friedrich A., Steube J., Neuba A., Schepper R., Müller P., Flörke U., Huber M., Lochbrunner S., Bauer M.. The Connection between NHC Ligand Count and Photophysical Properties in Fe(II) Photosensitizers: An Experimental Study. Inorg. Chem. 2018;57:360–373. doi: 10.1021/acs.inorgchem.7b02624. [DOI] [PubMed] [Google Scholar]
- Chábera P., Kjaer K. S., Prakash O., Honarfar A., Liu Y., Fredin L. A., Harlang T. C. B., Lidin S., Uhlig J., Sundström V., Lomoth R., Persson P., Wärnmark K.. FeII Hexa N-Heterocyclic Carbene Complex with a 528 ps Metal-to-Ligand Charge-Transfer Excited-State Lifetime. J. Phys. Chem. Lett. 2018;9:459–463. doi: 10.1021/acs.jpclett.7b02962. [DOI] [PubMed] [Google Scholar]
- Lindh L., Gordivska O., Persson S., Michaels H., Fan H., Chábera P., Rosemann N. W., Gupta A. K., Benesperi I., Uhlig J., Prakash O., Sheibani E., Kjaer K. S., Boschloo G., Yartsev A., Freitag M., Lomoth R., Persson P., Wärnmark K.. Dye-sensitized solar cells based on Fe N-heterocyclic carbene photosensitizers with improved rod-like push-pull functionality. Chem. Sci. 2021;12:16035–16053. doi: 10.1039/D1SC02963K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppermann M., Zinna F., Lacour J., Chergui M.. Chiral control of spin-crossover dynamics in Fe(II) complexes. Nat. Chem. 2022;14:739–745. doi: 10.1038/s41557-022-00933-0. [DOI] [PubMed] [Google Scholar]
- Liu Y., Harlang T., Canton S. E., Chábera P., Suárez-Alcántara K., Fleckhaus A., Vithanage D. A., Göransson E., Corani A., Lomoth R., Sundström V., Wärnmark K.. Towards longer-lived metal-to-ligand charge transfer states of iron(II) complexes: an N-heterocyclic carbene approach. Chem. Commun. 2013;49:6412–6414. doi: 10.1039/c3cc43833c. [DOI] [PubMed] [Google Scholar]
- Moll J., Naumann R., Sorge L., Förster C., Gessner N., Burkhardt L., Ugur N., Nuernberger P., Seidel W., Ramanan C.. et al. Pseudo-Octahedral Iron(II) Complexes with Near-Degenerate Charge Transfer and Ligand Field States at the Franck-Condon Geometry. Chem. Eur. J. 2022;28:e202201858. doi: 10.1002/chem.202201858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengel A. K. C., Förster C., Breivogel A., Mack K., Ochsmann J. R., Laquai F., Ksenofontov V., Heinze K.. A Heteroleptic Push–Pull Substituted Iron(II) Bis(Tridentate) Complex with Low-Energy Charge-Transfer States. Chem. Eur. J. 2015;21:704–714. doi: 10.1002/chem.201404955. [DOI] [PubMed] [Google Scholar]
- Prakash O., Chábera P., Kaul N., Hlynsson V. F., Rosemann N. W., Losada I. B., Hoang Hai Y. T., Huang P., Bendix J., Ericsson T., Häggström L., Gupta A. K., Strand D., Yartsev A., Lomoth R., Persson P., Wärnmark K.. How Rigidity and Conjugation of Bidentate Ligands Affect the Geometry and Photophysics of Iron N-Heterocyclic Complexes: A Comparative Study. Inorg. Chem. 2024;63:4461–4473. doi: 10.1021/acs.inorgchem.3c03972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger O. S.. A bright future for photosensitizers. Nat. Chem. 2020;12:323–324. doi: 10.1038/s41557-020-0448-x. [DOI] [PubMed] [Google Scholar]
- May A. M., Dempsey J. L.. A new era of LMCT: leveraging ligand-to-metal charge transfer excited states for photochemical reactions. Chem. Sci. 2024;15:6661–6678. doi: 10.1039/D3SC05268K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaltseva P., Wenger O. S.. Photocatalysis Gets Energized by Abundant Metals. Science. 2023;382:153–154. doi: 10.1126/science.adk5923. [DOI] [PubMed] [Google Scholar]
- Marcus R. A., Sutin N.. Electron transfers in chemistry and biology. Biochim. Biophys. Acta, Rev. Bioenerg. 1985;811:265–322. doi: 10.1016/0304-4173(85)90014-X. [DOI] [Google Scholar]
- Caspar J. V., Meyer T. J.. Application of the energy gap law to nonradiative, excited-state decay. J. Phys. Chem. 1983;87:952–957. doi: 10.1021/j100229a010. [DOI] [Google Scholar]
- Conti C., Castelli F., Forster L. S.. Photophysics of hexakis(cyano)chromate(3-) and hexakis(cyano)cobaltate(3-) in polyalcohol-water solutions at room temperature. J. Phys. Chem. 1979;83:2371–2376. doi: 10.1021/j100481a013. [DOI] [Google Scholar]
- Viaene L., D’Olieslager J.. Luminescence from and absorption by the 3T1g level of the hexacyanocobaltate(III) ion. Inorg. Chem. 1987;26:960–962. doi: 10.1021/ic00253a039. [DOI] [Google Scholar]
- Viaene L., D’Olieslager J., Ceulemans A., Vanquickenborne L. G.. Excited-state spectroscopy of hexacyanocobaltate(III) J. Am. Chem. Soc. 1979;101:1405–1409. doi: 10.1021/ja00500a009. [DOI] [Google Scholar]
- Mascarenhas E. J., Fondell M., Büchner R., Eckert S., Vaz da Cruz V., Föhlisch A.. The Role of the Lowest Excited Triplet State in Defining the Rate of Photoaquation of Hexacyanometalates. J. Phys. Chem. Lett. 2024;15:241–247. doi: 10.1021/acs.jpclett.3c02775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufhold S., Rosemann N. W., Chábera P., Lindh L., Bolaño Losada I., Uhlig J., Pascher T., Strand D., Wärnmark K., Yartsev A., Persson P.. Microsecond Photoluminescence and Photoreactivity of a Metal-Centered Excited State in a Hexacarbene–Co(III) Complex. J. Am. Chem. Soc. 2021;143:1307–1312. doi: 10.1021/jacs.0c12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarranton J. T., McCusker J. K.. Ligand-Field Spectroscopy of Co(III) Complexes and the Development of a Spectrochemical Series for Low-Spin d6 Charge-Transfer Chromophores. J. Am. Chem. Soc. 2022;144:12488–12500. doi: 10.1021/jacs.2c04945. [DOI] [PubMed] [Google Scholar]
- Ashley D. C., Jakubikova E.. Tuning the Redox Potentials and Ligand Field Strength of Fe(II) Polypyridines: The Dual π-Donor and π-Acceptor Character of Bipyridine. Inorg. Chem. 2018;57:9907–9917. doi: 10.1021/acs.inorgchem.8b01002. [DOI] [PubMed] [Google Scholar]
- Frenzel B. A., Hamaker C. G., Hightower S. E.. Synthesis, characterization, X-ray structure and DFT calculations of a bistridentate Co(III) complex based on the 2,6-bis(8′-quinolinyl)pyridine ligand. Inorg. Chem. Commun. 2014;50:51–53. doi: 10.1016/j.inoche.2014.10.024. [DOI] [Google Scholar]
- Pal A. K., Li C., Hanan G. S., Zysman-Colman E.. Blue-Emissive Cobalt(III) Complexes and Their Use in the Photocatalytic Trifluoromethylation of Polycyclic Aromatic Hydrocarbons. Angew. Chem., Int. Ed. 2018;57:8027–8031. doi: 10.1002/anie.201802532. [DOI] [PubMed] [Google Scholar]
- Frenzel B. A., Mergerdichian Z., Schumaker J. E., Kuester C. T., Hamaker C. G., Hightower S. E.. Synthesis, Structural Characterization and Spectroscopic Properties of Cobalt Complexes with the 2,6-Bis(8′-Quinolinyl)Pyridine Ligand. Polyhedron. 2014;81:653–660. doi: 10.1016/j.poly.2014.07.029. [DOI] [Google Scholar]
- Castellano F. N., Rosko M. C.. Steric and Electronic Influence of Excited-State Decay in Cu(I) MLCT Chromophores. Acc. Chem. Res. 2024;57:2872–2886. doi: 10.1021/acs.accounts.4c00476. [DOI] [PubMed] [Google Scholar]
- Ortiz R. J., Mondal R., McCusker J. K., Herbert D. E.. Leveraging Intramolecular π-Stacking to Access an Exceptionally Long-Lived 3MC Excited State in an Fe(II) Carbene Complex. J. Am. Chem. Soc. 2025;147:1694–1708. doi: 10.1021/jacs.4c12650. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Lee T. S., Favale J. M., Leary D. C., Petersen J. L., Scholes G. D., Castellano F. N., Milsmann C.. Delayed Fluorescence from a Zirconium(IV) Photosensitizer with Ligand-to-Metal Charge-Transfer Excited States. Nat. Chem. 2020;12:345–352. doi: 10.1038/s41557-020-0430-7. [DOI] [PubMed] [Google Scholar]
- Constable E. C., Harris K., Housecroft C. E., Neuburger M., Zampese J. A.. Turning {M(tpy)2}n+ embraces and CH···π interactions on and off in homoleptic cobalt(II) and cobalt(III) bis(2,2′: 6′,2″-terpyridine) complexes. CrystEngcomm. 2010;12:2949–2961. doi: 10.1039/c002834g. [DOI] [Google Scholar]
- Jäger M., Eriksson L., Bergquist J., Johansson O.. Synthesis and Characterization of 2,6-Di(quinolin-8-yl)pyridines. New Ligands for Bistridentate RuII Complexes with Microsecond Luminescent Lifetimes. J. Org. Chem. 2007;72:10227–10230. doi: 10.1021/jo7015373. [DOI] [PubMed] [Google Scholar]
- Hammarström L., Johansson O.. Expanded bite angles in tridentate ligands. Improving the photophysical properties in bistridentate RuII polypyridine complexes. Coord. Chem. Rev. 2010;254:2546–2559. doi: 10.1016/j.ccr.2010.01.006. [DOI] [Google Scholar]
- Parada G. A., Fredin L. A., Santoni M.-P., Jäger M., Lomoth R., Hammarström L., Johansson O., Persson P., Ott S.. Tuning the Electronics of Bis(tridentate)ruthenium(II) Complexes with Long-Lived Excited States: Modifications to the Ligand Skeleton beyond Classical Electron Donor or Electron Withdrawing Group Decorations. Inorg. Chem. 2013;52:5128–5137. doi: 10.1021/ic400009m. [DOI] [PubMed] [Google Scholar]
- Otto S., Förster C., Wang C., Resch-Genger U., Heinze K.. A Strongly Luminescent Chromium(III) Complex Acid. Chem. Eur. J. 2018;24:12555–12563. doi: 10.1002/chem.201802797. [DOI] [PubMed] [Google Scholar]
- Kupfer, S. ; Maisuradze, T. . Quantum Chemical Data: structural Control Of metal-Centered Excited States In cobalt(III) Complexes Via Bite Angle And π-π Interactions; Zenodo, 2025, 10.5281/zenodo.15365044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janiak C.. A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands. J. Chem. Soc., Dalton Trans. 2000:3885–3896. doi: 10.1039/b003010o. [DOI] [Google Scholar]
- Favre, H. A. ; Powell, W. H. . Nomenclature of Organic Chemistry; The Royal Society of Chemistry, 2013. [Google Scholar]
- Förster C., Dorn M., Reuter T., Otto S., Davarci G., Reich T., Carrella L., Rentschler E., Heinze K.. Ddpd as Expanded Terpyridine: Dramatic Effects of Symmetry and Electronic Properties in First Row Transition Metal Complexes. Inorganics. 2018;6:86. doi: 10.3390/inorganics6030086. [DOI] [Google Scholar]
- Nolan J. P., Jones T. W., Donne S. W., Wilson G. J.. Tuning the Electrochemistry of Homoleptic Cobalt 4,4′-Disubstituted-2,2′-Bipyridine Redox Mediators. Electrochim. Acta. 2013;108:690–697. doi: 10.1016/j.electacta.2013.07.016. [DOI] [Google Scholar]
- Tanabe Y., Sugano S.. On the Absorption Spectra of Complex Ions II. J. Phys. Soc. Jpn. 1954;9:766–779. doi: 10.1143/JPSJ.9.766. [DOI] [Google Scholar]
- Morselli G., Reber C., Wenger O. S.. Molecular Design Principles for Photoactive Transition Metal Complexes: A Guide for “Photo-Motivated” Chemists. J. Am. Chem. Soc. 2025;147:11608–11624. doi: 10.1021/jacs.5c02096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger M., Smeigh A., Lombeck F., Görls H., Collin J.-P., Sauvage J.-P., Hammarström L., Johansson O.. Cyclometalated RuII Complexes with Improved Octahedral Geometry: Synthesis and Photophysical Properties. Inorg. Chem. 2010;49:374–376. doi: 10.1021/ic9020788. [DOI] [PubMed] [Google Scholar]
- Pal A. K., Serroni S., Zaccheroni N., Campagna S., Hanan G. S.. Near Infra-Red Emitting Ru(II) Complexes of Tridentate Ligands: Electrochemical and Photophysical Consequences of a Strong Donor Ligand with Large Bite Angles. Chem. Sci. 2014;5:4800–4811. doi: 10.1039/C4SC01604A. [DOI] [Google Scholar]
- Brown A. M., McCusker C. E., McCusker J. K.. Spectroelectrochemical Identification of Charge-Transfer Excited States in Transition Metal-Based Polypyridyl Complexes. Dalton Trans. 2014;43:17635–17646. doi: 10.1039/C4DT02849J. [DOI] [PubMed] [Google Scholar]
- Larsen C. B., Braun J. D., Lozada I. B., Kunnus K., Biasin E., Kolodziej C., Burda C., Cordones A. A., Gaffney K. J., Herbert D. E.. Reduction of Electron Repulsion in Highly Covalent Fe-Amido Complexes Counteracts the Impact of a Weak Ligand Field on Excited-State Ordering. J. Am. Chem. Soc. 2021;143:20645–20656. doi: 10.1021/jacs.1c06429. [DOI] [PubMed] [Google Scholar]
- Reinhard M. E., Sidhu B. K., Lozada I. B., Powers-Riggs N., Ortiz R. J., Lim H., Nickel R., Lierop J. V., Alonso-Mori R., Chollet M., Gee L. B., Kramer P. L., Kroll T., Raj S. L., van Driel T. B., Cordones A. A., Sokaras D., Herbert D. E., Gaffney K. J.. Time-Resolved X-Ray Emission Spectroscopy and Synthetic High-Spin Model Complexes Resolve Ambiguities in Excited-State Assignments of Transition-Metal Chromophores: A Case Study of Fe-Amido Complexes. J. Am. Chem. Soc. 2024;146:17908–17916. doi: 10.1021/jacs.4c02748. [DOI] [PubMed] [Google Scholar]
- Garakyaraghi S., Danilov E. O., McCusker C. E., Castellano F. N.. Transient Absorption Dynamics of Sterically Congested Cu(I) MLCT Excited States. J. Phys. Chem. A. 2015;119:3181–3193. doi: 10.1021/acs.jpca.5b00901. [DOI] [PubMed] [Google Scholar]
- Scaiano J. C.. A Beginners Guide to Understanding the Mechanisms of Photochemical Reactions: Things You Should Know If Light Is One of Your Reagents. Chem. Soc. Rev. 2023;52:6330–6343. doi: 10.1039/D3CS00453H. [DOI] [PubMed] [Google Scholar]
- Pfund B., Wenger O. S.. Excited Organic Radicals in Photoredox Catalysis. JACS Au. 2025;5:426–447. doi: 10.1021/jacsau.4c00974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfund B., Gejsnæs-Schaad D., Lazarevski B., Wenger O. S.. Picosecond Reactions of Excited Radical Ion Super-Reductants. Nat. Commun. 2024;15(1):4738. doi: 10.1038/s41467-024-49006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley J. D., Zieleniewska A., Ripberger H. H., Shin N. Y., Lazorski M. S., Mast Z. J., Sayre H. J., McCusker J. K., Scholes G. D., Knowles R. R., Reid O. G., Rumbles G.. Ion-Pair Reorganization Regulates Reactivity in Photoredox Catalysts. Nat. Chem. 2022;14:746–753. doi: 10.1038/s41557-022-00911-6. [DOI] [PubMed] [Google Scholar]
- Zanzi J., Pastorel Z., Duhayon C., Lognon E., Coudret C., Monari A., Dixon I. M., Canac Y., Smietana M., Baslé O.. Counterion Effects in [Ru(bpy)3](X)2-Photocatalyzed Energy Transfer Reactions. JACS Au. 2024;4:3049–3057. doi: 10.1021/jacsau.4c00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger O. S.. Proton-Coupled Electron Transfer Originating from Excited States of Luminescent Transition-Metal Complexes. Chem. Eur. J. 2011;17:11692–11702. doi: 10.1002/chem.201102011. [DOI] [PubMed] [Google Scholar]
- Schmitz M., Bertrams M.-S., Sell A. C., Glaser F., Kerzig C.. Efficient Energy and Electron Transfer Photocatalysis with a Coulombic Dyad. J. Am. Chem. Soc. 2024;146:25799–25812. doi: 10.1021/jacs.4c08551. [DOI] [PubMed] [Google Scholar]
- Chantry N., Cotic A., De Kreijger S., Di Forti R., Elias B., Troian-Gautier L., Cadranel A.. Nature of Anti-Dissipative High-Energy Excited States in Quaterpyridine-Bridged Ruthenium Complexes. Angew. Chem., Int. Ed. 2025:e202507738. doi: 10.1002/anie.202507738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfund B., Wenger O. S.. Breaking Kasha’s Rule to Enable Higher Reactivity in Photoredox Catalysis. J. Am. Chem. Soc. 2025 doi: 10.1021/jacs.5c06115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke R. H., Hochstrasser R. M.. Location and Assignment of the Lowest Triplet State of Perylene. J. Mol. Spectrosc. 1969;32:309–319. doi: 10.1016/0022-2852(69)90225-2. [DOI] [Google Scholar]
- Fielding L.. Determination of Association Constants (Ka) from Solution NMR Data. Tetrahedron. 2000;56:6151–6170. doi: 10.1016/S0040-4020(00)00492-0. [DOI] [Google Scholar]
- Pysh E. S., Yang N. C.. Polarographic Oxidation Potentials of Aromatic Compounds. J. Am. Chem. Soc. 1963;85:2124–2130. doi: 10.1021/ja00897a019. [DOI] [Google Scholar]
- Turner J. M., Karl M. W., Kauffman J. F.. Spectroscopic Signatures of Protonated Perylene in Concentrated Sulfuric Acid. J. Photochem. Photobiol. A Chem. 2004;163:433–438. doi: 10.1016/j.jphotochem.2004.01.015. [DOI] [Google Scholar]
- Ni W., Sun L., Gurzadyan G. G.. Ultrafast Spectroscopy Reveals Singlet Fission, Ionization and Excimer Formation in Perylene Film. Sci. Rep. 2021;11(1):5220. doi: 10.1038/s41598-021-83791-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkel P. B., Luo P., Dinnocenzo J. P., Farid S.. Accurate Oxidation Potentials of Benzene and Biphenyl Derivatives via Electron-Transfer Equilibria and Transient Kinetics. J. Org. Chem. 2009;74:5163–5173. doi: 10.1021/jo9011267. [DOI] [PubMed] [Google Scholar]
- Guirado G., Fleming C. N., Lingenfelter T. G., Williams M. L., Zuilhof H., Dinnocenzo J. P.. Nanosecond Redox Equilibrium Method for Determining Oxidation Potentials in Organic Media. J. Am. Chem. Soc. 2004;126:14086–14094. doi: 10.1021/ja046946g. [DOI] [PubMed] [Google Scholar]
- Schmid L., Kerzig C., Prescimone A., Wenger O. S.. Photostable Ruthenium(II) Isocyanoborato Luminophores and Their Use in Energy Transfer and Photoredox Catalysis. JACS Au. 2021;1:819–832. doi: 10.1021/jacsau.1c00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malme J. T., Weaver J. N., Girolami G. S., Vura-Weis J.. Picosecond Metal-to-Ligand Charge-Transfer Deactivation in Co(ppy)3 via Jahn–Teller Distortion. Inorg. Chem. 2024;63:13825–13830. doi: 10.1021/acs.inorgchem.4c01959. [DOI] [PubMed] [Google Scholar]
- Braun J. D., Lozada I. B., Herbert D. E.. In Pursuit of Panchromatic Absorption in Metal Coordination Complexes: Experimental Delineation of the HOMO Inversion Model Using Pseudo-Octahedral Complexes of Diarylamido Ligands. Inorg. Chem. 2020;59:17746–17757. doi: 10.1021/acs.inorgchem.0c02973. [DOI] [PubMed] [Google Scholar]
- Mukherjee S., Torres D. E., Jakubikova E.. HOMO Inversion as a Strategy for Improving the Light-Absorption Properties of Fe(II) Chromophores. Chem. Sci. 2017;8:8115–8126. doi: 10.1039/C7SC02926H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha N., Pfund B., Wegeberg C., Prescimone A., Wenger O. S.. Cobalt(III) Carbene Complex with an Electronic Excited-State Structure Similar to Cyclometalated Iridium(III) Compounds. J. Am. Chem. Soc. 2022;144:9859–9873. doi: 10.1021/jacs.2c02592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz R. J., Braun J. D., Williams J. A. G., Herbert D. E.. Brightly Luminescent Platinum Complexes of N∧C – ∧ N Ligands Forming Six-Membered Chelate Rings: Offsetting Deleterious Ring Size Effects Using Site-Selective Benzannulation. Inorg. Chem. 2021;60:16881–16894. doi: 10.1021/acs.inorgchem.1c02551. [DOI] [PubMed] [Google Scholar]
- Steube J., Kruse A., Bokareva O. S., Reuter T., Demeshko S., Schoch R., Argüello Cordero M. A., Krishna A., Hohloch S., Meyer F., Heinze K., Kühn O., Lochbrunner S., Bauer M.. Janus-type emission from a cyclometalated iron(III) complex. Nat. Chem. 2023;15:468–474. doi: 10.1038/s41557-023-01137-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna A., Fritsch L., Steube J., Argüello Cordero M. A., Schoch R., Neuba A., Lochbrunner S., Bauer M.. Low Temperature Emissive Cyclometalated Cobalt(III) Complexes. Inorg. Chem. 2025;64:1401–1409. doi: 10.1021/acs.inorgchem.4c04479. [DOI] [PubMed] [Google Scholar]
- Jin T., Sinha N., Wagner D. S., Prescimone A., Häussinger D., Wenger O. S.. Making Mo(0) a Competitive Alternative to Ir(III) in Phosphors and Photocatalysts. J. Am. Chem. Soc. 2025;147:4587–4594. doi: 10.1021/jacs.4c16672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker C. E., Castellano F. N.. Design of a Long-Lifetime, Earth-Abundant, Aqueous Compatible Cu(I) Photosensitizer Using Cooperative Steric Effects. Inorg. Chem. 2013;52:8114–8120. doi: 10.1021/ic401213p. [DOI] [PubMed] [Google Scholar]
- Huang T., Du P., Lin Y.-M.. Recent Advances in Photoactive First-Row Transition Metal Complexes for Organic Synthesis. Chin. J. Chem. 2025;43:2566–2587. doi: 10.1002/cjoc.70135. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.