

Abstract
The tick-borne encephalitis (TBE) complex of viruses, genus Flavivirus, can cause severe encephalitis, meningitis, and/or hemorrhagic fevers. Effective interferon (IFN) responses are critical to recovery from infection with flaviviruses, and the mosquito-borne flaviviruses can inhibit this response. However, little is known about interactions between IFN signaling and TBE viruses. Langat virus (LGTV), a member of the TBE complex of viruses, was found to be highly sensitive to the antiviral effects of IFN. However, LGTV infection inhibited IFN-induced expression of a reporter gene driven by either IFN-α/β- or IFN-γ-responsive promoters. This indicated that LGTV can inhibit the IFN-mediated JAK-STAT (Janus kinase-signal transducer and activator of transcription) pathway of signal transduction. The mechanism of inhibition was due to blocks in the phosphorylation of both Janus kinases, Jak1 and Tyk2, during IFN-α signaling and at least a failure of Jak1 phosphorylation following IFN-γ stimulation. To determine the viral protein(s) responsible, we individually expressed all nonstructural (NS) proteins and examined their ability to inhibit signal transduction. Expression of NS5 alone inhibited STAT1 phosphorylation in response to IFN, thus identifying NS5 as a potential IFN antagonist. Examination of interactions between NS5 and cellular proteins revealed that NS5 associated with IFN-α/β and -γ receptor complexes. Importantly, inhibition of JAK-STAT signaling and NS5-IFN receptor interactions were demonstrated in LGTV-infected human monocyte-derived dendritic cells, important target cells for early virus replication. Because NS5 may interfere with both innate and acquired immune responses to virus infection, this protein may have a significant role in viral pathogenesis.
The tick-borne encephalitis (TBE) complex of viruses, genus Flavivirus, includes TBE virus (TBEV), Powassan virus, Omsk hemorrhagic fever virus, and Kyasanur Forest disease virus. These viruses cause encephalitis, meningitis, and/or hemorrhagic fevers with high mortality rates (6, 14, 15). Treatment for TBE virus infection is limited to supportive care. Similar to other flaviviruses, the TBE virus positive-sense RNA genome is transcribed as one large open reading frame and is co- and posttranslationally processed into three structural proteins (envelope [E], membrane [M], and capsid [C] proteins) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) (31). Several functions of the NS proteins in virus replication have been defined: NS5 is an RNA-dependent RNA polymerase, NS3 contains the viral serine protease, and NS2B is a cofactor for NS3 (31). However, little is known about the roles of individual nonstructural proteins in virus pathogenesis.
The interferons (IFN), IFN-α/β and IFN-γ, are important in recovery from infection by various flaviviruses (8, 30, 43, 51). However, use of IFN-α/β in clinical trials for treatment of flavivirus infection has not been uniformly successful (34, 50). This may be explained by the ability of some flaviviruses, including dengue virus serotype 2 (DEN-2), Japanese encephalitis virus, West Nile virus (WNV), and Kunjin virus (KUN) (16, 30, 33, 38, 39), to inhibit the JAK-STAT (Janus kinase-signal transducer and activator of transcription) pathway in response to IFN-α/β and/or IFN-γ. Efforts to determine the specific gene products of these viruses responsible for IFN antagonism have implicated the nonstructural proteins. Studies of DEN-2 antagonism of STAT1 phosphorylation have revealed NS4B as the primary antagonist, although NS2A and NS4A augment this response (38, 39). The function of NS4B as an IFN antagonist is conserved in both WNV and yellow fever virus (38). Recent studies of KUN (a close relative of WNV) suggested that NS2A, NS2B, NS3, NS4A, and NS4B were all capable of inhibiting JAK-STAT signaling (33). Despite the intense interest in this field, the precise mechanism of IFN antagonism is not well defined for any of the mosquito- or tick-borne flaviviruses.
Cells respond rapidly following stimulation with IFN via the JAK-STAT signal transduction pathway (3, 27, 49). Briefly, the specific receptor complex for each IFN-α/β and IFN-γ is composed of two major subunits (IFNAR1/IFNAR2 for IFN-α/β and IFNGR1/IFNGR2 for IFN-γ) and various JAK tyrosine kinases constitutively associated with the receptor. Jak1 and Tyk2 are required for IFN-α/β signaling; Jak1 and Jak2 are required for IFN-γ signaling. Following binding of the receptor subunits by IFN, the JAKs trans-phosphorylate each other and then phosphorylate critical tyrosine residues within the intracellular domains of the receptor subunits. These phosphorylated residues serve as recruitment sites for STAT proteins, which bind the activated receptor and are in turn phosphorylated by the JAKs. The phosphorylated STAT proteins then form homodimers, or heterodimers, with other STAT proteins and translocate to the nucleus, where they bind specific DNA sequences within the promoter regions of IFN-stimulated genes (ISGs). In the case of signaling via IFN-α/β, phosphorylated STAT1 and STAT2 bind each other as well as a third component, IFN regulatory factor 9 (ISGF3g/p48), to form the transcription factor ISGF3, which binds a promoter region called IFN-stimulated response element (ISRE). Signaling via IFN-γ results in the formation of phosphorylated STAT1 homodimers, which translocate to the nucleus to bind an IFN-γ-activated sequence (GAS) (25, 27). ISG expression induces an antiviral state within the cell, can modulate cell proliferation and cell death, and modulates immune responses via its roles in activation and maturation of antigen-presenting cells.
We examined the interactions of Langat virus (LGTV), a member of the TBE complex of viruses, with JAK-STAT signal transduction pathways. Our findings show that LGTV was sensitive to the antiviral effects of IFN. However, infection of cell lines with LGTV prior to the addition of IFN resulted in inhibited phosphorylation of STAT1 and STAT2, as well as Tyk2 and/or Jak1, in response to stimulation with either IFN-α or IFN-γ. To determine which viral protein was responsible, we analyzed the ability of the individual LGTV nonstructural proteins to antagonize JAK-STAT signaling. We found that NS5 blocked STAT1 phosphorylation in response to either IFN-β or IFN-γ. Furthermore, an association was observed between NS5 and both the IFN-α/β receptor subunit, IFNAR2 (IFNAR2-2 or IFNAR2c), and the IFN-γ receptor subunit, IFNGR1. LGTV-mediated inhibition of JAK-STAT signaling, as well as interactions between NS5 and IFNAR2, was demonstrated in infected human monocyte-derived dendritic cells (DC). As these cells are an early target for vector-borne flavivirus replication, this newly defined function of NS5 is likely to have a critical role in virus pathogenesis.
MATERIALS AND METHODS
Cells and viruses.
A Vero cell-derived virus stock of LGTV strain TP21 was generously provided by A. Pletnev (NIAID, NIH). The virus was further propagated in Vero cells beginning with a multiplicity of infection (MOI) of 0.005, as previously described (4, 46).
For virus titration by plaque assay, Vero cells were seeded at 3 × 105 cells in six-well plates in Dulbecco's minimal essential medium (DMEM) containing 10% fetal calf serum, 1% glutamine, and 50 μg/ml gentamicin at 37°C in 5% CO2. Cells were absorbed with LGTV titrated in 10-fold dilutions in complete DMEM for 60 min at 37°C. This inoculum was removed, the cells were washed in Dulbecco's phosphate-buffered saline (DPBS), and fresh DMEM containing 5% fetal calf serum and 0.75% methylcellulose was added. Infected cells were incubated for 7 days when the cell monolayers were fixed, and plaques were visualized using Giemsa stain.
For infections designed to examine interactions with JAK-STAT signaling, mouse neuroblastoma cells (MNB109) or Vero cells were seeded as described above in six-well plates or four-well Labtek slides (Fisher Scientific, Pittsburgh, PA), infected with LGTV TP21 at an MOI of 1 or 10, and incubated for 48 h at 37°C in 5% CO2. Cells were then treated with 1,000 U/ml of recombinant IFN-α or 10 ng/ml IFN-γ of the appropriate species for 15 min.
Monocyte-derived dendritic cells.
Human peripheral blood mononuclear cells were isolated from buffy coats (Biological Specialty Corp., Colmar, PA) by centrifugation through a Ficoll-Paque Plus density gradient (Amersham Pharmacia Biotech, Uppsala, Sweden). Cells were enriched for monocytes (CD14+ cells) by use of a RossetteSep monocyte enrichment kit (Stem Cell Technologies, Vancouver, Canada). Monocytes were resuspended at 106 cells/ml DC medium (RPMI medium supplemented with 5% fetal bovine serum, l-glutamine, HEPES, nonessential amino acids, sodium pyruvate, penicillin, and streptomycin) containing interleukin-4 (10 ng/ml) and granulocyte-macrophage colony-stimulating factor (10 ng/ml) and cultured for 6 days with replacement of half of the medium and addition of fresh cytokines every second day. The nonadherent cells were harvested by centrifugation and resuspended in DC medium without antibiotics. The resulting cells were determined to be >95% CD11c+/CD209+ by flow cytometry. For infections, 2 × 105 cells were seeded into each chamber of four-well Labtek slides (Fisher Scientific).
Cloning and transfection.
The LGTV E5 infectious cDNA clone kindly provided by A. Pletnev (NIAID, NIH) (4) served as a template for the cloning of LGTV genes. Each gene was PCR amplified and directionally cloned into Gateway entry vectors (Invitrogen, Carlsbad, CA), followed by subcloning into pcDNA6.2DEST/V5 (Invitrogen) to generate C-terminal V5 epitope-tagged LGTV genes. The primers used are detailed in Table 1. The sequence of each construct was verified by DNA sequencing. Constructs were transfected into cells in six-well plates using Lipofectamine 2000 and OptiMEM (Invitrogen) and allowed to express for 24 h prior to treatment for 15 min with IFN-β. Alternatively, the medium of cells expressing viral proteins was replaced with DMEM containing 10 μg/ml blasticidin for a further 24 to 48 h to select for cells containing expression constructs. Cells were then washed in DPBS before DMEM containing IFN-β was added for 15 min.
TABLE 1.
Sequences in the LGTV E5 cDNA used to PCR amplify viral genes
| Gene (nucleotide no.)a | Sequenceb
|
|
|---|---|---|
| 5′ Primerc | 3′ Primer | |
| NS1 (2380-3514) | CACCATGGGGAACCCAACCTTGTC | AGCCACTACCATGGATCTGAC |
| NS2A (3515-4204) | CACCATGGACAACGGGGCTCTACTCAGTGAAGGTGG | CGTCTCCCCCTGGAGGCCGCCAACTCCC |
| NS2B (4205-4597) | CACCATGTCCTTTAATGAGCCTATGACTGACATAGGAG | TCTCCTGGGTGAGCCAAGCATCTCCGA |
| NS3 (4598-6460) | CACCATGGGAACGGATCTTGTGTTTTCAGG | GCGTCTTCCAGAAGCATAGGACACAAACTC |
| NS4A (6461-6907) | CACCATGAGTGTGGGAGATGTGCTCATGGG | GGCAGCCACCATCCCGGCCAGGCCGC |
| NS4B (6908-7663) | CACCATGAATGAAATGGGCCTCTTGGAC | ACGCCGTGTCCCAGTGGTCCGGAGCC |
| NS5 (7664-10372) | CACCATGGGTGGATCCGAGGGAGAC | AAATATTGAGCTCTCCAGTTTGAGCTC |
Each cloned LGTV gene was confirmed by sequencing, and expression was checked by Western blotting and immunofluoresence.
Where appropriate, signal sequences from the preceding gene were included at the 5′ end of the target gene. Start codons were inserted at the 5′ ends of each gene, and 3′ ends were fused in frame to the V5 epitope sequence encoded in the plasmid which ended on a stop codon.
Bases in italics were added to the 5′ primers to allow proper cloning into the TOPO Gateway system (Invitrogen).
Antibodies and cytokines.
The following antibodies were used in this study: rabbit polyclonal anti-STAT1, anti-STAT2, anti-JAK1, anti-Tyk2, anti-IFNAR2, anti-IFNGR1 (C-20), and anti-IFNGR2 (H-310) (Santa Cruz Biotechnology, Santa Cruz, CA); antiphospho-STAT1 (Tyr701) and anti-phospho-Tyk2 (Tyr 1054/1055) (Cell Signaling Technology, Beverly, MA); anti-phospho-STAT2 (Tyr689) (Upstate Biotechnology, Chicago, IL); anti-phospho-Jak1 (Tyr1022/Tyr1023) (Biosource International, Camarillo, CA); monoclonal anti-IFNAR2 (R&D Systems, Minneapolis, MN); and mouse hyperimmune ascitic fluid specific for TBEV (Russian spring summer encephalitis virus or Far Eastern TBEV; ATCC, Manassas, VA). Monoclonal antibodies to LGTV E and NS1 proteins (18) were kindly provided by C. Schmaljohn (USAMRID, Fort Detrick, Frederick, MD). Recombinant human IFN-α, IFN-β, IFN-γ, tumor necrosis factor alpha (TNF-α), and murine IFN-α and IFN-γ were purchased from R&D Systems (Minneapolis, MN).
Luciferase reporter assays.
Vero cells were transfected with pISRE-luc, pGAS-luc, or pNFkB-luc reporter plasmids (Stratagene, La Jolla, CA) using Lipofectamine 2000 in OptiMEM (Invitrogen). Twenty-four hours following transfection, cells were infected with LGTV TP21 (MOI, 10) and incubated for a further 24 h, after which the appropriate cytokine was added. The cells were incubated for 6 to 7 h, the cell lysates were harvested, and the luciferase assay was performed using a luciferase kit as per the manufacturer's instructions (Stratagene). To examine the ability of each NS protein to inhibit luciferase activity, plasmids encoding NS1, NS4B, or NS5 or the vector backbone, pcDNA6.2/V5, was transfected into cells at the same time as the luciferase reporter plasmids. Twenty-four hours following transfection, the media were removed and replaced with fresh media containing the appropriate cytokine, and assays were continued as detailed above. All experiments were repeated four times.
Immunoprecipitation.
Examination of the phosphorylation status of various JAKs was achieved by immunoprecipition followed by Western blot analysis. The association of various LGTV NS proteins with cellular proteins was determined by coimmunoprecipitation. Vero cells or monocyte-derived human DC were left uninfected or were infected with LGTV (MOI of 10 or 1, respectively), incubated for 48 h, and then left untreated or treated with the appropriate IFN. Cells were washed twice with DPBS and lysed in lysis buffer according to the manufacturer's instructions (Roche, Indianapolis, IN), with the exception that the lysis and wash buffers contained 500 mM NaCl and 0.1% sodium dodecyl sulfate (SDS) to increase their stingency. Cells were subjected to three rounds each of freezing-thawing and sonication, followed by centrifugation for 10 min to remove cellular debris/nuclei. The lysates were cleared by the addition of protein G conjugated with agarose beads (Roche) and rotated at 4°C for 3 h. The beads were removed by centrifugation, and 2 μg of the antibody corresponding to the protein of interest was added to each lysate for 1 h with rotation at 4°C. Protein G-agarose beads were again added, and the lysates were incubated with rotation at 4°C overnight. The lysates were discarded after a brief centrifugation, and the beads were washed five times before the addition of SDS-polyacrylamide gel electrophoresis sample buffer and analysis by Western blotting.
Western blot analysis.
Cells were lysed in sample buffer (Tris-HCl [pH 6.8], SDS, glycerol, dithiothreitol, 1% bromophenol blue), boiled, and quantified using the Bradford protein assay (Bio-Rad Laboratories, Hercules, CA). Equal amounts of protein were separated by SDS-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ). Membranes were blocked for 1 h at room temperature (RT) in Tris-buffered saline (TBS) containing 3% bovine serum albumin, 0.5% Tween 20, 3% normal goat serum, and 50 mM sodium fluoride to inhibit phosphatase activity and then incubated overnight at 4°C in this buffer containing the appropriate primary antibody. All primary antibodies were used at a 1:1,000 dilution for Western blot analysis. Membranes were washed, incubated with horseradish peroxidase-conjugated secondary antibody for 1 h at RT, washed, and finally developed with ECL (Amersham Biosciences). Protein bands were visualized following exposure to X-ray film.
Immunofluorescence.
To examine virus protein expression and STAT1 phosphorylation in cells, Vero cells or human monocyte-derived DC treated with IFN were fixed in ice-cold 100% methanol for 10 min, rinsed twice in TBS, quenched in 50 mM glycine for 30 min at RT, washed three times for 5 min with TBS, and blocked in blocking buffer (TBS containing 1% normal goat serum and 1% bovine serum albumin) for 1 h at RT. The cells were rinsed once in TBS and then incubated with the primary antibodies (anti-phospho-STAT1 at a 1:100 dilution and/or anti-LGTV E protein or anti-V5 at a 1:1,000 dilution) overnight at 4°C. The cells were washed three times in TBS and incubated with secondary antibodies conjugated to Alexa Fluor 488 and Alexa Fluor 594 (1:500; Invitrogen) for 1 h at 37°C. Cells were washed, and coverslips were mounted using Prolong antifade mountant containing DAPI (4′,6′-diamidino-2-phenylindole; Invitrogen) and photographed using a Nikon Microphot SA microscope fitted with a Hamamatsu 5985 chilled digital camera.
RESULTS
Antiviral effects of IFN on LGTV replication.
Treatment of cells with IFN-α/β prior to infection inhibits the replication of flaviviruses, including DEN-2 and Japanese encephalitis virus (8, 30). However, the antiviral effect of IFN-α/β is less robust if added to cells following infection, which is due in part to the virus-mediated inhibition of IFN signaling (8, 30). To examine the effects of IFN on TBE viruses, cells were infected with LGTV (MOI, 1). LGTV titers from cell supernatants were measured at 72 hours postinfection (hpi) and following treatment at various times pre- and postinfection with IFN-α, IFN-γ, or both. These studies were performed with both mouse neuroblastoma (MNB) cells and Vero cells (not shown) with the same results.
IFN-α treatment of MNB cells for 24 h prior to infection reduced virus titers at 72 hpi approximately 1,000-fold compared to those in untreated cells (Fig. 1A). This inhibition occurred regardless of whether IFN was present throughout the course of infection or if it was removed prior to infection (data not shown). Cell viability measured by trypan blue exclusion was not different between treated and untreated cells (data not shown), suggesting that inhibition was not a result of IFN-mediated cytotoxicity over time. However, treatment of cells with IFN-α at 4 hpi led to a significantly (P < 0.05, Student's t test) less inhibitory effect. The inhibitory effect of IFN-α on virus titers was completely lost if cells were treated at 24 hpi (Fig. 1A). These results suggest that LGTV replication is sensitive to the action of IFN-α. The loss of IFN-α sensitivity suggests that LGTV may interfere with IFN functions, particularly if applied after virus replication is initiated. An alternative explanation for the loss of sensitivity consistent with these results is that IFN-α interferes with early steps in virus replication but has little effect on later steps.
FIG. 1.

Effect of IFN treatment on LGTV replication in MNB cells. MNB cells were infected with LGTV virus (strain TP21; MOI of 1) and incubated for 72 h, at which time the virus titer in the supernatant was determined. Cells were either not treated with IFN (NT), pretreated with IFN for 24 h (−24 hr), or treated with IFN 4 h (+4hr) or 24 h (+24hr) after infection. Cells were treated with (A) IFN-α (1,000 U/ml), (B) IFN-γ (10 ng/ml), or (C) IFN-α plus IFN-γ. The error bars represent standard errors of the mean (SEMs) for three experiments. *, P < 0.05.
Pretreatment of cells with IFN-γ for 24 h prior to infection reduced virus titers, but only approximately 10-fold, compared to untreated cells (P < 0.05) (Fig. 1B). Virus replication did not recover when IFN-γ was applied after replication was initiated, even when IFN-γ was first added at 24 hpi. These results suggest that IFN-γ had a marginal antiviral affect on LGTV replication, but it was not as effective as IFN-α. In contrast to the situation with IFN-α, LGTV does not appear to inhibit the small antiviral effect of IFN-γ.
The combination of IFN-α/β and IFN-γ has a synergistic effect on the inhibition of replication of some flaviviruses (8). Pretreatment of cells for 24 h with both IFN-α and IFN-γ (Fig. 1C) did not reduce LGTV titers below those observed following pretreatment with IFN-α alone. However, in contrast to the results shown in Fig. 1A, LGTV replication rebounded only when cells were treated with both IFN-α and IFN-γ at 24 hpi and not at 4 hpi. Thus, IFN-γ treatment augmented the anti-LGTV effects of IFN-α by increasing the time that the virus required to initiate replication before it was no longer sensitive to the effects of these cytokines.
LGTV replication inhibits the JAK-STAT pathway of signal transduction.
If LGTV inhibits IFN responses, interference could occur via global inhibition of the JAK-STAT signal transduction pathway or via inhibition of specific ISG products, such as protein kinase R or 2′,5′-oligoadenylate synthetase. To further examine the effect of LGTV replication on IFN responses, we examined luciferase reporter gene expression under the control of IFN-responsive promoters. These studies were done with Vero cells, which can respond to but do not produce interferon (9). Hence, luciferase expression can result only from exogenously added IFN. To determine if LGTV infection inhibits IFN-α- or IFN-γ-mediated gene expression, Vero cells were transfected with pISRE-luc or pGAS-luc plasmids, respectively, and then infected with LGTV (MOI, 10). Transfection of a pNFκB-luc plasmid was included as a control for surface receptor signaling pathways. The appropriate IFN, or TNF-α, was added 24 h later, and luciferase expression was measured following incubation for 6 to 7 h. LGTV infection alone did not induce luciferase expression (data not shown). Compared to uninfected cells, LGTV infection significantly reduced luciferase expression driven by both ISRE (Fig. 2A) and GAS (Fig. 2B) promoters (P < 0.01 and P < 0.05, respectively, Student's t test) but had no effect on NFκB-driven gene expression (Fig. 2C). These results suggest that LGTV replication specifically inhibited JAK-STAT signaling stimulated by both IFN-α and IFN-γ. The fact that NFκB-driven gene expression was not affected suggests that inhibition was not due to virus-mediated cell cytotoxicity or a general suppression of receptor-mediated signal transduction. Thus, LGTV can interfere with the JAK-STAT signaling pathway in response to both types of IFN, leading to inefficient gene expression.
FIG. 2.

Effect of LGTV (strain TP21) infection on luciferase reporter gene expression driven by ISRE, GAS, and NFκB promoters. Vero cells were transfected with (A) pISRE-luc, (B) pGAS-luc, or (C) pNFκB-luc reporter plasmids, infected with LGTV (MOI of 10) for 24 h, and treated with IFN-α (1,000 U/ml), IFN-γ (10 ng/ml), or TNF-α (100 μg/ml) for 6 to 7 h. Results are expressed as n-fold increases in luciferase activity over that of identical cultures not treated with cytokines (means ± SEMs for four experiments; *, P < 0.05).
Inhibition of STAT phosphorylation by LGTV.
Tyrosine phosphorylation of STAT2 and/or STAT1 is a major event after normal IFN ligation of cell surface receptors (49). To further analyze where in the JAK-STAT pathway virus-mediated inhibition occurs, we examined phosphorylation of STAT1 at Tyr701 (pY-STAT1) by immunoblot analysis of infected Vero cell lysates (Fig. 3). Steady-state levels of STAT1 were not affected by infection. However, the accumulation of pY-STAT1 was greatly reduced in IFN-α-treated cells infected with LGTV compared to that in uninfected cells. Similarly, pY-STAT1 accumulation in response to IFN-γ was inhibited in infected cells, albeit to a lesser extent than following IFN-α stimulation. The incomplete inhibition of STAT phosphorylation in response to IFN-γ by LGTV is consistent with two recent reports examining inhibition of JAK-STAT signaling by WNV and KUN (16, 33). Accumulation of Tyr689-phosphorylated STAT2 in response to IFN-α was also inhibited in LGTV-infected cells (Fig. 3, bottom panel), without an obvious change in the cellular STAT2 levels. pY-STAT inhibition was evident by 24 hpi (not shown) and maximal at 48 hpi (Fig. 3). These results were repeatable following infection of MNB cells (data not shown). Therefore, LGTV inhibited the JAK-STAT signaling pathway in response to either IFN-α or IFN-γ by preventing the phosphorylation of both STAT1 and STAT2.
FIG. 3.

Inhibition of tyrosine phosphorylation of STAT1 and STAT2 in response to IFN-α and IFN-γ by LGTV (strain TP21). Uninfected Vero cells or cells infected with LGTV for 48 h were treated with IFN-α or IFN-γ for 15 min or left untreated. The cell lysates were examined by Western blotting (shown in duplicate) with antibodies to STAT1, Tyr701-phosphorylated STAT1 (pY-STAT1), STAT2, or Tyr689-phosphorylated STAT2 (pY-STAT2; arrow), as indicated to the left of each panel.
Inhibition of Janus kinase phosphorylation by LGTV.
The Janus kinase Jak1 is a component of both the IFN-α/β and IFN-γ receptor-signaling complexes. Phosphorylation of Jak1 is required to initiate, or to amplify, the signal transduction cascade from the receptor (42). Thus, inhibition of Jak1 function could result in suppressed STAT phosphorylation in response to either IFN. We examined Jak1 phosphorylation by immunoprecipitation and Western blotting, utilizing antibodies that recognize both the phosphorylated (Tyr1022/Tyr1023) and the nonphosphorylated Jak1, respectively (Fig. 4). We saw no change in the steady-state levels of Jak1 following infection measured by Western blotting. As expected, pY-Jak1 was observed in the lysates of uninfected cells treated with IFN-α or IFN-γ but not in untreated cells. In contrast, phosphorylated Jak1 was not observed in the lysates of LGTV-infected cells treated with either IFN. Thus, LGTV inhibited the phosphorylation of Jak1 in response to both IFN-α and IFN-γ stimulations. This indicated that the inhibition of STAT phosphorylation might result from the failure of Jak1 to be phosphorylated in IFN-treated, LGTV-infected cells.
FIG. 4.
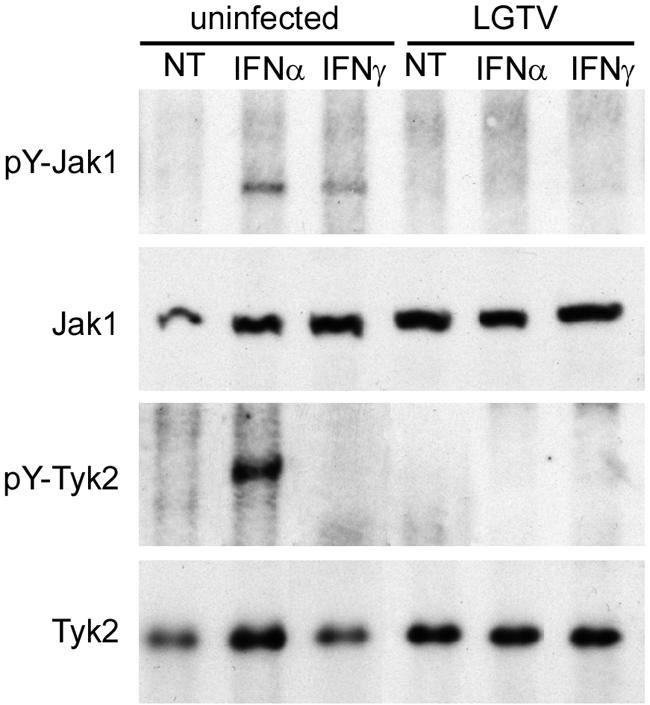
Inhibition of JAK-STAT signaling in response to IFN is associated with inhibition of Jak1 and Tyk2 phosphorylation. Vero cells were infected with LGTV strain TP21 for 48 h and treated with IFN-α or IFN-γ for 15 min. The cell lysates were examined by immunoprecipitation using antibodies to Tyr1022/Tyr1023-phosphorylated Jak1 (pY-Jak1) or Tyr1054/1055-phosphorylated Tyk2 (pY-Tyk2). The immunoprecipitates were analyzed by Western blotting using antibodies that recognize Jak1 or Tyk2. Whole-cell lysates were also examined by Western blotting to determine steady-state levels of Jak1 and Tyk2.
We also examined the phosphorylation levels of Tyk2 in LGTV-infected cells following IFN-α stimulation. Tyk2 is resident on the IFNAR1 subunit and is phosphorylated by Jak1 following stimulation. Phosphorylated Tyk2 was easily detected in lysates from uninfected cells stimulated for 15 min with IFN-α (Fig. 4). However, cell lysates from IFN-α-treated, LGTV-infected cells did not contain pY-Tyk2. These results suggest that LGTV inhibits phosphorylation, and thus activation, of Tyk2 in response to IFN-α. As Tyk2 is responsible for the phosphorylation of both the IFNAR2 subunit, to create a docking site for STAT2, and of STAT2 itself (which is required for any phosphorylation of STAT1 in response to IFN-α/β), this block is likely to result in the failure of STAT phosphorylation in response to IFN-α. We also examined Jak2 phosphorylation following treatment with IFN-γ but could not adequately resolve pY-Jak2 protein bands by immunoprecipitation and/or Western blotting to be definitive (data not shown). Thus, LGTV infection suppressed the activation of both Jak1 and Tyk2 in response to IFN-α and at least Jak1 in response to IFN-γ.
Identification of LGTV nonstructural proteins as IFN antagonists.
To determine the ability of individual virus proteins to antagonize the JAK-STAT pathway, we generated plasmids encoding the individual LGTV nonstructural proteins C-terminally fused to V5 epitope tags and expressed them in Vero cells. Western blot analysis indicated that all LGTV fusion proteins were expressed to similar, high levels following transfection of Vero cells (Fig. 7A). It must be noted that the NS4B construct used in these studies expresses the mature NS4B and does not contain the 2K signal peptide sequence. This sequence has recently been shown to be crucial for the IFN antagonist function of flavivirus NS4B (38).
FIG. 7.

Identification of LGTV NS proteins that associate with the IFN-α/β or IFN-γ receptor complexes in unstimulated (−) and IFN-β-stimulated (+) cells. (A) Western blot probed with anti-V5 antibody demonstrating the expression level of each NS-V5 fusion protein (arrowheads). The asterisk denotes a nonspecific anti-V5-reacting band present in all cell lysates. (B) Cell lysates from Vero cells expressing LGTV NS fusion proteins were immunoprecipitated with an anti-IFNAR2 monoclonal antibody (upper panel). Anti-V5-reacting, coprecipitating protein was detected by Western blot analysis (lower panel). Only the NS5 fusion protein coprecipitated with IFNAR2 (arrow). (C) Cell lysates from Vero cells expressing NS5 fusion protein were immunoprecipitated with anti-IFNGR1 or anti-IFNGR2 polyclonal antibodies. Anti-V5-reacting, coprecipitating protein was detected by Western blot analysis. The approximate molecular mass (kDa) is indicated. WB, Western blot; IP, immunoprecipitation.
To determine which LGTV NS protein(s) can inhibit JAK-STAT signaling, Vero cells were transfected with each construct and were selected with blasticidin to obtain a population of cells, the majority of which were expressing one viral protein. Cells were then treated with IFN-β and lysed. In addition to the Bradford assay (2a), the steady-state STAT1 level was used to normalize cell lysates, as LGTV-mediated inhibition of JAK-STAT signaling does not involve degradation or reduced expression of STAT1 (Fig. 5A, upper panel). Levels of phosphorylated STAT1 (pY-STAT1) in LGTV NS protein-containing cells were examined by Western blotting and were equivalent in cells transfected with the backbone plasmid, pcDNA6.2DEST/V5, and in cells expressing NS1, NS2A, NS2B, NS3, NS4A, or NS4B fusion proteins (Fig. 5A, lower panel). In contrast, pY-STAT1 was barely detectable in cells expressing the NS5 fusion protein. This suggested that NS5 can suppress STAT1 activation and directly implicated LGTV NS5 as a TBE virus antagonist of IFN-α/β-stimulated JAK-STAT signaling.
FIG. 5.

Identification of LGTV nonstructural proteins that function as IFN antagonists. (A) Vero cells expressing LGTV NS proteins fused to V5 epitope tags were treated with blasticidin to obtain a cell population, the majority of which expressed the individual NS protein indicated, or the backbone plasmid, pcDNA6.2/V5. Cells were then treated with IFN-β and examined for STAT1 phosphorylation by Western blot analysis. Steady-state STAT1 levels (upper panel) and tyrosine-phosphorylated STAT1 (pY-STAT1) (lower panel) are shown in cell populations expressing LGTV NS proteins and either left untreated (−) or treated (+) with IFN-β. (B) Vero cells were treated with IFN-β, fixed in methanol, and stained for anti-V5 (red) and anti-pY-STAT1 (green). Nuclei were counterstained with DAPI (blue). The upper panel demonstrates the localization of tyrosine-phosphorylated STAT1 in the nuclei of untransfected cells that were left untreated or treated with IFN-β. The lower panels represent cells that express NS4B, NS2A, NS4A, and NS5. Examples of V5-positive and either pY-STAT1-positive (arrows) or pY-STAT1-negative (arrowheads) cells are indicated.
To examine the potential of LGTV NS proteins to inhibit STAT1 phosphorylation at the level of the individual cell, we expressed each protein in Vero cells and treated the proteins with IFN-β. The presence of pY-STAT1 in cells expressing the NS proteins was examined by an immunofluorescence assay (IFA), using antibodies that specifically recognize pY-STAT1 and V5 (Fig. 5B). In normal cells treated with IFN-β, pY-STAT1 localized to the nucleus (Fig. 5B, upper panel). Phosphorylated STAT1 was observed in the majority of cells expressing NS1, NS2A, NS2B, NS3, NS4A, or NS4B. Representative images of cells expressing NS4B, NS2A, and NS4A are shown in Fig. 5B (lower panels). In contrast, cells expressing NS5 did not contain pY-STAT1 (Fig. 5B, bottom panel). The results of the IFA support those demonstrated by Western blotting in Fig. 5A and suggest that LGTV NS5 is an antagonist of IFN-α/β signaling.
Examination of JAK and STAT phosphorylation by Western blotting following infection with LGTV (Fig. 3 and 4) revealed that both IFN-α/β- and IFN-γ-mediated pathways were inhibited. To determine if NS5 was capable of inhibiting signaling following stimulation with both IFN-α/β and IFN-γ, we examined the ability of NS5 as well as NS1 and NS4B to block luciferase expression driven by either ISRE or GAS promoter elements in the presence of IFN-β or IFN-γ, respectively (Fig. 6A and 6B). The induction of luciferase activity was not affected by expression of NS1 or NS4B. However, NS5 expression significantly inhibited luciferase gene activity in response to either IFN-β (Fig. 6A) or IFN-γ (Fig. 6B) (P < 0.05, analysis of variance, followed by Tukey's honestly significant difference test). Interestingly, the relative level of inhibition observed was more pronounced in NS5-expressing cells treated with IFN-β than in those treated with IFN-γ.
FIG. 6.

Effect of LGTV NS proteins on luciferase reporter gene expression. Vero cells transfected with (A) pISRE-luc or (B) pGAS-luc were also transfected with the backbone plasmid, pcDNA6.2/V5, or with the NS1, NS4B, or NS5 expression plasmid and treated with IFN-α or IFN-γ. Results are expressed as n-fold increases in luciferase activity over that of identical cultures not treated with cytokines (means ± SEMs for four experiments; *, P < 0.05). (C) NS5-expressing Vero cells were treated with IFN-γ, fixed, and stained for anti-V5 (red) and anti-pY-STAT1 (green). Nuclei were counterstained with DAPI (blue). Examples of V5-positive and either pY-STAT1-positive (arrow) or pY-STAT1-negative (arrowheads) cells are indicated.
To examine the ability of NS5 to inhibit IFN-γ-induced STAT1 phosphorylation at the level of the individual cell, we treated NS5-expressing Vero cells with IFN-γ and examined STAT1 phosphorylation. In general, pY-STAT1 was not observed in IFN-γ-treated NS5-positive cells (Fig. 6C). However, the nuclei of some NS5-positive cells stained faintly positive for pY-STAT1 (Fig. 6C). This observation was in contrast to that found following stimulation with IFN-β, in which pY-STAT1 was undetectable in NS5-positive cells (Fig. 5B). Taken together, these results suggest that LGTV NS5 can function to antagonize both IFN-β- and IFN-γ-mediated JAK-STAT signaling, although NS5-mediated inhibition of IFN-β-stimulated signal transduction was more efficient.
Identification of NS5 interactions with the cellular IFN receptor complexes.
The inhibition of Jak1 and Tyk2 phosphorylation observed in LGTV-infected cells following IFN-α/β or IFN-γ stimulation suggests that the mechanism of virus interference involves the receptors and/or associated kinases. To determine if any of the LGTV NS proteins interact with the IFN receptor complex, we transfected Vero cells with each of the NS protein expression plasmids or the backbone plasmid, pcDNA6.2/V5. The expression levels of all NS proteins were verified by probing a Western blot with an anti-V5 monoclonal antibody (Fig. 7A). We then immunoprecipitated the IFN-α/β receptor subunit, IFNAR2, from cell lysates and examined them for coprecipitating anti-V5-reacting NS protein by Western blotting (Fig. 7B). NS5-V5 was the only NS protein detected in cell lysates immunoprecipitated with anti-IFNAR2 monoclonal antibody. Repeatedly, more NS5-V5 was coprecipitated from unstimulated cells than from IFN-β-stimulated cells. Similar results were obtained using a polyclonal antibody specific for IFNAR2 (data not shown). Furthermore, IFNAR2 was coimmunoprecipitated if hyperimmune mouse ascitic fluid against TBEV was used to immunoprecipitate NS5 (data not shown). We could not verify immunoprecipitation of IFNAR1 with any antibody we tested, and thus it is unknown if NS5 could associate with this molecule. These results further implicate NS5 as the protein responsible for disruption of IFN-β-stimulated JAK-STAT signaling and suggest that suppression may be mediated by an interaction with the IFN-α/β receptor complex.
To determine if NS5 also associates with IFN-γ receptor complexes, we expressed NS5 in Vero cells for 24 h (cells were not treated with IFN), followed by immunoprecipitation of the IFN-γ receptor subunits using antibodies specific for IFNGR1 or IFNGR2. Immunoprecipitates were examined for coprecipitating anti-V5-reacting NS protein by Western blotting (Fig. 7C). We observed an association between IFNGR1 and the NS5 fusion protein. We also detected a weak association between IFNGR2 and NS5. Hence, NS5-mediated antagonism of IFN-γ-stimulated JAK-STAT signaling may result from the association of NS5 with the IFN-γ receptor complex.
Inhibition of JAK-STAT signaling in infected primary DC.
In addition to direct antiviral effects, IFN serve as an important link between innate and adaptive immunity through their roles in the activation and maturation of antigen-presenting cells, including DC (2, 12). DC are potentially important target cells for tick-borne flavivirus infections (24), although whether human DC are infected by TBE viruses has not been investigated. Furthermore, DC are important target cells for DEN infection (52, 54). The pathogenesis of DEN is associated with aberrant immune activation (26). The proliferative responses of T lymphocytes from patients acutely infected with DEN are compromised, which is due primarily to the inability of antigen-presenting cells to provide adequate stimuli (35). If infection of DC by flaviviruses inhibits JAK-STAT signaling, then not only virus replication but also immune regulation may be affected. To test this possibility, we infected human monocyte-derived DC with LGTV (MOI, 1) for 24 h in vitro. Infection was verified by NS1 expression in infected cells (data not shown). Primary DC were stimulated with either IFN-β or IFN-γ and fixed in methanol. pY-STAT1, LGTV E protein, and nuclei were visualized by IFA (Fig. 8A, B, C, and D). pY-STAT1 accumulated in the nuclei of uninfected DC. However, of approximately 200 LGTV-infected DC observed, no single infected DC contained detectable pY-STAT1. Similarly, phosphorylated STAT2 was not detected in LGTV-infected DC stimulated with IFN-β (not shown). JAK-STAT signaling is therefore compromised in human DC infected with LGTV.
FIG. 8.

JAK-STAT signaling is inhibited in LGTV-infected human monocyte-derived dendritic cells. Human monocyte-derived dendritic cells were infected with LGTV TP21 (MOI of 1) for 24 h prior to the addition of IFN-α for 15 min. Cells were fixed in methanol and stained for (A) pY-STAT1 and (B) LGTV E protein and (C) counterstained with DAPI to visualize nuclei. Arrows in panel C indicate the nuclei of cells infected with LGTV. (D) Merged image of panels A and B showing no pY-STAT1 in LGTV-infected dendritic cells. (E) Monocyte-derived DC were infected with LGTV TP21 for 48 h and treated with IFN-α for 15 min. The cell lysates were immunoprecipitated using antibodies to Tyr1022/Tyr1023-phosphorylated Jak1 (pY-Jak1) or Tyr1054/1055-phosphorylated Tyk2 (pY-Tyk2) and analyzed by Western blotting using the same antibodies. (F) Cell lysates from human DC infected for 48 h with LGTV, and either left untreated (−) or treated (+) with IFN-β, were immunoprecipitated with antibodies specific for IFNAR2, Jak1, Tyk2, or STAT1 (upper panels). Western blots examining coprecipitating LGTV proteins were probed with anti-TBEV antisera, revealing an association between NS5 and IFNAR2. WB, Western blot; IP, immunoprecipitation.
In addition to STAT phosphorylation in infected DC, we examined Jak1 and Tyk2 phosphorylation in DC populations infected with LGTV for 48 h and treated with IFN-β. Similar to the observations in infected Vero cells, the activation of both kinases was effectively inhibited in LGTV-infected DC (Fig. 8E). Taken together, these results provide definitive evidence that LGTV inhibits IFN-induced JAK-STAT signaling in primary human DC.
We examined the potential for NS5 to interact with the IFN-α/β receptor complex in DC to determine if this interaction occurs during virus replication and to further define the NS5-IFN receptor complex interaction. Human monocyte-derived DC were infected with LGTV for 48 h and then left untreated or treated with IFN-β. DC lysates were immunoprecipitated with antibodies recognizing IFNAR2, Jak1, Tyk2, or STAT1. The coprecipitation of NS5 with these molecules was examined by probing Western blots of the precipitates with anti-TBEV ascitic fluid, which recognizes E, M, NS1, and NS5 (S. M. Best, D. N. Mitzel, and M. E. Bloom, unpublished). NS5 produced during virus replication coprecipitated with IFNAR2 but not with Jak1, Tyk2, or STAT1 (Fig. 8F). Equal amounts of NS5 were immunoprecipitated from IFN-β-treated and untreated cells. There was no coprecipitation of viral protein in uninfected cells or other viral proteins recognized by the anti-TBEV antiserum (data not shown). Identical results were obtained following infection of murine bone-derived dendritic cells (S. J. Robertson and S. M. Best, unpublished). Thus, NS5 interacts with IFNAR2 during LGTV replication. Furthermore, the interaction between NS5 and IFNAR2 occurred in primary DC from at least two different species, human and mouse.
DISCUSSION
In this report, we demonstrate that a tick-borne flavivirus, LGTV, antagonizes IFN signaling by inhibiting the JAK-STAT signal transduction pathway in response to both IFN-α/β and IFN-γ. Furthermore, we have demonstrated that LGTV NS5 alone can suppress STAT1 phosphorylation in response to IFN-β or IFN-γ stimulation (Fig. 5 and 6). Thus, NS5 is a direct antagonist of IFN signaling. This antagonistic function of NS5 is most likely to result from its association with the IFN receptor complexes (Fig. 7).
Human DC are important early target cells for flavivirus replication (24, 41, 52, 54). During LGTV replication in human DC, NS5 associated with the IFN-α/β receptor subunit, IFNAR2 (Fig. 8F). The direct association of NS5 with IFNAR2 in LGTV-infected DC is likely to result in the functional inhibition of JAK-STAT signal transduction observed in these cells. Consequently, there may be a dual role for LGTV NS5 in pathogenesis. First, inhibition of IFN will facilitate early virus replication at the level of the individual cell. Second, the inhibition of signaling in these cells may disrupt the bridge between innate and adaptive immunity by compromising DC activation and/or maturation, as IFN-α/β have critical roles in DC maturation, survival, and function (12). This hypothesis is supported by recent findings demonstrating inhibited maturation and T-cell-stimulatory capabilities of human DC infected with DEN-2 in vitro (41). If similar interactions occur between DC and the highly virulent TBE viruses, this may contribute significantly to the severity of some TBE virus infections.
Since both tick- and mosquito-borne flaviviruses prevent JAK activation in response to IFN-α/β and IFN-γ (16, 30, 32, 33, 39), it is possible that a common mechanism of inhibition exists. NS5 has 80 to 90% amino acid identity among members of the TBE virus serogroup, suggesting that the function of NS5 in IFN antagonism might be conserved in this group. However, previous studies investigating JAK-STAT signaling following DEN or WNV infection have implicated nonstructural proteins other than NS5 (32, 33, 39), and NS4B in particular (38, 39). The fact that we did not identify NS4B as an antagonist may be due to the expression in these studies of the mature LGTV NS4B that lacks the 23-amino-acid signal sequence. The presence of this sequence has recently been shown to be crucial to the inhibitory function of NS4B (38). Given the conservation in function of various proteins throughout the flaviviruses, it is surprising that NS5 has not been identified as an antagonist for other viruses. In particular, WNV infection prevented Jak1 and Tyk2 activation in response to IFN-α (33) in a manner very similar to that described here for LGTV, suggesting that the two viruses may utilize similar mechanisms to suppress JAK-STAT signaling pathways. Furthermore, while the polyprotein of LGTV shares only approximately 40% amino acid identity with WNV or DEN, LGTV NS5 shares a relatively high degree of amino acid identity (approximately 57 to 59%) with both WNV and DEN-2 NS5 proteins. The role of NS5 as an antagonist for LGTV may simply reflect a divergence in the IFN evasion strategies used by tick- and mosquito-borne flaviviruses. Therefore, it will be of interest to determine the relative contribution of NS5 in the IFN resistance of the different vector-borne flaviviruses.
This study suggests that NS5 has a role in viral pathogenesis as well as in virus replication. NS5 is the largest of the flavivirus NS proteins. It has a central function in RNA replication as it contains the viral RNA-dependent RNA polymerase and methyltransferase/RNA capping functions (10). The primary cellular location of NS5 is the endoplasmic reticulum, where virus replication occurs (48). However, the additional role for this protein in IFN antagonism suggests that some NS5 may localize to a position proximal to the plasma membrane of infected cells. The regulation of NS5 distribution most likely occurs through posttranslational modification, including phosphorylation (21, 47). Thus, further studies examining flavivirus NS5 expression, regulation, and distribution will be required to fully elucidate NS5 functions in both virus replication and IFN resistance.
The precise mechanism by which NS5 associates with the IFN receptor complexes and inhibits JAK-STAT signaling is not yet understood, although two broad possibilities exist. The simplest explanation is that NS5 prevents the receptors from functioning. This could be achieved by preventing ligation of receptor subunits through steric hindrance. Alternatively, NS5 may lock up or freeze receptor complexes through multivalent interactions with receptor components such that further signaling cannot occur. In unstimulated cells, the receptor components are preassembled. For example, IFNAR2 is associated with Jak1, STAT1, and STAT2; IFNAR1 is associated with Tyk2. Therefore, binding to a receptor subunit as well as additional components either preceding or following IFN stimulation may prevent further signal transduction.
The second possibility for NS5-mediated inhibition is that NS5 associates with receptors to specifically prevent JAK function. During normal JAK-STAT signal transduction, negative feedback mechanisms exist to regulate signaling. One mechanism operates through the expression of SOCS (suppressor of cytokine signaling proteins (1, 19, 20). SOCS proteins can bind IFN receptor subunits in order to access the catalytic domain of JAKs, thus preventing their activation. SOCS proteins can also bind phosphorylated JAKs and target them for degradation via the proteasome. In LGTV-infected cells, inhibition of the proteasome does not restore JAK-STAT signaling (S. M. Best and M. E. Bloom, unpublished), suggesting that this mechanism is not involved. However, LGTV NS5 may bind IFN receptors and function in an analogous way to SOCS proteins to prevent the phosphorylation of JAKs. Alternatively, NS5 might function as a virus equivalent of another cellular negative regulator, a protein tyrosine phosphatase, to remove phosphate groups from JAK catalytic domains (3, 40).
Both RNA and DNA viruses utilize a variety of mechanisms to inhibit IFN-stimulated JAK-STAT signaling (5, 11, 13, 22, 28). The paramyxoviruses interfere directly with STAT proteins, preventing their nuclear translocation, targeting them for degradation via the proteasome, or sequestering them in high-molecular-mass cytoplasmic complexes (17). Human cytomegalovirus degrades Jak1 and IRF-9 via the proteasome (36, 37), while the murine polyomavirus T antigen binds to and inhibits Jak1 activity (53). The E6 protein of human papillomavirus 18 binds to Tyk2 and prevents its normal association with the IFNAR1 subunit, thereby inhibiting JAK-STAT signaling (29). Inhibition of JAK phosphorylation has also been associated with suppression of JAK-STAT signaling following infection with human herpes simplex virus type 1 (inhibited phosphorylation of Jak1, Jak2, and Tyk2) (7, 56), Sendai virus (Tyk2) (23), and measles virus (Jak1) (55). The precise mechanism(s) by which flaviviruses circumvent JAK-STAT signaling remains to be determined.
The identification of NS5 as an inhibitor of JAK-STAT signaling may have implications for vaccine design and development of therapeutics. Currently, LGTV is being developed as a backbone for live-attenuated chimeric virus vaccines for protection against TBEV infection (44, 45). If NS5 of these chimeric viruses also interferes with IFN signaling, identification and alteration of the NS5 sequences responsible might further attenuate the viruses and increase immune responsiveness, thus reducing the risk of possible complications associated with a live vaccine. In addition, IFN-α/β are potential therapeutics for infection with various flaviviruses (34, 50). In this context, identification of the NS5 sequences that interact with the IFN receptor could lead to the design of additional therapeutic inhibitors for use in combination with, or instead of, IFN-α/β.
This newly defined role of NS5 as an IFN antagonist may promote virus survival by preventing the antiviral state from being established in infected cells. Furthermore, a consequence of the protein-protein interactions described, particularly in infected DC, may be to perturb adaptive immune responses to infection, thus delaying virus clearance. Because this interaction occurs in cells from mice and from humans, it may aid virus replication in hosts that serve to amplify the virus in the environment as well as in human hosts. Hence, in addition to its central role in viral RNA replication, NS5 is potentially an important virus virulence factor.
Acknowledgments
We thank Alexander Pletnev for providing LGTV TP21 virus and E5 cDNA clone, Connie Schmaljohn for providing monoclonal antibodies to LGTV proteins, Gary Hettrick and Anita Mora for graphical help, and members of the laboratory for critical review of the manuscript.
REFERENCES
- 1.Alexander, W. S., and D. J. Hilton. 2004. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu. Rev. Immunol. 22:503-529. [DOI] [PubMed] [Google Scholar]
- 2.Biron, C. A. 2001. Interferons alpha and beta as immune regulators-a new look. Immunity 14:661-664. [DOI] [PubMed] [Google Scholar]
- 2a.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 3.Brierley, M. M., and E. N. Fish. 2002. Review: IFN-alpha/beta receptor interactions to biologic outcomes: understanding the circuitry. J. Interferon Cytokine Res. 22:835-845. [DOI] [PubMed] [Google Scholar]
- 4.Campbell, M. S., and A. G. Pletnev. 2000. Infectious cDNA clones of Langat tick-borne flavivirus that differ from their parent in peripheral neurovirulence. Virology 269:225-237. [DOI] [PubMed] [Google Scholar]
- 5.Cebulla, C. M., D. M. Miller, and D. D. Sedmak. 1999. Viral inhibition of interferon signal transduction. Intervirology 42:325-330. [DOI] [PubMed] [Google Scholar]
- 6.Charrel, R. N., H. Attoui, A. M. Butenko, J. C. Clegg, V. Deubel, T. V. Frolova, E. A. Gould, T. S. Gritsun, F. X. Heinz, M. Labuda, V. A. Lashkevich, V. Loktev, A. Lundkvist, D. V. Lvov, C. W. Mandl, M. Niedrig, A. Papa, V. S. Petrov, A. Plyusnin, S. Randolph, J. Suss, V. I. Zlobin, and X. de Lamballerie. 2004. Tick-borne virus diseases of human interest in Europe. Clin. Microbiol. Infect. 10:1040-1055. [DOI] [PubMed] [Google Scholar]
- 7.Chee, A. V., and B. Roizman. 2004. Herpes simplex virus 1 gene products occlude the interferon signaling pathway at multiple sites. J. Virol. 78:4185-4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diamond, M. S., T. G. Roberts, D. Edgil, B. Lu, J. Ernst, and E. Harris. 2000. Modulation of dengue virus infection in human cells by alpha, beta, and gamma interferons. J. Virol. 74:4957-4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diaz, M. O., S. Ziemin, M. M. L. Beau, P. Pitha, S. D. Smith, R. R. Chilcote, and J. D. Rowley. 1988. Homozygous deletion of the alpha- and beta 1-interferon genes in human leukemia and derived cell lines. Proc. Natl. Acad. Sci. USA 85:5259-5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Egloff, M. P., D. Benarroch, B. Selisko, J. L. Romette, and B. Canard. 2002. An RNA cap (nucleoside-2′-O-)-methyltransferase in the flavivirus RNA polymerase NS5: crystal structure and functional characterization. EMBO J. 21:2757-2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Sastre, A. 2002. Mechanisms of inhibition of the host interferon alpha/beta-mediated antiviral responses by viruses. Microbes Infect. 4:647-655. [DOI] [PubMed] [Google Scholar]
- 12.Gauzzi, M. C., I. Canini, P. Eid, F. Belardelli, and S. Gessani. 2002. Loss of type I IFN receptors and impaired IFN responsiveness during terminal maturation of monocyte-derived human dendritic cells. J. Immunol. 169:3038-3045. [DOI] [PubMed] [Google Scholar]
- 13.Goodbourn, S., L. Didcock, and R. E. Randall. 2000. Interferons: cell signalling, immune modulation, antiviral response and virus countermeasures. J. Gen. Virol. 81:2341-2364. [DOI] [PubMed] [Google Scholar]
- 14.Gritsun, T. S., V. A. Lashkevich, and E. A. Gould. 2003. Tick-borne encephalitis. Antivir. Res. 57:129-146. [DOI] [PubMed] [Google Scholar]
- 15.Gritsun, T. S., P. A. Nuttall, and E. A. Gould. 2003. Tick-borne flaviviruses. Adv. Virus Res. 61:317-371. [DOI] [PubMed] [Google Scholar]
- 16.Guo, J.-T., J. Hayashi, and C. Seeger. 2005. West Nile virus inhibits the signal transduction pathway of alpha interferon. J. Virol. 79:1343-1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horvath, C. M. 2004. Weapons of STAT destruction. Eur. J. Biochem. 271:4621-4628. [DOI] [PubMed] [Google Scholar]
- 18.Iacono-Connors, L. C., J. F. Smith, T. G. Ksiazek, C. L. Kelley, and C. S. Schmaljohn. 1996. Characterization of Langat virus antigenic determinants defined by monoclonal antibodies to E, NS1 and preM and identification of a protective, non-neutralizing preM-specific monoclonal antibody. Virus Res. 43:125-136. [DOI] [PubMed] [Google Scholar]
- 19.Ilangumaran, S., S. Ramanathan, and R. Rottapel. 2004. Regulation of the immune system by SOCS family adaptor proteins. Semin. Immunol. 16:351-365. [DOI] [PubMed] [Google Scholar]
- 20.Ilangumaran, S., and R. Rottapel. 2003. Regulation of cytokine receptor signaling by SOCS1. Immunol. Rev. 192:196-211. [DOI] [PubMed] [Google Scholar]
- 21.Kapoor, M., L. Zhang, M. Ramachandra, J. Kusukawa, K. E. Ebner, and R. Padmanabhan. 1995. Association between NS3 and NS5 proteins of dengue virus type 2 in the putative RNA replicase is linked to differential phosphorylation of NS5. J. Biol. Chem. 270:19100-19106. [DOI] [PubMed] [Google Scholar]
- 22.Katze, M. G., Y. He, and M. Gale, Jr. 2002. Viruses and interferon: a fight for supremacy. Nat. Rev. Immunol. 2:675-687. [DOI] [PubMed] [Google Scholar]
- 23.Komatsu, T., K. Takeuchi, J. Yokoo, Y. Tanaka, and B. Gotoh. 2000. Sendai virus blocks alpha interferon signaling to signal transducers and activators of transcription. J. Virol. 74:2477-2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Labuda, M., J. M. Austyn, E. Zuffova, O. Kozuch, N. Fuchsberger, J. Lysy, and P. A. Nuttall. 1996. Importance of localized skin infection in tick-borne encephalitis virus transmission. Virology 219:357-366. [DOI] [PubMed] [Google Scholar]
- 25.Lau, J. F., and C. M. Horvath. 2002. Mechanisms of type I interferon cell signaling and STAT-mediated transcriptional responses. Mt. Sinai J. Med. 69:156-168. [PubMed] [Google Scholar]
- 26.Lei, H. Y., T. M. Yeh, H. S. Liu, Y. S. Lin, S. H. Chen, and C. C. Liu. 2001. Immunopathogenesis of dengue virus infection. J. Biomed. Sci. 8:377-388. [DOI] [PubMed] [Google Scholar]
- 27.Levy, D. E., and J. E. Darnell, Jr. 2002. Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 3:651-662. [DOI] [PubMed] [Google Scholar]
- 28.Levy, D. E., and A. García-Sastre. 2001. The virus battles: IFN induction of the antiviral state and mechanisms of viral evasion. Cytokine Growth Factor Rev. 12:143-156. [DOI] [PubMed] [Google Scholar]
- 29.Li, S., S. Labrecque, M. C. Gauzzi, A. R. Cuddihy, A. H. Wong, S. Pellegrini, G. J. Matlashewski, and A. E. Koromilas. 1999. The human papilloma virus (HPV)-18 E6 oncoprotein physically associates with Tyk2 and impairs Jak-STAT activation by interferon-alpha. Oncogene 18:5727-5737. [DOI] [PubMed] [Google Scholar]
- 30.Lin, R.-J., C.-L. Liao, E. Lin, and Y.-L. Lin. 2004. Blocking of the alpha interferon-induced Jak-Stat signaling pathway by Japanese encephalitis virus infection. J. Virol. 78:9285-9294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lindenbach, B. D., and C. M. Rice. 2003. Molecular biology of flaviviruses. Adv. Virus Res. 59:23-61. [DOI] [PubMed] [Google Scholar]
- 32.Liu, W. J., H. B. Chen, X. J. Wang, H. Huang, and A. A. Khromykh. 2004. Analysis of adaptive mutations in Kunjin virus replicon RNA reveals a novel role for the flavivirus nonstructural protein NS2A in inhibition of beta interferon promoter-driven transcription. J. Virol. 78:12225-12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu, W. J., X. J. Wang, V. V. Mokhonov, P.-Y. Shi, R. Randall, and A. A. Khromykh. 2005. Inhibition of interferon signaling by the New York 99 strain and Kunjin subtype of West Nile virus involves blockage of STAT1 and STAT2 activation by nonstructural proteins. J. Virol. 79:1934-1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manns, M. P., J. G. McHutchison, S. C. Gordon, V. K. Rustgi, M. Shiffman, R. Reindollar, Z. D. Goodman, K. Koury, M. Ling, and J. K. Albrecht. 2001. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 358:958-965. [DOI] [PubMed] [Google Scholar]
- 35.Mathew, A., I. Kurane, S. Green, D. W. Vaughn, S. Kalayanarooj, S. Suntayakorn, F. A. Ennis, and A. L. Rothman. 1999. Impaired T cell proliferation in acute dengue infection. J. Immunol. 162:5609-5615. [PubMed] [Google Scholar]
- 36.Miller, D. M., B. M. Rahill, J. M. Boss, M. D. Lairmore, J. E. Durbin, J. W. Waldman, and D. D. Sedmak. 1998. Human cytomegalovirus inhibits major histocompatibility complex class II expression by disruption of the Jak/Stat pathway. J Exp. Med. 187:675-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller, D. M., Y. Zhang, B. M. Rahill, W. J. Waldman, and D. D. Sedmak. 1999. Human cytomegalovirus inhibits IFN-alpha-stimulated antiviral and immunoregulatory responses by blocking multiple levels of IFN-alpha signal transduction. J. Immunol. 162:6107-6113. [PubMed] [Google Scholar]
- 38.Muñoz-Jordán, J. L., M. Laurent-Rolle, J. Ashour, L. Martínez-Sobrido, M. Ashok, W. I. Lipkin, and A. García-Sastre. 2005. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J. Virol. 79:8004-8013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muñoz-Jordán, J. L., G. G. Sanchez-Burgos, M. Laurent-Rolle, and A. García-Sastre. 2003. Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. USA 100:14333-14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neel, B. G., and N. K. Tonks. 1997. Protein tyrosine phosphatases in signal transduction. Curr. Opin. Cell Biol. 9:193-204. [DOI] [PubMed] [Google Scholar]
- 41.Palmer, D. R., P. Sun, C. Celluzzi, J. Bisbing, S. Pang, W. Sun, M. A. Marovich, and T. Burgess. 2005. Differential effects of dengue virus on infected and bystander dendritic cells. J. Virol. 79:2432-2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pellegrini, S., and I. Dusanter-Fourt. 1997. The structure, regulation and function of the Janus kinases (JAKs) and the signal transducers and activators of transcription (STATs). Eur. J. Biochem. 248:615-633. [DOI] [PubMed] [Google Scholar]
- 43.Perelygin, A. A., S. V. Scherbik, I. B. Zhulin, B. M. Stockman, Y. Li, and M. A. Brinton. 2002. Positional cloning of the murine flavivirus resistance gene. Proc. Natl. Acad. Sci. USA 99:9322-9327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pletnev, A. G., M. Bray, K. A. Hanley, J. Speicher, and R. Elkins. 2001. Tick-borne Langat/mosquito-borne dengue flavivirus chimera, a candidate live attenuated vaccine for protection against disease caused by members of the tick-borne encephalitis virus complex: evaluation in rhesus monkeys and in mosquitoes. J. Virol. 75:8259-8267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pletnev, A. G., G. G. Karganova, T. I. Dzhivanyan, V. A. Lashkevich, and M. Bray. 2000. Chimeric Langat/dengue viruses protect mice from heterologous challenge with the highly virulent strains of tick-borne encephalitis virus. Virology 274:26-31. [DOI] [PubMed] [Google Scholar]
- 46.Pletnev, A. G., and R. Men. 1998. Attenuation of the Langat tick-borne flavivirus by chimerization with mosquito-borne flavivirus dengue type 4. Proc. Natl. Acad. Sci. USA 95:1746-1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reed, K. E., A. E. Gorbalenya, and C. M. Rice. 1998. The NS5A/NS5 proteins of viruses from three genera of the family Flaviviridae are phosphorylated by associated serine/threonine kinases. J. Virol. 72:6199-6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salonen, A., T. Ahola, and L. Kaariainen. 2005. Viral RNA replication in association with cellular membranes. Curr. Top. Microbiol. Immunol. 285:139-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shuai, K., and B. Liu. 2003. Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 3:900-911. [DOI] [PubMed] [Google Scholar]
- 50.Solomon, T., N. M. Dung, B. Wills, R. Kneen, M. Gainsborough, T. V. Diet, T. T. Thuy, H. T. Loan, V. C. Khanh, D. W. Vaughn, N. J. White, and J. J. Farrar. 2003. Interferon alfa-2a in Japanese encephalitis: a randomised double-blind placebo-controlled trial. Lancet 361:821-826. [DOI] [PubMed] [Google Scholar]
- 51.Sumpter, R., Jr., C. Wang, E. Foy, Y.-M. Loo, and M. Gale, Jr. 2004. Viral evolution and interferon resistance of hepatitis C virus RNA replication in a cell culture model. J. Virol. 78:11591-11604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tassaneetrithep, B., T. H. Burgess, A. Granelli-Piperno, C. Trumpfheller, J. Finke, W. Sun, M. A. Eller, K. Pattanapanyasat, S. Sarasombath, D. L. Birx, R. M. Steinman, S. Schlesinger, and M. A. Marovich. 2003. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J. Exp. Med. 197:823-829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weihua, X., S. Ramanujam, D. J. Lindner, R. D. Kudaravalli, R. Freund, and D. V. Kalvakolanu. 1998. The polyoma virus T antigen interferes with interferon-inducible gene expression. Proc. Natl. Acad. Sci. USA 95:1085-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu, S. J., G. Grouard-Vogel, W. Sun, J. R. Mascola, E. Brachtel, R. Putvatana, M. K. Louder, L. Filgueira, M. A. Marovich, H. K. Wong, A. Blauvelt, G. S. Murphy, M. L. Robb, B. L. Innes, D. L. Birx, C. G. Hayes, and S. S. Frankel. 2000. Human skin Langerhans cells are targets of dengue virus infection. Nat. Med. 6:816-820. [DOI] [PubMed] [Google Scholar]
- 55.Yokota, S., H. Saito, T. Kubota, N. Yokosawa, K. Amano, and N. Fujii. 2003. Measles virus suppresses interferon-alpha signaling pathway: suppression of Jak1 phosphorylation and association of viral accessory proteins, C and V, with interferon-alpha receptor complex. Virology 306:135-146. [DOI] [PubMed] [Google Scholar]
- 56.Yokota, S., N. Yokosawa, T. Kubota, T. Suzutani, I. Yoshida, S. Miura, K. Jimbow, and N. Fujii. 2001. Herpes simplex virus type 1 suppresses the interferon signaling pathway by inhibiting phosphorylation of STATs and janus kinases during an early infection stage. Virology 286:119-124. [DOI] [PubMed] [Google Scholar]