

Abstract
A mass spectroscopic analysis of proteins from human herpesvirus 6 (HHV-6)-infected cells showed that the HHV-6 U14 protein coimmunoprecipitated with the tumor suppressor p53. The binding of U14 to p53 was verified by coimmunoprecipitation experiments in both Molt-3 cells infected with HHV-6 and 293 cells cotransfected with U14 and p53 expression vectors. Indirect immunofluorescence assays (IFAs) showed that by 18 h postinfection (hpi) U14 localized to the dot-like structures observed in both the nucleus and cytoplasm where p53 was partly accumulated. Despite Northern blotting evidence that U14 follows late kinetics, the U14 protein was detected immediately after infection (at 3 hpi) by IFA. In addition, by Western blotting, U14 was detected at 0 hpi or in the presence of cycloheximide which completely abolished the expression of IE1 protein. In addition to U14, p53 was detected at 0 hpi although it was not detected in mock-infected cells. Furthermore, both U14 and p53 were clearly detected in the viral particles by Western blotting and immunoelectron microscopy, supporting the idea that U14 and p53 are incorporated into virions. Our study provides the first evidence of the incorporation of cellular p53 into viral particles and suggests that p53 may play an important role in viral infection.
Human herpesviruses (HHVs) are large, enveloped DNA viruses that carry a double-stranded DNA genome of approximately 120 to ∼230 kbp. On the basis of a diverse collection of in vivo and in vitro biological properties, HHVs are divided into three subgroups: alpha, beta, and gamma (27, 28, 39). Human herpesvirus 6 (HHV-6) is a ubiquitous betaherpesvirus that was first isolated in 1986 from the peripheral blood of patients with lymphoproliferative disorders (41) and AIDS (18, 54). HHV-6 utilizes the cellular CD46 molecule as an entry receptor (42), predominantly infects and replicates in CD4+ lymphocytes (25, 51), and may establish latency in the monocyte/macrophage lineage (22).
The tumor suppressor protein p53 is a negative regulator that functions in multiple scenarios, including the cell cycle, apoptosis, and senescence, in response to several genotoxic stresses including viral infections. Like small DNA tumor viruses, HHVs encode their own proteins that interact with p53 and inhibit its innate functions of arresting cell proliferation and inducing apoptosis (5, 12, 15, 33-35, 37, 38, 44, 47, 59). However, HHVs other than HHV-6 and Epstein-Barr virus induce the accumulation of p53 in the nucleus, especially in the viral DNA replication compartment (11, 20, 56, 60), which leads us to suspect that the HHVs exploit p53 for their replication. Although HHV-6 has also been reported to encode a p53-binding protein, open reading frame 1 (ORF-1), which binds to p53 and suppresses its transcriptional activity in NIH 3T3 cells transfected with the SalI L fragment of the HHV-6 genome (19), p53 accumulates not in the nucleus but in the cytoplasm of HHV-6-infected cells, unlike other HHVs, as we previously reported (53). Why HHV-6 causes p53 to accumulate in the cytoplasm and not in the replication compartment as other HHVs do and what mechanism underlies that event remain to be investigated.
Here we used amino acid sequencing to determine the identity of a peptide fragment that coimmunoprecipitated with p53 from HHV-6-infected cell lysates that were incubated with an anti-p53 monoclonal antibody (MAb). The sequence matched that of HHV-6 U14. Although the U14 gene was expressed with late kinetics in Northern blot analysis, the protein was detected as dot-like structures at the immediate-early (IE) phase (3 h postinfection [hpi]) by immunocytochemistry. Western blot analysis revealed that U14 existed at 0 hpi and, furthermore, that p53 increased at 0 hpi, which led us to hypothesize that U14 and p53 were structural proteins delivered from the virion into the cells. We successfully detected both proteins in Western blots of the virion, which was purified from the culture supernatant, and in immunoelectron microscopy of HHV-6 strain HST-infected Molt-3 cells. This is the first report that p53 is incorporated into viral particles, although it remains to be further investigated whether the virion-associated p53 plays a role in the viral life cycle.
MATERIALS AND METHODS
Cells and viruses.
The human T-lymphoblastoid cell lines Molt-3 and MT-4 were cultured in RPMI 1640 medium containing 10% fetal calf serum (FCS). 293 cells were maintained in Dulbecco's modified Eagle's medium (Nissui) supplemented with 10% FCS.
The methods for preparing viral stocks of HHV-6A strain U1102 and HHV-6B strain HST from umbilical cord blood mononuclear cells and infection with these viruses were described previously (53).
To monitor the expression of RNA when de novo protein synthesis and viral DNA replication are inhibited, HST-infected MT-4 cells were treated with 50 μg of cycloheximide (CHX) (Sigma) per ml and 200 μg of phosphonoformic acid (PFA) (Sigma) per ml. For the CHX-treated samples, the cells were pretreated with CHX for 1 h prior to the infection.
Antibodies.
Anti-p53 MAb (DO-1) and anti-p53 goat polyclonal antibody (PAb) (FL-393) were purchased from Santa Cruz Biotechnology. The MAbs against α-tubulin (B-5-1-2) and topoisomerase II α were from Sigma and MBL, respectively. The anti-IE1 rabbit PAb and the anti-glycoprotein L (gL) MAbs have been described previously (30, 32).
The U14 MAb was generated by immunizing mice with recombinant U14 protein according to a method described elsewhere (32). The immunogen was prepared as follows. A PCR product generated with the primer set 6U14-1B (5′-CATGGATCCATGGAAGGGTCGAAGACATTC-3′) and 6U14-2H (5′-CATAAGCTTAATCCTCGAACTCTATTAGGT-3′) was digested with BamHI and HindIII and inserted into a pMAL-c2 vector (New England Biolabs) at these sites. Histidine-tagged protein, His-U14, expressed in Escherichia coli was purified with Ni-nitrilotriacetic acid agarose resin (QIAGEN) and used for immunization.
Immunoprecipitation.
The cells were lysed in TNE (10 mM Tris [pH 7.8], 1% NP-40 0.15 M NaCl, 1 mM EDTA, and 2 μg/ml each of leupeptin, aprotinin, and pepstatin) for 1 h at 4°C and then spun in a centrifuge. The supernatants of the cell lysates were preincubated with protein G-Sepharose that had already been blocked with 0.1% FCS to remove proteins nonspecifically bound to Sepharose beads. Subsequently, the supernatants were incubated with anti-p53 or anti-U14 MAb-bound protein G-Sepharose at 4°C overnight. The Sepharose beads were rinsed four times with TNE buffer, resuspended in Laemmli reducing sample buffer (LRSB) (50 mM Tris [pH 6.8], 2% SDS, 10% glycerol, 50 mM dithiothreitol, 0.5% bromophenol blue), and boiled for 5 min. The immunoprecipitants were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE; 10% polyacrylamide).
Mass spectrometry.
The products of coimmunoprecipitation with anti-p53 MAb were resolved by 6% SDS-PAGE and stained with Coomassie brilliant blue. The protein band of interest was excised from the gel and subjected to in-gel digestion with trypsin. The resulting digest was spotted onto a normal phase chip and analyzed by quadruple time-of-flight mass spectrometry (MS) using a Q-star system (Applied Biosystems) operated in single MS mode. Peptide mass fingerprints were determined by comparison with the database included with Mascot software (Micromass; Waters Corp). Tandem MS (MS/MS) analysis was performed in using MassLynx software (Micromass; Waters Corp).
Northern blotting.
Total RNAs isolated from HST-infected MT-4 cells at various time points and in the presence of CHX (8 hpi) or PFA (72 hpi) using RNeasy (QIAGEN) were fractionated by electrophoresis in denaturing 1% agarose gels containing 18% formaldehyde and blotted onto GeneScreen plus (New England Nuclear Corp) in 20× SSC (1× SSC contains 0.15 M NaCl plus 15 mM sodium citrate [pH 7.0]), cross-linked to the membrane by UV irradiation, and prehybridized for 1 h at 42°C in a hybridization buffer containing 50% formamide, 5× SSPE (0.9 M NaCl, 50 mM NaH2PO4, and 5 mM EDTA [pH 7.4]), 5× Denhardt's solution (0.1% Ficoll, 0.1% polyvinylpyrrolidone, and 0.1% bovine serum albumin [BSA]), 0.5% SDS, and 100 μg of denatured salmon sperm DNA per ml. To make the template for the probes, U14 and U12 cDNAs were excised from the expression vectors, pEF-U14 and pcDNA-U12, at the EcoRI or BamHI-KpnI sites, respectively, and U13 cDNA was generated by PCR using the primer pair 6U13KpnF (5′-CAGGGTACCATGGCGCACGCTAAAAAGCGG-3′) and 6U13BamR (5′-CAGGGATCCTTATAAGAAAGGTGATAGGGT-′). The genomic HST DNA was the template. The probes were labeled with [α-32P]dCTP using a random primer labeling kit (TaKaRa), purified on a Sephadex column, and then hybridized to the blots overnight at 42°C. The membrane was rinsed twice at room temperature for 5 min with 2× SSC-0.1% SDS and once at 65°C for 20 min with 0.1× SSC-0.1% SDS. Subsequently, the membranes were allowed to expose Kodak XAR film, with an intensifying screen.
Western blotting and silver staining.
Cells were resuspended in LRSB at a concentration of 104/μl of buffer and then boiled for 5 min. Equal volumes of whole-cells lysate were loaded onto SDS-polyacrylamide gels, fractionated by electrophoresis, and Western blotted onto polyvinylidene difluoride membranes (Bio-Rad). The blots were blocked with 3% skim milk in Tris-buffered saline (TBS) overnight at 4°C, washed twice briefly with TBST (TBS containing 0.02% Tween 20), and incubated for 1 h at room temperature with primary antibody in TBS containing 3% BSA. The blots were then washed three times with TBST, incubated for 1 h at room temperature with a 1:2,500 dilution of the appropriate secondary antibody conjugated with horseradish peroxidase (Amersham-Pharmacia) in TBS containing 3% skim milk, washed three times with TBST, and developed using an ECL detection kit (Amersham-Pharmacia), according to the manufacturer's instructions.
For nuclear and cytoplasmic fractionation, mock- and HST-infected Molt-3 cells (2 × 106) were collected at 24 hpi and were resuspended in cytoplasmic buffer (10 mM Tris-Hcl [pH 8.0], 10 mM Kcl). Cells were swelled for 2 min on ice and centrifuged after the addition of NP-40 to a final concentration of 0.4%, and the supernatant was used as the soluble cytoplasmic fraction. The pellets, washed five times with the cytoplasmic buffer, were resuspended in RIPA buffer (50 mM Tris-HCl [pH 8.0], 5 mM EDTA, 400 mM NaCl, 1% NP-40, 1% deoxycholate, and 0.025% SDS), and the resuspension was used as the nuclear fraction.
To prepare a time course sample at 0 hpi, Molt-3 cells resuspended in the virus stock were immediately harvested, rinsed twice with phosphate-buffered saline (PBS), and lysed with LRSB.
Silver staining analysis of an SDS-PAGE gel was performed by using two-dimensional silver stain II “Daiichi” (Daiichi Pure Chemicals), according to the manufacture's instructions.
Plasmids and transfection.
The reverse transcription-PCR product amplified with the primer pair 6U14-1K (5′-CGCGGTACCGATGGAAGGGTCGAAGACATT-3′) and 6U14-4B (5′-CGCGGATCCGAATTGGCCAGATAAAGCACT-3′) was subcloned into the pCRII TA cloning vector (Invitrogen), and a clone that contained the wild-type sequence was isolated. The U14 expression vectors, pEF-U14 and pFlag-U14, were generated by inserting the cDNA fragment excised from pCRII into the EcoRI site of pEFBOS-EX and pCMV2-Flag (Sigma), respectively, and determining their direction. The transfection of 293 cells with these vectors was carried out using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Immunocytochemistry.
Cells were plated on glass coverslips, fixed in cold acetone for 5 min, and incubated with the primary antibody in PBS containing 3% BSA for 1 h at room temperature. The coverslips were then washed with PBS-Tween (PBST) for 5 min, followed by a wash with PBS for 5 min, and incubated for 30 min at room temperature with the appropriate secondary antibody labeled with fluorescein isothiocyanate (FITC) or tetramethyl rhodamine isothiocyanate (DAKO). After being washed as above, the coverslips were mounted in glycerol and examined by fluorescence microscopy. The confocal images were captured using a Zeiss LSM410 confocal microscope and software provided by the manufacturer.
Purification of HHV-6 virions.
A total of 5 × 108 Molt-3 cells infected with HST were harvested at 72 hpi and spun twice at 5,000 rpm for 10 min to remove cells and cell debris. HHV-6 virions contained in the culture supernatant were collected by precipitation with polyethylene glycol (PEG) 20000 as described below. An equal volume of PEG solution (20% [wt/vol] PEG 20000, 0.9% [wt/vol] NaCl) was thoroughly mixed with the culture medium. The sample was then incubated at 4°C for 16 h and spun at 5,000 rpm for 2 h. The pellet was resuspended in PBS and spun again at 20,000 rpm for 2 h through a 15% sucrose cushion to concentrate the virions, resuspended in PBS, and layered onto a 15 to 60% sucrose gradient. After centrifugation at 27,000 rpm for 1 h, 500-μl fractions collected from the bottom of the tube were subjected to PCR to detect viral DNA. Positive fractions were gathered and layered onto a discontinuous CsCl gradient (10, 15, 20, 25, 30, 35, and 40% [wt/wt] CsCl). After centrifugation at 30,000 rpm for 48 h, all 500-μl fractions were subjected to Western blotting and PCR.
Immunoelectron microscopy.
Immunoelectron microscopy was performed as described previously (31). Briefly, Molt-3 cells infected with HST were fixed at 48 hpi with 0.1% glutaraldehyde and 4% paraformaldehyde for 10 min, left in 4% parformaldehyde for another 12 h, and subjected to the cryo-thin-section immunogold method. The ultrathin sections, which were labeled with 5-nm colloidal gold particles, were fixed in 2% glutaraldehyde, postfixed in 1% OsO4, embedded in London resin white, and examined with a Hitachi H-7100 electron microscope. For control experiments, sections were directly incubated with the secondary antibodies without treatments by the first antibodies. Some sections were incubated with the nonimmunized mouse serum diluted 1:100, followed by the respective secondary antibodies.
RESULTS
Identifying HHV-6 U14 as the p53-binding protein.
Our previous study showed that the tumor suppressor p53 protein accumulates and is retained in the cytoplasm in response to HHV-6 infection, indicating that HHV-6 encodes a protein(s) that binds p53 and blocks its nuclear localization. To analyze the p53-binding protein in the infected cells, we performed a coimmunoprecipitation experiment using an anti-p53 MAb and Molt-3 cells infected with strains HHV-6 U1102 (variant A) and HST (variant B). The products, which were coimmunoprecipitated from the cell lysates extracted at 24 hpi, were analyzed by SDS-PAGE and silver staining (Fig. 1). Six bands common to both variants and three bands specific to HHV-6B HST (Fig. 1) were detected. Of these bands, we focused on the strongest one, the variant-common 75-kDa band. We collected this protein as a gel band from a large number of HST-infected Molt-3 cells and subjected it to MS/MS to determine its amino acid sequence. By matching a fragment of 10 amino acids to sequence derived from HHV-6 U14 (Fig. 1), we identified the band as the U14 protein. This result suggested that U14 binds p53 in the infected cells.
FIG. 1.
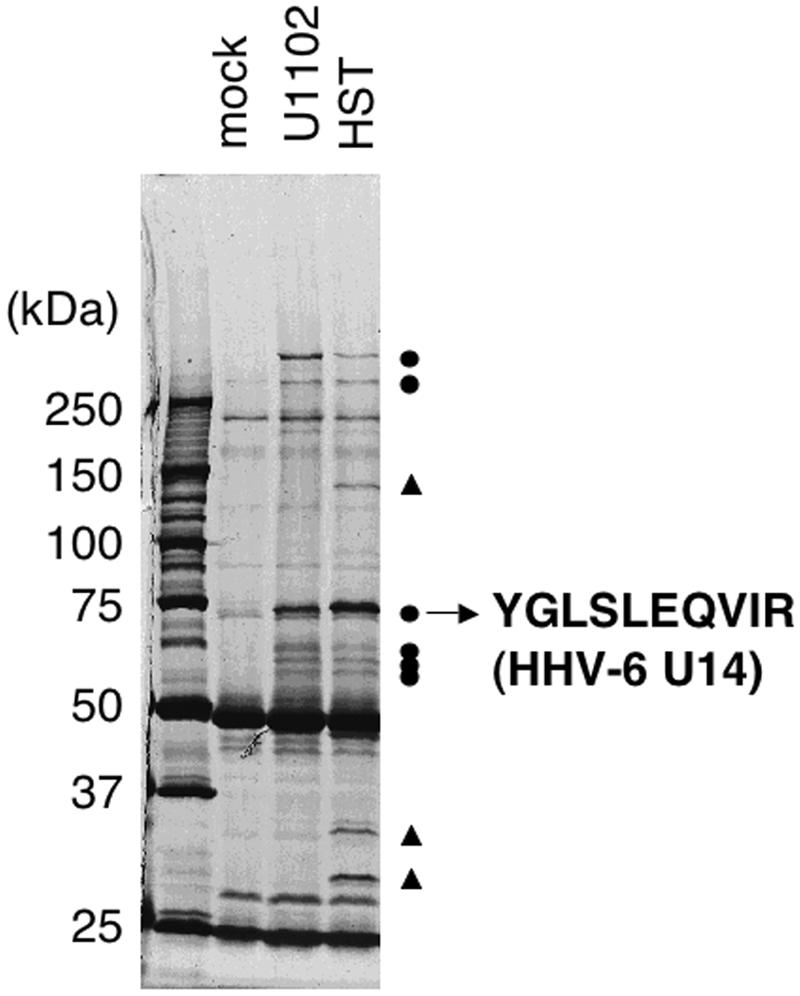
Coimmunoprecipitation experiments with the anti-p53 MAb and identification of the 75-kDa protein as HHV-6 U14. Molt-3 cells infected with the U1102 or HST strain were lysed with TNE buffer at 24 hpi and subjected to coimmunoprecipitation. Samples were resolved on 6% SDS-PAGE, the proteins were stained with Coomassie brilliant blue, and the protein band of interest was excised from the gel (the photo is of a silver-stained gel). Circles and triangles indicate six bands common to both variants and three bands specific to HHV-6B HST, respectively.
U14 binds to p53 in both infected and transfected cells.
To further investigate the U14 protein, we generated a MAb against U14 by immunizing mice with purified His-tagged U14 protein with a deletion of the carboxy terminus. Using this MAb, we first confirmed the binding of U14 to p53 in the infected cells by coimmunoprecipitation experiments. Molt-3 cells infected with HST were lysed in TNE buffer at 48 hpi and subjected to coimmunoprecipitation with anti-U14 and anti-p53 MAbs, respectively, followed by Western blotting for these proteins. As expected, the anti-U14 MAb reacted with a self-precipitated 75-kDa band, the predicted molecular mass of U14, which was specific to the HST-infected cells (Fig. 2A, lanes 3 and 4). In addition, p53 was coimmunoprecipitated by the anti-U14 MAb (Fig. 2A, lanes 1 and 2), and U14 was coimmunoprecipitated by the anti-p53 MAb (Fig. 2A, lanes 5 and 6). The coimmunoprecipitated p53 was successfully detected by using a goat PAb against p53 (FL393) although a little cross-reactivity of anti-goat secondary antibody conjugated with horseradish peroxidase against the mouse immunoglobulin heavy chain of the anti-U14 MAb was present (Fig. 2A, lane 1). These experiments confirmed the binding of U14 to p53 in the infected cells.
FIG. 2.

Confirmation of U14 and p53 binding in infected and transfected cells. (A) Mock (m)- and HST (i)-infected Molt-3 cells were lysed with TNE buffer at 48 hpi and subjected to coimmunoprecipitation with anti-U14 and anti-p53 MAbs. (B) 293 cells were cotransfected with pC53-SN3 and pFlag-BAP or pC53-SN3 and pFlag-U14, lysed with TNE buffer at 24 h posttransfection, and subjected to coimmunoprecipitation using anti-Flag M2-agarose (Sigma). To detect p53, anti-p53 goat PAb was used in both experiments. IP, immunoprecipitation; IB, immunoblot.
Furthermore, to examine whether another viral protein is required for the binding between U14 and p53, we performed the coimmunoprecipitation using transfected cell extracts. 293 cells were cotransfected with the pC53-SN3 and pFlag-U14 plasmids, which express wild-type p53 and Flag-tagged U14, respectively, and the cells were lysed at 24 h posttransfection. As shown in Fig. 2B, when the cells were cotransfected with a commercially available control plasmid, pFlag-BAP, which expresses Flag-tagged bacterial alkaline phosphatase (BAP), p53 was not coprecipitated from the cell lysates expressing the Flag-BAP with anti-Flag MAb, while p53 was coprecipitated from cell lysates expressing the Flag-U14 with anti-Flag MAb, thus demonstrating that no other viral factor mediates the interaction between U14 and p53.
Classification and structure of the U14 gene.
Because the HHV-6-encoded ORF U14 has not been studied to date, no information is available except that it belongs to the human cytomegalovirus (HCMV) UL25/35 gene family. To characterize the U14 gene, we analyzed its kinetics in the course of infection by Northern blot analysis using total RNAs extracted from HST-infected MT-4 cells at different time points. A blot probed with a random-primed 32P-labeled cDNA fragment displayed several bands specific to the U14 probe that had lengths of 2.0 kb, 2.9 kb, and 4.3 kb, among others (Fig. 3B). When we considered the molecular size of the U14 ORF and protein (1.8 kb and 70 kDa, respectively), only the smallest band, not the others, was expected to be the U14 mRNA although the other bands appeared to encompass the U14 ORF. We hypothesized that the 4.3-kb and 2.9-kb bands were probably generated by the utilization of the U14 polyadenylation signal by the U12 and U13 transcripts, respectively. To address this possibility, we performed an additional Northern analysis using the U12, U13, and U14 cDNAs as probes. Since the 4.3-kb band was detected in all three blots (Fig. 3C), it was suggested that the 4.3-kb transcript encoded a sequence from the beginning of U12 to the end of the U14 ORF, as shown in Fig. 3A. In contrast, the 2.9-kb band was not detected by the U13 or U12 probes. The 2.0-kb transcript that was thought to encode the U14 protein was expressed after 16 hpi (Fig. 3B), which represented the kinetics of a typical late gene. To confirm this, we examined the expression of U14 mRNA in the presence of CHX, an inhibitor of de novo protein synthesis, and PFA, a specific inhibitor of viral DNA polymerase. CHX was used to determine whether the U14 gene is an immediate-early gene, and PFA was used for inhibition of the late gene. As shown in Fig. 3B, all bands were significantly reduced in the presence of PFA; therefore, U14 is indeed a late gene.
FIG. 3.

Northern blot analysis. (A) Schematic organization showing ORFs, the template for random primer synthesis of the probe (gray bars), and the transcripts detected (arrows). (B) Twenty micrograms of total RNAs extracted from HST-infected MT-4 cells at the indicated time points (hpi) and in the presence of 50 μg of CHX per ml (8 hpi) and 200 μg of PHA per ml (72 hpi) was loaded and hybridized with 32P-labeled U14 (upper panel) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) probes. (C) Twenty micrograms total RNAs from mock (m)- and HST (i)-infected MT-4 cells at 48 hpi was used to hybridize with probes against U12 and U13, and 10 μg was used for the U14 probe.
U14 localizes to dot-like structures in the nucleus and cytoplasm.
Our previous data showing the cytoplasmic localization of p53 in HHV-6-infected cells led us to suspect that the viral p53-binding protein would also show a similar distribution. To examine whether U14 is localized as we had speculated, we performed a time course immunofluorescence assay (IFA) of HST-infected Molt-3 cells and found that U14 unexpectedly localized as fluorescent dots (Fig. 4A). As shown in Fig. 4A, p53 did not seem to form any dots until 36 hpi; however, a confocal image of HST-infected cells at 18 hpi revealed that p53 slightly colocalized with the U14 dots in the nucleus (arrowheads) although the perinuclear staining was predominant (Fig. 4B). The punctate structures of U14 were also found in the cytoplasm (arrows) where p53 was more intensively found (Fig. 4B), although it is difficult to distinguish exactly the nuclear periphery from the cytoplasm in lymphocytes because of the very thin rims of the cytoplasm. To investigate more precisely, the intracellular localization of U14 was analyzed in 293 cells transfected with a U14 expression plasmid, pEF-U14. As seen in Fig. 4C, the punctate structures of U14 were observed in either the nucleus (arrowheads) or cytoplasm (arrows) in 293 cells, demonstrating that U14 makes dot-like aggregates in both the nucleus and cytoplasm without the other viral proteins.
FIG. 4.

Intracellular distribution of U14. (A) Time course IFA of HST-infected Molt-3 cells harvested at the indicated time points (hpi). Acetone-fixed cells were incubated with anti-U14 and anti-p53 (DO-1) MAbs and then labeled with FITC-conjugated goat anti-mouse immunoglobulin G. (B) Confocal microscopy of HST-infected Molt-3 cells after 18 h. Anti-p53 goat PAb (FL393) and anti-U14 MAb were reacted with FITC- and tetramethyl rhodamine isothiocyanate-labeled secondary antibodies, respectively. Arrows and arrowheads indicate the cytoplasmic and nuclear dots, respectively. (C) 293 cells were transfected with the U14 expression plasmid, pEF-U14, and subjected to IFA at 24 h posttransfection. U14 was labeled with FITC-conjugated secondary antibody. Arrows and arrowheads indicate the cytoplasmic and nuclear dots, respectively. (D) Distribution of U14 and p53 in cytoplasmic and nuclear fractions. Mock (m)- and HST (i)-infected Molt-3 cells were harvested at 24 hpi and subjected to cytoplasmic and nuclear fractionation. Topoisomerase II α and α-tubulin were used as positive controls for the nuclear and cytoplasmic proteins, respectively.
Furthermore, to understand the amount of U14 and p53 proteins distributed in the nucleus and cytoplasm, we performed Western blot analyses by using nuclear and cytoplasmic extracts fractionated from mock- or HST-infected Molt-3 cells at 24 hpi. The purity of the cytoplasmic and nuclear fractions was confirmed by the absence of topoisomerase II α and α-tubulin, respectively. As shown in Fig. 4D, the amount of U14 was higher in the cytoplasm than in the nucleus, but on the other hand, high amounts of p53 were equally detected in both extracts. This result conflicted with the results of IFA in the present study and our previous report (53) that the majority of p53 was found in the cytoplasm in HHV-6-infected cells. The localization of p53 remains to be determined more precisely.
Kinetics of the U14 protein.
Figure 4A shows the unexpected result that U14 was detected in HST-infected Molt-3 cells at low levels at 3 hpi by IFA even though U14 had been defined as a late gene by Northern blot analysis (Fig. 3B). The results led us to suspect that U14 might be a component of HHV-6 virions. To address this issue, the nature of the U14 protein was investigated by a time course Western blot analysis. As expected, U14 was already present at 0 hpi when IE1, the one viral protein expressed immediately after infection, was not detected, and the expression of U14 started to increase markedly after 16 hpi. The true time course of U14 expression was most likely represented by the elevation at 16 hpi since the Northern blot result showed that the U14 transcript was expressed only after 16 hpi (Fig. 3B). This result indicated that U14 was expressed in virions and delivered from the virions into the cells. To confirm this result, we next examined whether U14 was detected in the absence of de novo protein synthesis by treatment with CHX. Figure 5B shows that U14 was still detectable in the presence of CHX, whereas IE1 was not detected; this finding demonstrated that the U14 at the IE phase was expressed without new protein synthesis, unlike IE1, and strongly supported the idea that the U14 at the IE phase was derived from viral particles. The U14 level at 1 to 6 hpi in the presence of CHX was higher than that in the absence of CHX, and the reason for this difference at present is unknown. Interestingly, the level of p53 was also found to be elevated at 0 hpi (Fig. 5A), which raised the possibility that p53 was derived from the virions as well as U14, although the possibility that p53 was rapidly induced in response to the infection cannot be eliminated.
FIG. 5.

Kinetics of U14 protein expression. (A) Time course expression of U14, IE1, p53, and α-tubulin proteins in HST-infected Molt-3 cells. Whole-cell lysates collected at the indicated time points (hpi) were resolved by SDS-PAGE and followed by Western blot analysis. (B) Time course expression of viral antigens at the IE stage in the presence of CHX. Molt-3 cells untreated or pretreated with 50 μg of CHX per ml for 1 h prior to the infection were cultured in the absence or presence of CHX, respectively, harvested at the indicated time points (hpi), and subjected to Western blot analysis.
U14 and p53 are recruited to viral particles.
The above findings that U14 was detected at 0 hpi without protein synthesis and that the p53 level was upregulated at 0 hpi led us to hypothesize that U14 and p53 are components of the HHV-6 virions. To confirm this hypothesis, we purified virions from culture supernatants of HST-infected Molt-3 cells collected at 72 hpi. The first step of purification was carried out by sucrose gradient centrifugation. As a result of PCR of 20 fractions collected from the bottom of the tube after centrifugation, viral DNA was detected in fractions 6 to 10 (Fig. 6A). To examine the purity of the sucrose gradient fractions, fraction 9 was subjected to Western blotting for IE1 and gL as negative and positive controls, respectively. As shown in Fig. 6B, while IE1 was only present in the lysate of infected cells, gL was contained in cell extracts and with more intensity in sucrose fraction 9. These results indicated that the virions were successfully purified by sucrose gradient centrifugation. Fractions 6 to 10 in the sucrose gradient were collected and subjected to the second step of purification by discontinuous CsCl gradient centrifugation. All 20 fractions collected from the bottom of the tube were subjected to Western blotting and PCR. As shown in Fig. 6C, U14, p53, gL, and viral DNA were detected in the same fractions 7 to 12 although U14 and p53 were still positive in fractions 13 and 14. IE1 was not detected in any fractions at all. Fractions 8 and 20 were analyzed by SDS-PAGE followed by silver staining to visualize the polypeptides included in the purified virion (Fig. 6D). We could detect at least 26 polypeptides ranging from 25 to 250 kDa, which were comparable to the 29 polypeptides reported previously (45).
FIG. 6.

Purification of virions and Western blotting for U14 and p53. (A) PCR assay to detect viral DNA in fractions from the first step of purification by sucrose gradient centrifugation. Fractions of 500 μl each were collected from the bottom, diluted 1:100 in Tris-EDTA buffer, and subjected to PCR using the primer pair 6U14-1B and 6U14-2H amplifying part of the U14 ORF. (B) The virions purified from the sucrose gradient centrifugation were subjected to Western blotting to confirm the purity. Fraction 9 was resolved by SDS-PAGE with whole-cell lysates of mock (m)- and HST (i)-infected Molt-3 cells at 72 hpi. Molecular weights of protein markers are shown at the left side of each gel. (C) Virions purified by the second step of purification by discontinuous CsCl gradient centrifugation were subjected to Western blotting with antibodies against U14, p53, gL, and IE1 and to PCR using the same primer set used above. (D) Fractions 8 and 20 were analyzed by silver staining. Molecular weight markers (lanes M) are indicated at the left of the gel. IB, immunoblot.
For further confirmation, HST-infected Molt-3 cells were fixed at 48 hpi and analyzed by immunoelectron microscopy. The photographs in Fig. 7 show that the immunogold particles that reacted with the anti-U14 MAb were frequently localized to the tegument or nucleocapsid substructure of the virion (Fig. 7A) and that the anti-p53 MAb clearly labeled a similar substructure, although the signal appeared less frequently and clearly than that of the anti-U14 MAb (Fig. 7B and C). In contrast, immunogold particles specific to the virions were not observed in either section incubated with the second antibodies without the first antibodies (Fig. 7D) or with preincubation with the nonimmunized mouse serum diluted 1:100 (Fig. 7E). In conclusion, the U14-p53 complex is recruited to the HHV-6 virions.
FIG. 7.

Immunoelectron microscopy for U14 and p53. Molt-3 cells infected with HST were fixed at 48 hpi, incubated with anti-U14 MAb (A) or anti-p53 MAb (B and C), immunogold labeled, and examined by electron microscopy. For control experiments, sections were directly incubated with the second antibodies without treatments with the first antibodies (D). Some sections were incubated with the nonimmunized mouse serum diluted 1:100 (E), followed by the respective second antibodies. Arrows indicate virions containing U14 or p53. Scale bar, 0.2 μm.
DISCUSSION
We previously reported that the p53 level increases in various cell types infected with HHV-6 strains U1102 (variant A) and HST (variant B) and begins to be elevated immediately after the infection of Molt-3 cells through the reduction of its ubiquitination and subsequent degradation (53). p53 that gradually accumulates in the cytoplasm during lytic infection does not translocate into the nucleus in response to UV irradiation, but it accumulates markedly in the nucleus of uninfected cells following irradiation, which results in the acquisition of resistance to UV-induced apoptosis (53). De Bolle et al. (6) and Oster and colleagues (36) have recently reported the accumulation of p53 in relation to the cell cycle arrest at G2/M phase caused by HHV-6. One candidate molecule for interfering with the subcellular relocalization of p53 is the HHV-6 DR7 protein, which was reported previously (as the ORF-1 protein) to suppress the transcriptional activity of p53 by direct binding in NIH 3T3 cells transfected with the SalI L fragment of the HHV-6 genome (19). However, it remains to be demonstrated that the interaction between p53 and DR7 indeed occurs in the infected cells. In addition, no information about the kinetics, the subcellular localization, or even the molecular weight of DR7 in the context of HHV-6 infection has been provided to date. In the present study, therefore, we took a different approach to defining the molecular mechanism by which the virus impairs the p53 function by looking for a viral protein from infected cells that bound p53.
We identified HHV-6 U14 as a p53-binding protein, using MS/MS analysis to determine the sequence of a trypsin-digested, HHV-6-specific, 75-kDa peptide fragment that was among the coimmunoprecipitants brought down with an anti-p53 MAb. While it has been reported that the HCMV UL35 (24) and HHV-7 U14 (49) proteins, which are homologs of HHV-6 U14, comprise the viral particles, no information about HHV-6 U14 was available. We demonstrated that U14 bound to p53 in HST-infected Molt-3 cells and 293 cells that had been cotransfected with expression vectors for both molecules and that they colocalized in the cytoplasm in the late phase of infection. However, we attempted unsuccessfully to coimmunoprecipitate U14 and p53 when only these two proteins, derived from in vitro translation, were mixed. There are two possibilities that could explain this result: (i) the binding of U14 to p53 is not direct, and some other cellular protein mediates their interaction; and (ii) some modifications, such as phosphorylation and acetylation, of either or both U14 or p53 may be requisite for their binding. These possibilities remain to be investigated.
Because the original aim of the present study was to define the molecular mechanism by which HHV-6 impairs the redistribution and function of p53, we tested whether U14 suppressed the transcriptional activity of p53, using the reporter gene p53-luc. When Jurkat cells were cotransfected with wild-type p53 expression vector pC53-SN3 and pEF-U14, the luciferase activity of p53-luc did not decrease but, rather, increased compared with its activity in cells transfected with pC53-SN3 alone. In contrast, a p53 dominant-negative plasmid, pC53-SCX3, significantly suppressed reporter gene expression in Jurkat cells (data not shown). We also performed this assay using pcDNA-U14, which was constructed so that the promoter controlling the U14 expression was the same strength as the promoter in pC53-SCX3; under these conditions we did see a reduction of the luciferase activity, but it was very slight (data not shown). The fact that U14 localizes to both the cytoplasm and the nucleus (Fig. 4) conflicts with the hypothesis that p53 is retained in the cytoplasm of HHV-6-infected cells by U14. For the present, therefore, we conclude that U14 does not prevent p53 activity, despite the binding of U14 and p53 in HHV-6-infected cells; the possibility remains that some other viral factor is involved in the U14 and p53 complex and functions to inhibit p53.
In Northern blot analysis, we could not identify the origin of a 2.9-kb band by using U13 or U12 probes (Fig. 3C). It is unlikely that the transcript that we originally hypothesized to encode both U13 and U14 ORFs exists, although it still remains possible that we may have failed to detect it by using the U13 probe because the activity of the probe was rather low given the difficulty of labeling such a short ORF (300 bases). There is also the possibility that the 2.9-kb transcript may be derived from a spliced event and, thus, that the 5′ end of the transcript may initiate upstream of U12 and may not encompass the U12 and U13 sequences; alternatively, the 3′ end of the transcript may be elongated downstream of U14.
Confocal immunofluorescence assays demonstrated that U14 partly concentrates in some structures like nuclear dots which are exemplified by nuclear domain 10 (ND10) structures, as shown in Fig. 4. The ND10s are nuclear bodies that contain promyelocytic leukemia (PML) protein as a structural marker and are implicated in the regulation of transcription, apoptosis, tumor suppression, and the antiviral response (reviewed in reference 7). PML is frequently targeted and degraded by some IE proteins encoded by herpesviruses such as herpes simplex virus type 1 ICP0 (10), Epstein-Barr virus EBNA-5 (50), BZLF1 (1), HCMV IE1 and IE2 (2, 23), and HHV-6 IE1 (14, 48). Recently, it has been demonstrated that the HCMV tegument pp71 protein also targets PML (16, 17, 26) and recruits the UL35 protein, a homolog of HHV-6 U14, to the ND10 structures (43). However, double staining for U14 and PML revealed that the nuclear dots labeled with the anti-HHV-6 U14 MAb are not ND10s and that U14 does not colocalize with IE2, which is concentrated in other nuclear bodies (data not shown). The identification of subcellular structures targeted by U14 may be a clue to its functional role in the IE phase.
The intracellular localization of p53 also remained unclear. Oster et al. reported that p53 was strongly induced in HHV-6-infected cells and strongly detected in nuclear extracts (36). In this study, Western blotting of nuclear and cytoplasmic fractions showed that both fractions contained almost equivalent amounts of p53 (Fig. 4D). These data conflict with our IFA results that p53 is intensively detected in the perinuclear or cytoplasmic regions in HHV-6-infected Molt-3 cells. One possibility is that p53 is concentrated in the cytoplasm although it is detected in the nucleus at a similar level, since the relative volume of cytoplasm is much smaller than that of nucleus in lymphocytes. Another possibility is that p53 associates with the perinuclear organelles which are insoluble to NP-40. Despite the data for nuclear and cytoplasmic fractions, the intracellular distribution of p53 still remains to be further investigated.
So far, several host cellular proteins have been identified in viral particles and found to contribute to increasing infectivity. For example, CD55 and CD59 associate with virions of human immunodeficiency virus type 1, human T-cell leukemia virus type 1, and HCMV and protect these viruses from C-mediated lysis (40, 46). Annexin II (57), cytosolic phospholipase A2 (3), and serine/threonine protein phosphatases (PP1 and PP2A) (29) are incorporated into the HCMV virion and seem to promote various aspects of HCMV infectivity, e.g., respectively, attachment, receptor signaling, and hypophosphorylation of cellular proteins before IE gene expression. In this study, we found that p53 is recruited with U14 to the nucleocapsid or tegument substructure of the HHV-6 virion. To our knowledge, this is the first report that shows the incorporation of the cellular tumor suppressor p53 into viral particles. Recently, proteins composing virions of HCMV (55), murine CMV (MCMV) (21), and Kaposi's sarcoma-associated herpesvirus (KSHV) (4, 61) have been comprehensively identified by using the proteomics approach. Although many cellular proteins incorporated into their virions have been revealed, p53 is unlikely involved in the particles of such herpesviruses as HCMV, MCMV, and KSHV. p53 is a critical regulator of cell proliferation and apoptosis in response to DNA damage; its function is mostly exerted by direct transcriptional activation of its responsive genes. Nuclear import of p53 is a prerequisite for transactivation and is carried out by the microtubule network (13). Considering that the tegument and nucleocapsid proteins function to direct the viral capsid to the nucleus using the microtubule network (8, 9, 58) and to transactivate IE genes, it can be hypothesized that virion-associated p53 may be involved in such events. However, p53 has an antiviral activity of inducing apoptosis in virus-infected cells that is dependent on alpha interferon, as recently reported (52), and therefore the possibility that p53 may suppress viral infectivity, unlike the other cellular proteins described above, should be also noted. Despite certain experimental limitations, for example, that HHV-6 cannot infect mouse cells (preventing us from making use of p53−/− cells from knockout mice to generate virus lacking p53) and that the strategy of knocking out a specific gene encoded by HHV-6 has not yet been established, further investigation using new technologies like RNA interference instead of conventional knockouts may elucidate the role of virion-associated p53 in the life cycle of HHV-6.
Acknowledgments
This study was supported in part by a grant-in-aid for scientific research from the Society for the Promotion of Science of Japan and a grant-in-aid for specially promoted research from the Ministry of Education, Culture, Sports, Science and Technology of Japan. This study was also supported by a grant-in-aid for scientific research on priority areas, MEXT of Japan.
REFERENCES
- 1.Adamson, A. L., and S. Kenney. 2001. Epstein-barr virus immediate-early protein BZLF1 is SUMO-1 modified and disrupts promyelocytic leukemia bodies. J. Virol. 75:2388-2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahn, J. H., and G. S. Hayward. 1997. The major immediate-early proteins IE1 and IE2 of human cytomegalovirus colocalize with and disrupt PML-associated nuclear bodies at very early times in infected permissive cells. J. Virol. 71:4599-4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allal, C., C. Buisson-Brenac, V. Marion, C. Claudel-Renard, T. Faraut, P. Dal Monte, D. Streblow, M. Record, and J. L. Davignon. 2004. Human cytomegalovirus carries a cell-derived phospholipase A2 required for infectivity. J. Virol. 78:7717-7726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bechtel, J. T., R. C. Winant, and D. Ganem. 2005. Host and viral proteins in the virion of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:4952-4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonin, L. R., and J. K. McDougall. 1997. Human cytomegalovirus IE2 86-kilodalton protein binds p53 but does not abrogate G1 checkpoint function. J. Virol. 71:5861-5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Bolle, L., S. Hatse, E. Verbeken, E. De Clercq, and L. Naesens. 2004. Human herpesvirus 6 infection arrests cord blood mononuclear cells in G(2) phase of the cell cycle. FEBS Lett. 560:25-29. [DOI] [PubMed] [Google Scholar]
- 7.Dellaire, G., and D. P. Bazett-Jones. 2004. PML nuclear bodies: dynamic sensors of DNA damage and cellular stress. Bioessays 26:963-977. [DOI] [PubMed] [Google Scholar]
- 8.Douglas, M. W., R. J. Diefenbach, F. L. Homa, M. Miranda-Saksena, F. J. Rixon, V. Vittone, K. Byth, and A. L. Cunningham. 2004. Herpes simplex virus type 1 capsid protein VP26 interacts with dynein light chains RP3 and Tctex1 and plays a role in retrograde cellular transport. J. Biol. Chem. 279:28522-28530. [DOI] [PubMed] [Google Scholar]
- 9.Elliott, G., and P. O'Hare. 2000. Cytoplasm-to-nucleus translocation of a herpesvirus tegument protein during cell division. J. Virol. 74:2131-2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Everett, R. D., and G. G. Maul. 1994. HSV-1 IE protein Vmw110 causes redistribution of PML. EMBO J. 13:5062-5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fortunato, E. A., and D. H. Spector. 1998. p53 and RPA are sequestered in viral replication centers in the nuclei of cells infected with human cytomegalovirus. J. Virol. 72:2033-2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friborg, J., Jr., W. Kong, M. O. Hottiger, and G. J. Nabel. 1999. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 402:889-894. [DOI] [PubMed] [Google Scholar]
- 13.Giannakakou, P., D. L. Sackett, Y. Ward, K. R. Webster, M. V. Blagosklonny, and T. Fojo. 2000. p53 is associated with cellular microtubules and is transported to the nucleus by dynein. Nat. Cell Biol. 2:709-717. [DOI] [PubMed] [Google Scholar]
- 14.Gravel, A., J. Gosselin, and L. Flamand. 2002. Human herpesvirus 6 immediate-early 1 protein is a sumoylated nuclear phosphoprotein colocalizing with promyelocytic leukemia protein-associated nuclear bodies. J. Biol. Chem. 277:19679-19687. [DOI] [PubMed] [Google Scholar]
- 15.Hobbs, W. E., II, and N. A. DeLuca. 1999. Perturbation of cell cycle progression and cellular gene expression as a function of herpes simplex virus ICP0. J. Virol. 73:8245-8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hofmann, H., H. Sindre, and T. Stamminger. 2002. Functional interaction between the pp71 protein of human cytomegalovirus and the PML-interacting protein human Daxx. J. Virol. 76:5769-5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishov, A. M., O. V. Vladimirova, and G. G. Maul. 2002. Daxx-mediated accumulation of human cytomegalovirus tegument protein pp71 at ND10 facilitates initiation of viral infection at these nuclear domains. J. Virol. 76:7705-7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Josephs, S. F., S. Z. Salahuddin, D. V. Ablashi, F. Schachter, F. Wong-Staal, and R. C. Gallo. 1986. Genomic analysis of the human B-lymphotropic virus (HBLV). Science 234:601-603. [DOI] [PubMed] [Google Scholar]
- 19.Kashanchi, F., J. Araujo, J. Doniger, S. Muralidhar, R. Hoch, S. Khleif, E. Mendelson, J. Thompson, N. Azumi, J. N. Brady, M. Luppi, G. Torelli, and L. J. Rosenthal. 1997. Human herpesvirus 6 (HHV-6) ORF-1 transactivating gene exhibits malignant transforming activity and its protein binds to p53. Oncogene 14:359-367. [DOI] [PubMed] [Google Scholar]
- 20.Katano, H., K. Ogawa-Goto, H. Hasegawa, T. Kurata, and T. Sata. 2001. Human-herpesvirus-8-encoded K8 protein colocalizes with the promyelocytic leukemia protein (PML) bodies and recruits p53 to the PML bodies. Virology 286:446-455. [DOI] [PubMed] [Google Scholar]
- 21.Kattenhorn, L. M., R. Mills, M. Wagner, A. Lomsadze, V. Makeev, M. Borodovsky, H. L. Ploegh, and B. M. Kessler. 2004. Identification of proteins associated with murine cytomegalovirus virions. J. Virol. 78:11187-11197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kondo, K., T. Kondo, T. Okuno, M. Takahashi, and K. Yamanishi. 1991. Latent human herpesvirus 6 infection of human monocytes/macrophages. J. Gen. Virol. 72:1401-1408. [DOI] [PubMed] [Google Scholar]
- 23.Korioth, F., G. G. Maul, B. Plachter, T. Stamminger, and J. Frey. 1996. The nuclear domain 10 (ND10) is disrupted by the human cytomegalovirus gene product IE1. Exp. Cell Res. 229:155-158. [DOI] [PubMed] [Google Scholar]
- 24.Liu, Y., and B. J. Biegalke. 2002. The human cytomegalovirus UL35 gene encodes two proteins with different functions. J. Virol. 76:2460-2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lusso, P., P. D. Markham, E. Tschachler, F. di Marzo Veronese, S. Z. Salahuddin, D. V. Ablashi, S. Pahwa, K. Krohn, and R. C. Gallo. 1988. In vitro cellular tropism of human B-lymphotropic virus (human herpesvirus-6). J. Exp. Med. 167:1659-1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marshall, K. R., K. V. Rowley, A. Rinaldi, I. P. Nicholson, A. M. Ishov, G. G. Maul, and C. M. Preston. 2002. Activity and intracellular localization of the human cytomegalovirus protein pp71. J Gen. Virol. 83:1601-1612. [DOI] [PubMed] [Google Scholar]
- 27.McGeoch, D. J. 1990. Evolutionary relationships of virion glycoprotein genes in the S regions of alphaherpesvirus genomes. J Gen. Virol. 71:2361-2367. [DOI] [PubMed] [Google Scholar]
- 28.McGeoch, D. J. 1989. The genomes of the human herpesviruses: contents, relationships, and evolution. Annu. Rev. Microbiol. 43:235-265. [DOI] [PubMed] [Google Scholar]
- 29.Michelson, S., P. Turowski, L. Picard, J. Goris, M. P. Landini, A. Topilko, B. Hemmings, C. Bessia, A. Garcia, and J. L. Virelizier. 1996. Human cytomegalovirus carries serine/threonine protein phosphatases PP1 and a host-cell derived PP2A. J. Virol. 70:1415-1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mori, Y., P. Akkapaiboon, X. Yang, and K. Yamanishi. 2003. The human herpesvirus 6 U100 gene product is the third component of the gH-gL glycoprotein complex on the viral envelope. J. Virol. 77:2452-2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mori, Y., P. Akkapaiboon, S. Yonemoto, M. Koike, M. Takemoto, T. Sadaoka, Y. Sasamoto, S. Konishi, Y. Uchiyama, and K. Yamanishi. 2004. Discovery of a second form of tripartite complex containing gH-gL of human herpesvirus 6 and observations on CD46. J. Virol. 78:4609-4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mori, Y., T. Seya, H. L. Huang, P. Akkapaiboon, P. Dhepakson, and K. Yamanishi. 2002. Human herpesvirus 6 variant A but not variant B induces fusion from without in a variety of human cells through a human herpesvirus 6 entry receptor, CD46. J. Virol. 76:6750-6761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muganda, P., O. Mendoza, J. Hernandez, and Q. Qian. 1994. Human cytomegalovirus elevates levels of the cellular protein p53 in infected fibroblasts. J. Virol. 68:8028-8034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muralidhar, S., J. Doniger, E. Mendelson, J. C. Araujo, F. Kashanchi, N. Azumi, J. N. Brady, and L. J. Rosenthal. 1996. Human cytomegalovirus mtrII oncoprotein binds to p53 and down-regulates p53-activated transcription. J. Virol. 70:8691-8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakamura, H., M. Li, J. Zarycki, and J. U. Jung. 2001. Inhibition of p53 tumor suppressor by viral interferon regulatory factor. J. Virol. 75:7572-7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oster, B., B. Bundgaard, and P. Hollsberg. 2005. Human herpesvirus 6B induces cell cycle arrest concomitant with p53 phosphorylation and accumulation in T cells. J. Virol. 79:1961-1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park, J., T. Seo, S. Hwang, D. Lee, Y. Gwack, and J. Choe. 2000. The K-bZIP protein from Kaposi's sarcoma-associated herpesvirus interacts with p53 and represses its transcriptional activity. J. Virol. 74:11977-11982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rivas, C., A. E. Thlick, C. Parravicini, P. S. Moore, and Y. Chang. 2001. Kaposi's sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J. Virol. 75:429-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roizmann, B., R. C. Desrosiers, B. Fleckenstein, C. Lopez, A. C. Minson, M. J. Studdert, et al. 1992. The family Herpesviridae: an update. Arch. Virol. 123:425-449. [DOI] [PubMed] [Google Scholar]
- 40.Saifuddin, M., C. J. Parker, M. E. Peeples, M. K. Gorny, S. Zolla-Pazner, M. Ghassemi, I. A. Rooney, J. P. Atkinson, and G. T. Spear. 1995. Role of virion-associated glycosylphosphatidylinositol-linked proteins CD55 and CD59 in complement resistance of cell line-derived and primary isolates of HIV-1. J Exp. Med. 182:501-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salahuddin, S. Z., D. V. Ablashi, P. D. Markham, S. F. Josephs, S. Sturzenegger, M. Kaplan, G. Halligan, P. Biberfeld, F. Wong-Staal, B. Kramarsky, et al. 1986. Isolation of a new virus, HBLV, in patients with lymphoproliferative disorders. Science 234:596-601. [DOI] [PubMed] [Google Scholar]
- 42.Santoro, F., P. E. Kennedy, G. Locatelli, M. S. Malnati, E. A. Berger, and P. Lusso. 1999. CD46 is a cellular receptor for human herpesvirus 6. Cell 99:817-827. [DOI] [PubMed] [Google Scholar]
- 43.Schierling, K., T. Stamminger, T. Mertens, and M. Winkler. 2004. Human cytomegalovirus tegument proteins ppUL82 (pp71) and ppUL35 interact and cooperatively activate the major immediate-early enhancer. J. Virol. 78:9512-9523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seo, T., J. Park, D. Lee, S. G. Hwang, and J. Choe. 2001. Viral interferon regulatory factor 1 of Kaposi's sarcoma-associated herpesvirus binds to p53 and represses p53-dependent transcription and apoptosis. J. Virol. 75:6193-6198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shiraki, K., T. Okuno, K. Yamanishi, and M. Takahashi. 1989. Virion and nonstructural polypeptides of human herpesvirus-6. Virus Res. 13:173-178. [DOI] [PubMed] [Google Scholar]
- 46.Spear, G. T., N. S. Lurain, C. J. Parker, M. Ghassemi, G. H. Payne, and M. Saifuddin. 1995. Host cell-derived complement control proteins CD55 and CD59 are incorporated into the virions of two unrelated enveloped viruses. Human T cell leukemia/lymphoma virus type I (HTLV-I) and human cytomegalovirus (HCMV). J. Immunol. 155:4376-4381. [PubMed] [Google Scholar]
- 47.Speir, E., R. Modali, E. S. Huang, M. B. Leon, F. Shawl, T. Finkel, and S. E. Epstein. 1994. Potential role of human cytomegalovirus and p53 interaction in coronary restenosis. Science 265:391-394. [DOI] [PubMed] [Google Scholar]
- 48.Stanton, R., J. D. Fox, R. Caswell, E. Sherratt, and G. W. Wilkinson. 2002. Analysis of the human herpesvirus-6 immediate-early 1 protein. J Gen. Virol. 83:2811-2820. [DOI] [PubMed] [Google Scholar]
- 49.Stefan, A., P. Secchiero, T. Baechi, W. Kempf, and G. Campadelli-Fiume. 1997. The 85-kilodalton phosphoprotein (pp85) of human herpesvirus 7 is encoded by open reading frame U14 and localizes to a tegument substructure in virion particles. J. Virol. 71:5758-5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Szekely, L., K. Pokrovskaja, W. Q. Jiang, H. de The, N. Ringertz, and G. Klein. 1996. The Epstein-Barr virus-encoded nuclear antigen EBNA-5 accumulates in PML-containing bodies. J. Virol. 70:2562-2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takahashi, K., S. Sonoda, K. Higashi, T. Kondo, H. Takahashi, M. Takahashi, and K. Yamanishi. 1989. Predominant CD4 T-lymphocyte tropism of human herpesvirus 6-related virus. J. Virol. 63:3161-3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takaoka, A., S. Hayakawa, H. Yanai, D. Stoiber, H. Negishi, H. Kikuchi, S. Sasaki, K. Imai, T. Shibue, K. Honda, and T. Taniguchi. 2003. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 424:516-523. [DOI] [PubMed] [Google Scholar]
- 53.Takemoto, M., Y. Mori, K. Ueda, K. Kondo, and K. Yamanishi. 2004. Productive human herpesvirus 6 infection causes aberrant accumulation of p53 and prevents apoptosis. J Gen. Virol. 85:869-879. [DOI] [PubMed] [Google Scholar]
- 54.Tedder, R. S., M. Briggs, C. H. Cameron, R. Honess, D. Robertson, and H. Whittle. 1987. A novel lymphotropic herpesvirus. Lancet 2:390-392. [DOI] [PubMed] [Google Scholar]
- 55.Varnum, S. M., D. N. Streblow, M. E. Monroe, P. Smith, K. J. Auberry, L. Pasa-Tolic, D. Wang, D. G. Camp, 2nd, K. Rodland, S. Wiley, W. Britt, T. Shenk, R. D. Smith, and J. A. Nelson. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J. Virol. 78:10960-10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wilcock, D., and D. P. Lane. 1991. Localization of p53, retinoblastoma and host replication proteins at sites of viral replication in herpes-infected cells. Nature 349:429-431. [DOI] [PubMed] [Google Scholar]
- 57.Wright, J. F., A. Kurosky, E. L. Pryzdial, and S. Wasi. 1995. Host cellular annexin II is associated with cytomegalovirus particles isolated from cultured human fibroblasts. J. Virol. 69:4784-4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ye, G. J., K. T. Vaughan, R. B. Vallee, and B. Roizman. 2000. The herpes simplex virus 1 UL34 protein interacts with a cytoplasmic dynein intermediate chain and targets nuclear membrane. J. Virol. 74:1355-1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang, Q., D. Gutsch, and S. Kenney. 1994. Functional and physical interaction between p53 and BZLF1: implications for Epstein-Barr virus latency. Mol. Cell. Biol. 14:1929-1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhong, L., and G. S. Hayward. 1997. Assembly of complete, functionally active herpes simplex virus DNA replication compartments and recruitment of associated viral and cellular proteins in transient cotransfection assays. J. Virol. 71:3146-3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu, F. X., J. M. Chong, L. Wu, and Y. Yuan. 2005. Virion proteins of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:800-811. [DOI] [PMC free article] [PubMed] [Google Scholar]