

ABSTRACT
Hypertensive cardiac hypertrophy (HCH) is a compensatory response to chronic pressure overload, ultimately progressing to heart failure if left unmanaged. Emerging evidence highlights the critical role of mitochondrial dysfunction in HCH pathogenesis, with impaired mitophagy—a selective autophagic process that removes damaged mitochondria—contributing to cardiomyocyte death, oxidative stress, and fibrosis. Protective mitophagy eliminates damaged mitochondria, averting reactive oxygen species (ROS)/calcium overload in HCH. Conversely, its dysregulation—either insufficient clearance or excessive removal—exacerbates mitochondrial dysfunction, driving pathological hypertrophy, fibrosis, and bioenergetic crisis. This dual nature presents a therapeutic paradox demanding contextual modulation. This review comprehensively examines the molecular mechanisms underlying mitophagy dysregulation in HCH, focusing on key pathways such as PINK1/Parkin, BNIP3/NIX, and FUNDC1. We also discuss the interplay between mitophagy and other cellular processes, including mitochondrial biogenesis, inflammasome activation, and metabolic remodeling. Furthermore, we explore potential therapeutic strategies targeting mitophagy to ameliorate HCH, including pharmacological agents, lifestyle interventions, and gene therapy approaches. Understanding the dual role of mitophagy in HCH—both protective and detrimental—may pave the way for novel precision medicine strategies in cardiovascular disease.
Keywords: hypertensive cardiac hypertrophy, mitochondrial dysfunction, mitophagy, oxidative stress, therapeutic targets
1. Introduction
Hypertensive cardiac hypertrophy (HCH) features cardiomyocyte enlargement, interstitial fibrosis, and diastolic dysfunction from chronic pressure overload [1]. Initially, compensatory, sustained stress causes pathological remodeling progressing to heart failure and arrhythmias [2]. Mitochondria maintain cardiac energetics, calcium homeostasis, and redox balance [3], but pressure overload in HCH induces mitochondrial dysfunction: excessive ROS production, calcium overload, and impaired ATP synthesis [4]. These dysfunctions promote oxidative stress, apoptosis, and myocardial damage. Mitochondrial DNA mutations and defective mitophagy further impair repair mechanisms, accelerating disease progression [5]. Understanding these pathways is crucial for developing targeted mitochondrial therapies to mitigate HCH complications.
Mitophagy, the selective autophagy of dysfunctional mitochondria, is essential for cardiomyocyte viability and cardiac function [6]. In HCH, chronic pressure overload induces mitochondrial damage (ROS excess, calcium dysregulation, ATP deficiency). Dysregulated mitophagy—either insufficient (causing toxic accumulation) or excessive (depleting organelles)—exacerbates mitochondrial dysfunction, bioenergetic failure, and cell death [7]. This review examines: (1) molecular mechanisms: Hypertension disrupts mitochondrial dynamics and membrane potential, impairing mitophagic flux via recognition/lysosomal defects; (2) key pathways: dysfunctional PINK1/Parkin (ubiquitin‐dependent), BNIP3/NIX (hypoxia‐responsive), and FUNDC1 (hypoxia‐regulated) pathways; (3) cellular cross‐talk: mitophagy integration with apoptosis, NLRP3 inflammasome activation, and metabolism; (4) therapeutic strategies: pharmacological modulators, mitochondrial protectants, gene therapy, and exercise to restore homeostasis and halt heart failure progression.
2. Molecular Mechanisms in HCH
2.1. Mitochondrial Damage in Hypertensive Stress
Sustained hypertension imposes profound biomechanical/neurohormonal stress, disrupting mitochondrial homeostasis through aberrant dynamics and membrane compromise. Elevated pressure and ROS/calcium dysregulation impair fission–fusion balance via dysregulated DRP1 activation and suppressed MFN1/2/OPA1 function. Concurrently, membrane potential destabilizes due to ROS‐damaged ETC components and mPTP opening [8, 9, 10]. Fragmented, depolarized mitochondria trigger mitophagy: depolarization stabilizes PINK1 on the outer membrane, recruiting Parkin to ubiquitinate proteins for autophagosome engulfment [11].
2.2. Defective Mitophagic Flux in HCH
Mitophagic failure in HCH occurs sequentially: Initial recognition impairment stems from oxidative Parkin inactivation, altered PINK1 kinetics, or damage saturation. Subsequently, lysosomal degradation defects manifest through reduced acidification, diminished cathepsin activity, and impaired autophagosome–lysosome fusion due to Rab GTPase/SNARE protein dysregulation, collectively preventing mitochondrial clearance [12, 13].
2.3. Mitochondrial Fragmentation Triggers Mitophagy in HCH
In HCH, dysregulated mitochondrial dynamics—characterized by excessive DRP1‐mediated fission and impaired MFN1/2/OPA1‐dependent fusion—generate fragmented, depolarized organelles that recruit PINK1/Parkin to activate mitophagy. Mitophagic flux impairment occurs through dual mechanisms: Recognition failure due to Parkin oxidation and damage saturation, coupled with lysosomal dysfunction characterized by diminished acidification and defective autophagosome–lysosome fusion.
2.4. Pathological Consequences of Failed Clearance
Accumulated mitochondria fuel mitochondrial dysfunction via ROS overproduction, calcium mishandling, and ATP deficiency, creating a self‐amplifying damage cycle. This mitophagic stalling accelerates cardiomyocyte death and drives disease progression from hypertension to metabolic dysfunction, bioenergetic failure, myocyte loss, and pathological cardiac remodeling (Figure 1).
FIGURE 1.
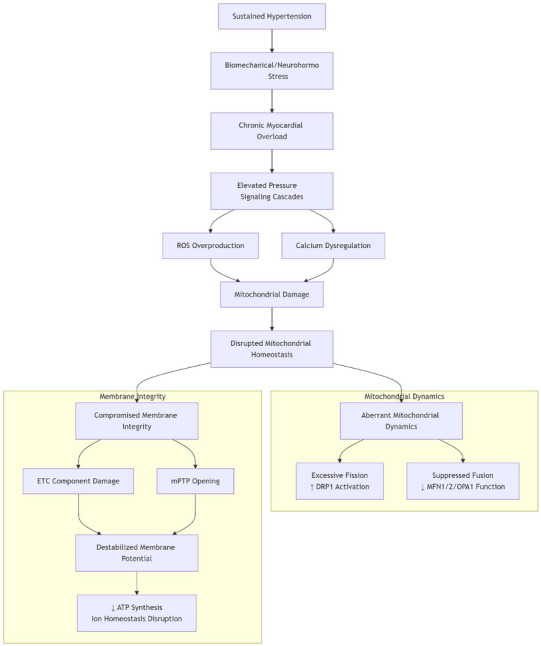
Pathophysiological mechanisms of sustained hypertension on myocardial mitochondria.
3. Mechanisms of Mitophagy in HCH
3.1. PINK1/Parkin‐Dependent Mitophagy
HCH imposes chronic biomechanical and neurohormonal stress on the heart, leading to sustained mitochondrial damage characterized by depolarization, ROS overproduction, and calcium overload [14]. Efficient clearance of these damaged mitochondria via mitophagy is crucial for cardiomyocyte survival and function [15]. The PINK1/Parkin pathway represents the most extensively studied mechanism for targeted mitochondrial removal [16]. However, this critical quality control system is profoundly impaired in HCH, significantly contributing to mitochondrial dysfunction accumulation and disease progression.
Mitochondrial depolarization, a frequent consequence of hypertensive stress, serves as the primary trigger. Depolarization prevents the import and processing of PTEN‐induced putative kinase 1 (PINK1), leading to its stabilization on the outer mitochondrial membrane (OMM) [17]. OMM‐bound PINK1 phosphorylates ubiquitin and recruits the cytosolic E3 ubiquitin ligase Parkin. Activated Parkin then ubiquitinates multiple OMM proteins, including voltage‐dependent anion channels (VDACs, e.g., VDAC1) and Mitofusins (MFN1, MFN2) [18]. This polyubiquitination acts as a recognition signal for autophagy adaptor proteins (e.g., p62/SQSTM1, OPTN, NDP52), which simultaneously bind the ubiquitinated proteins and the lipidated form of LC3 (LC3‐II) embedded in the phagophore membrane [19]. This linkage facilitates engulfment of the damaged mitochondrion by the expanding autophagosome, ultimately leading to lysosomal degradation.
Chronic hypertension impairs PINK1/Parkin mitophagy at the following three critical junctures: (1) Transcriptional/Translational downregulation via angiotensin II/β‐adrenergic signaling reduces PINK1/Parkin expression, limiting mitophagy initiation despite damage [20]. (2) ROS‐mediated Parkin inactivation: H2O2 oxidizes critical cysteines (Cys431/Cys95) in Parkin's RING domains, abolishing E3 ligase activity and ubiquitination signaling [21]. (3) Calcium overload: Elevated cytosolic Ca2⁺ (from RyR2 leak/SERCA2a downregulation) activates calpain, which cleaves OMM‐stabilized PINK1, preventing Parkin recruitment. Severe Ca2⁺ overload also induces sustained mPTP opening, causing catastrophic depolarization [22].
Collectively, these disruptions cause dysfunctional mitochondrial accumulation, triggering a ROS‐Ca2⁺ vicious cycle that promotes bioenergetic failure, cardiomyocyte death, sterile inflammation, and accelerated remodeling toward heart failure. Restoring PINK1/Parkin function represents a therapeutic avenue to interrupt this cascade (Figure 2).
FIGURE 2.
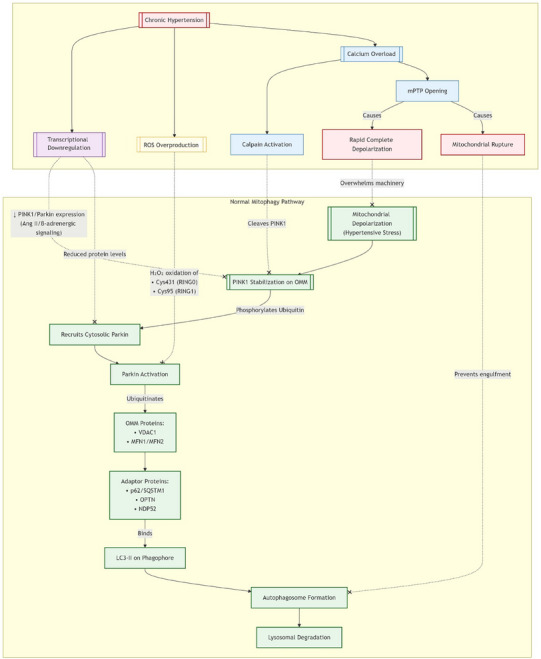
The mitophagy pathway and disruption mechanisms in hypertension.
3.2. Receptor‐Mediated Mitophagy: BNIP3, NIX, and FUNDC1
HCH engages ubiquitin‐independent mitophagy via OMM receptors: (1) Stress‐induced BNIP3/NIX anchors to OMM via C‐terminal domains, exposing N‐terminal LIR motifs to directly bind LC3‐II for mitochondrial engulfment [23]. They also disrupt Beclin‐1/Bcl‐2(Bcl‐XL) complexes, enhancing autophagy initiation. (2) Constitutive FUNDC1 undergoes phosphoregulation: Normoxic phosphorylation (Tyr18/Ser13) inhibits LC3 binding, while hypoxic dephosphorylation activates mitophagy [18]. Chronic hypertension induces maladaptive alterations in these pathways—BNIP3/NIX overexpression shifts from protective mitophagy to apoptosis promotion, while FUNDC1 deficiency impairs hypoxia‐responsive clearance. Combined with PINK1/Parkin defects, this disrupts mitochondrial quality control, exacerbating HCH pathology.
3.2.1. BNIP3/NIX: The Double‐Edged Sword
BNIP3/NIX upregulation by HIF‐1α/NF‐κB/FoxO enhances mitophagy in hypertensive cardiomyopathy. However, sustained overexpression: (1) sensitizes mPTP opening via ROS/Ca2⁺, causing depolarization [24]; (2) directly induces apoptosis via OMM pores/Bcl‐2 interactions [25]; (3) saturates autophagic machinery, impairing engulfment [26]. Paradoxically, while moderate levels aid clearance, supraphysiological expression drives mitochondrial catastrophe and cardiac dysfunction.
3.2.2. FUNDC1 Deficiency and Therapeutic Potential
In hypertensive cardiomyopathy, FUNDC1 reduction due to chronic stress/persistent phosphorylation [27] impairs hypoxia‐responsive mitophagy, accelerating mitochondrial dysfunction [28]. Enhancing FUNDC1 (overexpression, phosphomimetics, kinase inhibition) protects against HCH by improving clearance and contractility [28, 29]. BNIP3/NIX dysfunction converts mitophagy to pro‐apoptotic instability, while FUNDC1 deficiency cripples hypoxia clearance. Combined with PINK1/Parkin defects, this causes catastrophic QC failure. Damaged mitochondria fuel oxidative stress, inflammation, and cardiomyocyte loss, accelerating heart failure. Therapeutically, modulating BNIP3/NIX and restoring FUNDC1 enhance clearance to mitigate disease.
3.3. Crosstalk Between Mitophagy and Other Pathways
The dysregulation of mitophagy in HCH does not occur in isolation. It critically disrupts interconnected cellular pathways, exacerbating cardiac pathology through multifaceted crosstalk.
3.3.1. Imbalance With Mitochondrial Biogenesis (PGC‐1α/NRF1/TFAM)
Mitophagy and mitochondrial biogenesis are normally coupled processes, dynamically regulated by signals like energy demand and oxidative stress, primarily orchestrated by the PGC‐1α/NRF1/TFAM axis [30]. PGC‐1α, the master co‐activator, stimulates NRF1/2, which in turn upregulates TFAM, driving mitochondrial DNA replication and transcription. In HCH, impaired mitophagic flux disrupts this equilibrium. The failure to remove damaged mitochondria generates inhibitory signals (e.g., sustained ROS, accumulated damaged proteins) that suppress PGC‐1α expression and activity. Consequently, despite the presence of damaged organelles, the generation of new, functional mitochondria via biogenesis is blunted. This leads to the net accumulation of defective mitochondria, further compromising cellular bioenergetics and redox balance.
3.3.2. Activation of the NLRP3 Inflammasome
Mitochondria accumulating from failed mitophagy release damage‐associated molecular patterns (DAMPs). Mitochondrial DNA (mtDNA), cardiolipin, and excessive mtROS act as danger signals in the cytosol [31]. These activate the NLRP3 inflammasome in cardiomyocytes and cardiac macrophages. NLRP3 oligomerization triggers caspase‐1, cleaving pro‐inflammatory cytokines IL‐1β and IL‐18. This sterile inflammation promotes cardiomyocyte dysfunction, hypertrophy, and death. Persistent cytokine release drives fibroblast activation, causing excessive extracellular matrix deposition (fibrosis), myocardial stiffening, and diastolic dysfunction—hallmarks of HCH progression.
3.3.3. Maladaptive Metabolic Switching
Healthy adult cardiomyocytes rely predominantly on fatty acid β‐oxidation (FAO) in mitochondria for efficient ATP generation. Mitophagy impairment and the resultant mitochondrial dysfunction severely compromise FAO capacity. Damaged mitochondria exhibit reduced fatty acid import and impaired β‐oxidation enzyme function [32]. The ensuing energy deficit and accumulation of toxic lipid intermediates trigger a compensatory shift towards glycolysis. While glycolysis provides ATP faster, it is significantly less efficient per glucose molecule than oxidative phosphorylation. This shift to glycolytic metabolism worsens cardiac efficiency, contributing to the energetic starvation state observed in HCH. Moreover, sustained glycolytic flux can promote further pathological remodeling through mechanisms involving hexosamine biosynthesis pathway activation and altered signaling [33].
3.4. Detrimental and Protective Roles of Mitophagy in HCH
Persistent mitophagic impairment permits damaged mitochondrial accumulation, releasing ROS and mtDNA DAMPs that activate NF‐κB and TGF‐β signaling. This stimulates cardiomyocyte hypertrophy through ANP/BNP upregulation and promotes fibrosis via fibroblast‐to‐myofibroblast transition with excessive collagen deposition, driving pathological remodeling in HCH. Protective mitophagy eliminates damaged mitochondria, averting ROS/calcium overload in HCH. Conversely, its dysregulation—either insufficient clearance or excessive removal—exacerbates mitochondrial dysfunction, driving pathological hypertrophy, fibrosis, and bioenergetic crisis. This dual nature presents a therapeutic paradox demanding contextual modulation. Targeting the restoration of mitophagic flux in HCH holds promise not only for direct mitochondrial quality control but also for rebalancing biogenesis, quenching inflammasome‐driven inflammation, and promoting a more favorable metabolic substrate utilization, collectively ameliorating cardiac dysfunction and remodeling.
4. Therapeutic Implications of Targeting Mitophagy in HCH
4.1. Lifestyle and Dietary Interventions
Lifestyle and dietary modifications enhance mitophagy and ameliorate HCH, synergistically boosting mitochondrial quality control [34].
4.1.1. Caloric Restriction and Exercise
It activates AMPK/SIRT1 signaling, stimulating mitophagy (via ULK1/PINK1/FOXO) and mitochondrial biogenesis (via PGC‐1α). In HCH, this improves damaged organelle clearance, reduces oxidative stress, and enhances cardiac compliance.
4.1.2. Polyphenols
Resveratrol activates SIRT1/PINK1/Parkin‐mediated mitophagy and biogenesis [35]. Quercetin stabilizes cardiolipin, preserves ΔΨm, and boosts autophagic flux. Both reduce inflammation, inhibit fibrosis, and improve systolic function in preclinical HCH models.
4.2. Pharmacological Approaches
Restoring dysfunctional mitophagy represents a promising HCH therapeutic strategy.
4.2.1. Urolithin A
It enhances PINK1/Parkin activity, promoting damaged mitochondrial clearance. Preclinical studies show improved mitochondrial quality, reduced cardiomyocyte stress, and enhanced cardiac function in hypertension [36].
4.2.2. Metformin
It activates AMPK (stimulating mitophagy via ULK1 phosphorylation) while suppressing mTOR signaling [37]. This dual action reduces pathological hypertrophy and improves myocardial energetics.
4.2.3. SS‐31 (Elamipretide)
This mitochondria‐targeted peptide binds cardiolipin, stabilizing inner membrane integrity [38]. By preventing cardiolipin externalization/peroxidation, it preserves ΔΨm, enhances mitophagic efficiency, reduces oxidative stress, and attenuates ventricular remodeling.
4.3. Gene Therapy and Future Directions
Targeting mitophagy through advanced molecular strategies offers significant therapeutic potential for HCH:
AAV‐mediated gene therapy: Cardiotropic AAV serotypes (e.g., AAV9) enable myocardial delivery. Preclinical studies show that AAV‐mediated PINK1/Parkin overexpression restores mitophagic flux, reduces dysfunctional mitochondrial accumulation, attenuates fibrosis, and improves cardiac function in hypertensive models—directly countering transcriptional downregulation in HCH [39].
CRISPR‐based modulation: CRISPR‐Cas9 technologies allow precise mitophagy modulation through novel approaches [40]: (i) CRISPRa systems (dCas9‐SAM) upregulate endogenous genes (PINK1/Parkin/FUNDC1); (ii) gene editing corrects loss‐of‐function mutations; (iii) epigenetic editing enhances stress‐responsive gene expression via promoter modifications. Proof‐of‐concept studies demonstrate reduced cellular hypertrophy and enhanced mitochondrial turnover despite delivery challenges.
Future work needs optimized delivery systems, long‐term safety assessments, and combinatorial strategies targeting multiple mitophagy pathways.
5. Conclusion
Mitophagy exerts a dual role in HCH, acting as a protective mechanism under physiological conditions but becoming detrimental when dysregulated. Therapeutic targeting of mitophagy pathways presents promising opportunities, though precise modulation is essential to avoid potential adverse effects. Future research should prioritize the following: (1) identifying reliable biomarkers of mitophagy efficiency in HCH, (2) developing tissue‐specific mitophagy enhancers to improve selectivity, and (3) exploring combinatorial approaches, such as pairing mitophagy inducers with antifibrotic agents. This review highlights mitophagy as a pivotal regulatory mechanism in HCH, laying the groundwork for innovative therapeutic strategies in hypertensive heart disease.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgments
I would like to express my sincere gratitude to all the participants who made this study possible. I also extend my thanks to the technical staff for their assistance.
Li S. and Li X., “Mitophagy in Hypertensive Cardiac Hypertrophy: Mechanisms and Therapeutic Implications.” The Journal of Clinical Hypertension 27, no. 8 (2025): 27, e70127. 10.1111/jch.70127
Funding: The authors received no specific funding for this work.
Data Availability Statement
No new data were generated.
References
- 1. Nakashima H., Shinohara K., Matsumoto S., et al., “Establishment of a HFpEF Model Using Female Dahl Salt‐Sensitive Rats: A Valuable Tool for Elucidating the Pathophysiology of HFpEF in Women,” Hypertension Research 48 (2025): 672–680. [DOI] [PubMed] [Google Scholar]
- 2. Sigle M., Rohlfing A. K., Cruz Santos M., et al., “Targeting Cyclophilin A in the Cardiac Microenvironment Preserves Heart Function and Structure in Failing Hearts,” Circulation Research 135 (2024): 758–773. [DOI] [PubMed] [Google Scholar]
- 3. Ljubojević‐Holzer S., Kraler S., Djalinac N., et al., “Loss of Autophagy Protein ATG5 Impairs Cardiac Capacity in Mice and Humans Through Diminishing Mitochondrial Abundance and Disrupting Ca2+ Cycling,” Cardiovascular Research 118 (2022): 1492–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Garbincius J. F., Luongo T. S., Jadiya P., et al., “Enhanced NCLX‐dependent Mitochondrial Ca2+ Efflux Attenuates Pathological Remodeling in Heart Failure,” Journal of Molecular and Cellular Cardiology 167 (2022): 52–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Orekhov A. N., Poznyak A. V., Sobenin I. A., et al., “Mitochondrion as a Selective Target for the Treatment of Atherosclerosis: Role of Mitochondrial DNA Mutations and Defective Mitophagy in the Pathogenesis of Atherosclerosis and Chronic Inflammation,” Current Neuropharmacology 18 (2020): 1064–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rabinovich‐Nikitin I., Rasouli M., Reitz C. J., et al., “Mitochondrial Autophagy and Cell Survival Is Regulated by the Circadian Clock Gene in Cardiac Myocytes During Ischemic Stress,” Autophagy 17 (2021): 3794–3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tong M., Zablocki D., and Sadoshima J., “The Role of Drp1 in Mitophagy and Cell Death in the Heart,” Journal of Molecular and Cellular Cardiology 142 (2020): 138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li Y., Xu Y. F., Chi H. L., et al., “Testis‐Specific Protein, Y‐Encoded‐Like 2 Activates JAK2/STAT3 Pathway in Hypothalamic Paraventricular Nucleus to Sustain Hypertension,” American Journal of Hypertension 37 (2024): 682–691. [DOI] [PubMed] [Google Scholar]
- 9. Basu U., Case A. J., Liu J., et al., “Redox‐Sensitive Calcium/Calmodulin‐Dependent Protein Kinase IIα in Angiotensin II Intra‐Neuronal Signaling and Hypertension,” Redox Biology 27 (2019): 101230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zorov D. B., Juhaszova M., and Sollott S. J., “Mitochondrial ROS‐Induced ROS Release: An Update and Review,” Biochimica Et Biophysica Acta 1757 (2006): 509–517. [DOI] [PubMed] [Google Scholar]
- 11. Liu S., Li C., Fu X., et al., “Regulation on Mitophagy in Adenomyosis by Guizhi Fuling Wan,” Journal of Ethnopharmacology 344 (2025): 119570. [DOI] [PubMed] [Google Scholar]
- 12. Zhou J., Li X. Y., Liu Y. J., et al., “Full‐Coverage Regulations of Autophagy by ROS: From Induction to Maturation,” Autophagy 18 (2022): 1240–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang H., Ye J., Peng Y., et al., “CKLF Induces Microglial Activation via Triggering Defective Mitophagy and Mitochondrial Dysfunction,” Autophagy 20 (2024): 590–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mongelli A., Mengozzi A., Geiger M., et al., “Mitochondrial Epigenetics in Aging and Cardiovascular Diseases,” Frontiers in Cardiovascular Medicine 10 (2023): 1204483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kleele T., Rey T., Winter J., et al., “Distinct Fission Signatures Predict Mitochondrial Degradation or Biogenesis,” Nature 593 (2021): 435–439. [DOI] [PubMed] [Google Scholar]
- 16. Mao Z., Tian L., Liu J., et al., “Ligustilide Ameliorates Hippocampal Neuronal Injury After Cerebral Ischemia Reperfusion Through Activating PINK1/Parkin‐Dependent Mitophagy,” Phytomedicine 101 (2022): 154111. [DOI] [PubMed] [Google Scholar]
- 17. Xian H. and Liou Y. C., “Loss of MIEF1/MiD51 Confers Susceptibility to BAX‐Mediated Cell Death and PINK1‐PRKN‐Dependent Mitophagy,” Autophagy 15 (2019): 2107–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu Y., Zhang H., Liu Y., et al., “Hypoxia‐Induced GPCPD1 Depalmitoylation Triggers Mitophagy via Regulating PRKN‐Mediated Ubiquitination of VDAC1,” Autophagy 19 (2023): 2443–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou J., Chuang Y., Redding‐Ochoa J., et al., “The Autophagy Adaptor TRIAD3A Promotes Tau Fibrillation by Nested Phase Separation,” Nature Cell Biology 26 (2024): 1274–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Belosludtseva N. V., Starinets V. S., Mikheeva I. B., et al., “Effect of Chronic Treatment With Uridine on Cardiac Mitochondrial Dysfunction in the C57BL/6 Mouse Model of High‐Fat Diet‐Streptozotocin‐Induced Diabetes,” International Journal of Molecular Sciences 23 (2022): 10633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Todisco S., Musio B., and Pesce V., “Targeting Mitochondrial Impairment for the Treatment of Cardiovascular Diseases: From Hypertension to Ischemia‐Reperfusion Injury, Searching for New Pharmacological Targets,” Biochemical Pharmacology 208 (2023): 115405. [DOI] [PubMed] [Google Scholar]
- 22. Haustein M., Hannes T., Trieschmann J., et al., “Excitation‐Contraction Coupling in Zebrafish Ventricular Myocardium Is Regulated by Trans‐Sarcolemmal Ca2+ Influx and Sarcoplasmic Reticulum Ca2+ Release,” PLoS One 10 (2015): e0125654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wei J., Xie J., He J., et al., “Active Fraction of Polyrhachis vicina (Roger) Alleviated Cerebral Ischemia/Reperfusion Injury by Targeting SIRT3‐Mediated Mitophagy and Angiogenesis,” Phytomedicine 121 (2023): 155104. [DOI] [PubMed] [Google Scholar]
- 24. Olgar Y., Billur D., Tuncay E., et al., “MitoTEMPO Provides an Antiarrhythmic Effect in Aged‐Rats Through Attenuation of Mitochondrial Reactive Oxygen Species,” Experimental Gerontology 136 (2020): 110961. [DOI] [PubMed] [Google Scholar]
- 25. Singh R., Letai A., and Sarosiek K., “Regulation of Apoptosis in Health and Disease: The Balancing Act of BCL‐2 Family Proteins,” Nature Reviews Molecular Cell Biology 20 (2019): 175–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yuan Z., Cai K., Li J., et al., “ATG14 Targets Lipid Droplets and Acts as an Autophagic Receptor for Syntaxin18‐Regulated Lipid Droplet Turnover,” Nature Communications 15 (2024): 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liang Q., Wan J., Liu H., et al., “A Plant Nonenveloped Double‐Stranded RNA Virus Activates and Co‐Opts BNIP3‐Mediated Mitophagy to Promote Persistent Infection in Its Insect Vector,” Autophagy 19 (2023): 616–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhuang Z., Zhu Y., Tao J., et al., “UCF101 Rescues Against Diabetes‐Evoked Cardiac Remodeling and Contractile Anomalies Through AMP‐Activated Protein Kinase‐Mediated Induction of Mitophagy,” Pharmacology 16 (2024): 1–14. [DOI] [PubMed] [Google Scholar]
- 29. Fu T., Ma Y., Li Y., et al., “Mitophagy as a Mitochondrial Quality Control Mechanism in Myocardial Ischemic Stress: From Bench to Bedside,” Cell Stress & Chaperones 28 (2023): 239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhao Y., Jia Q., Hao G., et al., “JiangyaTongluo Decoction Ameliorates Tubulointerstitial Fibrosis via Regulating the SIRT1/PGC‐1α/Mitophagy Axis in Hypertensive Nephropathy,” Frontiers in Pharmacology 15 (2024): 1491315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rashmi K. C., Harsha Raj M., Paul M., et al., “A New Pyrrole Based Small Molecule From Tinospora cordifolia Induces Apoptosis in MDA‐MB‐231 Breast Cancer Cells via ROS Mediated Mitochondrial Damage and Restoration of p53 Activity,” Chemico‐Biological Interactions 299 (2019): 120–130. [DOI] [PubMed] [Google Scholar]
- 32. Cardoso A. C., Lam N. T., Savla J. J., et al., “Mitochondrial Substrate Utilization Regulates Cardiomyocyte Cell Cycle Progression,” Nature Metabolism 2 (2020): 167–178. [PMC free article] [PubMed] [Google Scholar]
- 33. Johswich A., Longuet C., Pawling J., et al., “N‐Glycan Remodeling on Glucagon Receptor Is an Effector of Nutrient Sensing by the Hexosamine Biosynthesis Pathway,” Journal of Biological Chemistry 289 (2014): 15927–15941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu L., Li Y., Chen G., et al., “Crosstalk Between Mitochondrial Biogenesis and Mitophagy to Maintain Mitochondrial Homeostasis,” Journal of Biomedical Science 30 (2023): 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu L., Chen Q., Dong B., et al., “Resveratrol Alleviates Lipopolysaccharide‐Induced Liver Injury by Inducing SIRT1/P62‐Mediated Mitophagy in Gibel Carp (Carassius gibelio),” Frontiers in Immunology 14 (2023): 1177140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang Y., Jasper H., Toan S., et al., “Mitophagy Coordinates the Mitochondrial Unfolded Protein Response to Attenuate Inflammation‐Mediated Myocardial Injury,” Redox Biology 45 (2021): 102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang F., Zhou W., Huang L., et al., “Artesunate Alleviates Cerebral Ischemia/Reperfusion Injury by Suppressing FUNDC1‐Mediated Excessive Mitophagy,” Brain Research Bulletin 228 (2025): 111407. [DOI] [PubMed] [Google Scholar]
- 38. Gumpper‐Fedus K., Park K. H., Ma H., et al., “MG53 Preserves Mitochondrial Integrity of Cardiomyocytes During Ischemia Reperfusion‐Induced Oxidative Stress,” Redox Biology 54 (2022): 102357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang X. J., Qi L., Cheng Y. F., et al., “PINK1 Overexpression Prevents Forskolin‐Induced Tau Hyperphosphorylation and Oxidative Stress in a Rat Model of Alzheimer's Disease,” Acta Pharmacologica Sinica 43 (2022): 1916–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hoshino A., Wang W. J., Wada S., et al., “The ADP/ATP Translocase Drives Mitophagy Independent of Nucleotide Exchange,” Nature 575 (2019): 375–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data were generated.