

Abstract
The biological extinction that occurred at the Permian–Triassic boundary represents the most extensive loss of species of any known event of the past 550 million years. There have been a wide variety of explanations offered for this extinction. In the present paper, a number of the more popular recent hypotheses are evaluated in terms of predictions that they make, or that they imply, concerning the global carbon cycle. For this purpose, a mass balance model is used that calculates atmospheric CO2 and oceanic δ13C as a function of time. Hypotheses considered include: (i) the release of massive amounts of CO2 from the ocean to the atmosphere resulting in mass poisoning; (ii) the release of large amounts of CO2 from volcanic degassing; (iii) the release of methane stored in methane hydrates; (iv) the decomposition and oxidation of dead organisms to CO2 after sudden mass mortality; and (v) the long-term reorganization of the global carbon cycle. The modeling indicates that measured short-term changes in δ13C at the boundary are best explained by methane release with mass mortality and volcanic degassing contributing in secondary roles. None of the processes result in excessively high levels of atmospheric CO2 if they occurred on time scales of more than about 1,000 years. The idea of poisoning by high levels of atmospheric CO2 depends on the absence of subthermocline calcium carbonate deposition during the latest Permian. The most far-reaching effect was found to be reorganization of the carbon cycle with major sedimentary burial of organic matter shifting from the land to the sea, resulting in less burial overall, decreased atmospheric O2, and higher atmospheric CO2 for the entire Triassic Period.
The biological extinction that occurred at the Permian–Triassic (P/Tr) boundary represents the most extensive loss of species of any known event of the past 550 million years (1). Both marine and terrestrial fauna and flora were affected. Many hypothetical processes have been offered to explain this event, including changes in sea level, climate change, large scale volcanism, overturn of the ocean with the release of toxic gases, massive methane release from methane hydrates, a bolide impact, plus many others of an historical interest (see refs. 1 and 2 for a review). There is some confusion over whether certain geochemical signatures at the P/Tr boundary represent causes or effects of the extinction. For example, a sudden cataclysm caused by the impact of an asteroid or comet (3, 4) or passage of the earth through a supernova (5), could have resulted in mass mortality, which in turn would have brought about changes in the geochemical properties, such as carbon isotopic composition, of the earth-air-ocean system. In this case, some geochemical changes would represent effects, not causes of the extinction. This situation agrees with the findings of Twitchett et al. (6) of excursion in carbon isotopic composition after the extinction of both plants and animals.
It is not the purpose of this paper to evaluate all of the many hypotheses, but rather to examine some of the more recent ones and to see what consequences involving the global carbon cycle would result. In this way, both causes and effects will be considered. It has been well established that there was a large and rapid drop at this time in oceanic δ13C, as recorded in carbonates and organic matter (refs. 6–10 and many other studies). All of the theories discussed in the present paper address this sharp drop in δ13C. A plot taken from Bowring et al. (11) is shown in Fig. 1. By constructing a global model for both total carbon and δ13C, in an attempt to match the large drops in δ13C, some quantitative limits on the hypotheses can be placed. In this paper the following hypotheses will be examined.
Figure 1.

Measurements of δ13C of carbonates vs. stratigraphic position at the Meishan, China, Permo–Triassic boundary locality. Horizontal scale is δ13C in ‰; large numbers are ages in million years. After Bowring et al. (11).
(i) The idea (12) that severe ocean stratification over an extended period resulted in the buildup of very high concentrations of isotopically light dissolved CO2 in the subthermocline ocean as a result of the transfer downward of the remains of surface-dwelling organisms followed by their oxidation to CO2 (“the biological pump”). When the ocean overturned because of climate change, this large mass of CO2 was degassed into the atmosphere, resulting in the poisoning and mass mortality of a wide variety of organisms.
(ii) The idea that a burst of very isotopically light CH4 from the breakdown of methane hydrates in sediments resulted in greenhouse warming or methane poisoning (9, 10).
(iii) The idea that volcanogenic CO2 released with the massive eruptions of Siberian basalts could have introduced enough isotopically light CO2 to both lower the oceanic δ13C appreciably (13) and raise CO2 to induce greenhouse warming and/or produce sulfate aerosols to induce global cooling and toxicity (13, 14).
(iv) The idea that sudden mass mortality of terrestrial and marine organisms led to a “strangelove” (dead) ocean and a landscape littered with rotting vegetation that led to a global fungal spike. Oxidation of the biogenic remains contributed to a rapid drop in oceanic δ13C (2) and rise in atmospheric CO2.
(v) The idea that sudden mass mortality of the terrestrial and marine organisms resulted in a long term irreversible reorganization of the global carbon cycle (15). Total global organic carbon burial decreased, and the locus of major deposition shifted from the terrestrial to the marine environments with a consequent drop in atmospheric oxygen (16). The cause of the mass mortality is left open, but it should have occurred rapidly.
Carbon Model Description
The global carbon cycle calculations of the present paper are based on the blag model (17) as simplified by Beerling and Berner (18). Separate mass balance expressions for total carbon, calcium (plus magnesium), and 13C are included in the modeling. A diagrammatic presentation is shown in Fig. 2. The model calculates the variation with time of all of the fluxes shown in Fig. 2 along with the concentrations of CO2 in the atmosphere, dissolved inorganic carbon and calcium in the oceans, and the δ13C value of the oceans. Starting conditions for fluxes and masses were those for the late Permian derived from geocarb modeling (19) amended to include data on δ13C for the latest Permian (ref. 20; E. Grossman, personal communication). Before each introduced perturbation, a steady-state cycle was assumed. Also, oceanic concentrations of dissolved Ca and inorganic carbon similar to the present ocean were assumed at the start of each run. Because of the “short” times involved, several factors normally included in models of the long-term carbon cycle, such as the geocarb model, when applied over millions of years, are not considered. These include changes in continental size, position, and topography, and changes in the hydrological cycle and solar radiation. By contrast, such geocarb subjects as changes in the rates of global sedimentary organic matter burial, rates of weathering accompanying changes in CO2 greenhouse-controlled temperature, and rates of volcanic CO2 degassing, are considered in the present paper.
Figure 2.

Diagram for the carbon cycle box model used in the present paper. Fv = flux of volcanic CO2; Fm = flux of methane from methane hydrates (the methane is assumed to be oxidized to CO2 essentially instantaneously); Fwc = uptake of CO2 by means of the weathering of carbonates (twice this value is the flux of carbon to the oceans from carbonate weathering); Fwsi = uptake of atmospheric CO2 by means of the weathering of Ca–Mg silicates with transfer of the carbon to the oceans; Fbg = burial flux of organic carbon in sediments; Fwg = weathering flux of ancient sedimentary organic carbon (kerogen); Fbio = flux of CO2 caised by the mass mortality of terrestrial biota; Fbc = burial flux of marine carbonates ( = flux of CO2 from ocean to the atmosphere). Modified from Beerling and Berner (18).
Results
Ocean Overturn.
Knoll et al. (12) showed that if the oceans accumulated as much dissolved CO2 as they assumed, then a sudden upwelling or turnover could release toxic levels of CO2 to the atmosphere. However, there is a serious problem with this scenario. Their assumed value of atmospheric PCO2 = 10,000 μatm (1 atm = 101.3 kPa), which before turnover is equivalent to a dissolved CO2 concentration of 400 μM in the subthermocline ocean, would lead to highly acidic conditions, and any CaCO3 existing on the seafloor would dissolve. To test the hypothesis of Knoll et al., a simple calculation, whereby 400 μM dissolved CO2 reacts with CaCO3 to equilibrium, was done in an ocean with original contents of dissolved Ca2+ and HCO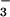 as at present. The effective equilibrium constant for the reaction was the value obtained from the masses of CO2, and dissolved Ca2+ and HCO
as at present. The effective equilibrium constant for the reaction was the value obtained from the masses of CO2, and dissolved Ca2+ and HCO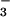 present in the modern ocean (17, 21). Exchange with the atmosphere was ignored. The final equilibrium value of dissolved CO2 is equivalent to a PCO2 of only 1,400 μatm, not 10,000 μatm. This result is not materially affected by accounting for the extra alkalinity added during bacterial sulfate reduction. Inclusion of the addition of an extra 1,000 μM HCO
present in the modern ocean (17, 21). Exchange with the atmosphere was ignored. The final equilibrium value of dissolved CO2 is equivalent to a PCO2 of only 1,400 μatm, not 10,000 μatm. This result is not materially affected by accounting for the extra alkalinity added during bacterial sulfate reduction. Inclusion of the addition of an extra 1,000 μM HCO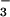 from sulfate reduction, as assumed by Knoll et al., results, after equilibration with CaCO3, in a PCO2 of 1,700 μatm. (This is a maximum value because sulfate reduction is assumed by Knoll et al. to produce only H2S and not HS−, which is the dominant sulfide species in present seawater.) Thus, inclusion of sulfate reduction makes little difference in the result.
from sulfate reduction, as assumed by Knoll et al., results, after equilibration with CaCO3, in a PCO2 of 1,700 μatm. (This is a maximum value because sulfate reduction is assumed by Knoll et al. to produce only H2S and not HS−, which is the dominant sulfide species in present seawater.) Thus, inclusion of sulfate reduction makes little difference in the result.
The question then arises whether an atmospheric PCO2 of about 1,500 μatm would be sufficient to bring about mass poisoning of marine organisms. It is unlikely, judging that this and higher levels of atmospheric CO2 appeared to have been maintained during long periods of the Mesozoic (22) without major adverse effects on shallow-water fauna. The only way that the Knoll et al. hypothesis (12) could work is if the ocean floor up to a few hundred meters depth was devoid of calcium carbonate sediment and that negligible CaCO3 was sedimented past the thermocline over the long period during which the high level of CO2 was built up. To neutralize 400 μM dissolved CO2 would require the dissolution of about 5 m of CaCO3 depositing, or already deposited on, the seafloor during the period of CO2 increase; this is equivalent to a deposition rate of only 0.25 cm per thousand years (kyr) for two million years, and includes not just the “deep sea” but all seafloor within about 500 m of the surface, where carbonate sedimentation by such processes as turbidity currents could have been much higher. Was the subthermocline seafloor devoid of carbonates at this time? The great decrease during the late Permian in shelf area available for shallow water CaCO3 deposition (23) suggests that at this time there must have been appreciable CaCO3 deposition onto the deep sea floor simply to balance the global calcium and carbon budgets.
Methane Release.
The carbon cycle model was run with the perturbation being a hypothetical release of methane with δ13C = −65‰ sufficient to rapidly drop the oceanic δ13C value by 7–8‰ (Fig. 3A). Oxidation of CH4 to CO2, either in the oceans (by bacteria) or in the atmosphere (by OH radicals), was assumed to be instantaneous. The CH4 release was assumed to occur over 20,000 years, with a peak at 10,000 years after the start of the experiment. This assumption agrees roughly with the suggested time span of the extinction (24). This sudden release is capable of reproducing the maximum measured short term changes in δ13C of the oceans ranging up to 8‰ at the P/Tr boundary as reported by Baud et al. (8) and Bowring et al. (11). However, the total integrated mass of released CH4 needed is about 4,200 Gt C (1 Gt C = 1015 g of carbon), which is several times higher than that postulated to explain the δ13C drop for the Paleocene thermal maximum (25). Suggestions of much higher P/Tr boundary drops in δ13C up to 10‰ (6, 10) require about twice as much methane. Note that after the initial drop, the value of δ13C rises to a steady state as the light carbon is mixed with heavier carbon derived from carbonate weathering. Also, allowing the methane to be given off directly to the oceans, rather than to the atmosphere, makes no major difference in the results (Fig. 3A). This alternative formulation was done simply by replacing the flux between CH4 and the atmosphere in Fig. 2 by a flux from CH4 directly to the oceans.
Figure 3.

Plots of oceanic δ13C and atmospheric CO2 vs. time as a result of the input of methane hydrate-derived CH4 to the atmosphere or oceans. It is assumed that the methane is oxidized essentially instantaneously to CO2 in either case. (A) δ13C. (B) CO2.
Quick release of such large quantities of methane, after oxidation to CO2 (mean residence time of CH4 in the atmosphere is only 10 years), results in only a relatively small rise in atmospheric CO2 (Fig. 3B). This rise is not nearly sufficient to induce mass mortality by means of greenhouse warming. The effect of CH4 release, thus, is to drag down δ13C, but not to add excessive CO2 to the atmosphere. The large required mass of methane hydrate needed to produce the measured δ13C excursions could have been supplied by the extensive burial of methane hydrate during the latest Permian, as evidenced by a sustained high value of δ13C in the oceans at that time. As will be shown below, derivation of light carbon from any other terrestrial source, such as organic matter or mantle CO2, does not even approach these changes without invoking unreasonably high total masses of biomass or volcanic rocks. Thus, unless the light carbon is extraterrestrially derived, it appears that CH4 degassing may have played a prominent role in the changes accompanying the Permo–Triassic extinction. If the degassing was so rapid that it swamped the oxidizing capacity of the ocean and atmosphere, then a possible drop in stratospheric ozone, caused by reaction with excessive CH4, could have accounted for excessive UV-B radiation and the drastic changes in the terrestrial flora that occurred across the P/Tr boundary (26).
Volcanic CO2 Release from Siberian Volcanism.
According to Renne et al. (13) between 2 and 3 million cubic kilometers of volcanic material was ejected over a period of about 1 million years near the P/Tr boundary in the Siberian region. When the estimates of 3.5 × 1012 and 1.6 × 1013 g of CO2 per km3 for the release of CO2 from modern basalts (27, 28) are used, this amounts to a total CO2 degassing between 2,000 and 13,000 Gt C. To test how this degassing would affect the δ13C of seawater and the CO2 level of the atmosphere, a model calculation was done with the excess volcanic CO2 serving as the sole perturbation of the otherwise steady-state carbon cycle. The results are shown in Fig. 4A for a release of 11,000 Gt C as CO2 with δ13C = −6‰. As can be seen it is impossible to match the measured drops in δ13C as found by many investigators (e.g., Fig. 1). Also, the rise in atmospheric CO2 is only moderate going from 300 ppm to about 700 ppm at the peak value (Fig. 4B). In order for CO2 to rise to higher values and δ13C to drop more, the degassing from volcanism must have occurred over much shorter time scales. The results of letting degassing occur over just 30,000 years is also shown in Fig. A and B. As can be seen, the drop in δ13C is still insufficient and the rise in CO2 is only moderately higher. (To get CO2 to rise above 1,000 ppm it is necessary to have the entire degassing occur within 3,000 years, which is highly unlikely). Volcanic degassing by itself is clearly insufficient to explain the P/Tr changes in δ13C documented in Fig. 1.
Figure 4.

Plots of oceanic δ13C and atmospheric CO2 vs. time as a result of the input of volcanically derived CO2. The terms fast and slow refer to inputs lasting approximately 30,000 and 200,000 years, respectively. (A) δ13C. (B) CO2.
Sudden Mass Mortality.
If there had been mass mortality of terrestrial vegetation as the result of an impact, solar flare, etc., then there would be a rapid input of isotopically light CO2 to the atmosphere as the result of rotting of the vegetation and the oxidation of soil carbon that was not being replenished by litterfall. Evidence for mass mortality at the P/Tr boundary is the presence of a fungal pollen “spike” (29–31), representing the massive global decomposition of woody material. The total carbon in present day terrestrial vegetation plus soils is approximately 2,000 Gt C. If we assume the same value for the latest Permian terrestrial biota, then one can examine the consequences of adding this much carbon as CO2 to the global carbon cycle. To represent sudden mass mortality of land plants the carbon cycle model was run with an input of 2,000 GtC as CO2 to the atmosphere, with a δ13C value of −25 ‰, over a very short period of less than 1,000 years. Results for atmospheric CO2 and δ13C for the oceans as a function of time are shown in Fig. 5. As can be seen, even oxidizing all terrestrial biota and soil carbon (an unrealistic scenario) over a very short time is insufficient to drop the δ13C value to the values recorded by the measurements of many investigators. Also, the rise in atmospheric CO2 is only about a doubling, which would not lead to a massive lethal greenhouse effect. If the CO2 release from biogenic decay is extended over a longer period, the drop in δ13C is the same but the rise in CO2 is less, showing this calculation to be for a maximum effect.
Figure 5.

Plots of δ13C and atmospheric CO2 vs. time for the sudden mass mortality of terrestrial vegetation with all vegetation plus soil carbon converted to CO2. Note the much shorter time scale compared with Figs. 3 and 4.
Mass mortality would also have affected marine plankton. The killing of marine plankton would stop the “biological pump,” whereby photosynthetically fixed and isotopically light carbon is transferred to the deep ocean by settling of dead plankton and its oxidation back to CO2 in deep water. This situation has been referred to as a “strangelove” ocean. The drop in δ13C of dissolved inorganic carbon in the surface ocean as a result of oceanic overturn following cessation of the biological pump has been shown by D'Hondt et al. (32) for the Cretaceous–Tertiary mass extinction to be about 1–1.5‰. Also, cessation of the present-day biological pump has been shown by Sarmiento and Orr (33) to result, after exchange with the atmosphere, in a rise in atmospheric CO2 from 280 ppm to about 500 ppm. If these results are added to those of Fig. 6 for the mass mortality of terrestrial vegetation, we are still left with an insufficient drop in δ13C to match the larger changes shown in Fig. 1 and a rise in CO2 insufficient to cause a major lethal greenhouse effect. Thus, the mass mortality hypothesis as a sole cause of the drop in δ13C appears to be untenable.
Figure 6.

Plots of oceanic δ13C and atmospheric CO2 vs. time as a result of a sudden drop in global organic C burial rate from 60 Gt C/kyr to 24 Gt C/kyr with a constantly maintained organic C weathering rate of 60 Gt C/kyr.
Reorganization of the Carbon Cycle.
As emphasized by Broecker and Peacock (15) and Berner and Raiswell (34), the large drop in the global rate of burial of organic matter occurring between the Permian and Triassic periods was likely caused by a drop in terrestrial production resulting in a shift of the major focus of burial from the terrestrial-plus-marine environments to the marine environment alone. The drop in global organic burial is further documented by the large drop in coal deposition between the Permian and Triassic (35, 36) and to a shift in major production from (high biomass) arborescent to (low biomass) herbaceous flora (29, 31). Furthermore, Broecker and Peacock (15) suggest that this shift occurred suddenly at the P/Tr boundary as a result of a sudden mass mortality of terrestrial organisms combined with burial of marine organic matter under more highly anoxic bottom waters because of a decrease in the efficiency of the marine food web. In support of the latter, an increase in anoxic black shales at the boundary has been documented by several authors (e.g., refs. 37–39).
A drop in the total global burial rate of organic matter in sediments between the Permian and Triassic periods has been documented by sedimentary rock analyses (35) and by carbon and sulfur isotope mass balance modeling (15, 16). If the high oceanic δ13C (+5‰) values found for the early and mid-Permian are maintained right up to near the P/Tr boundary (ref. 20; E. Grossman, personal communication), then modeling calculations (40) using rapid recycling and the 13C fractionation reported by Hayes et al. (41) result in a drop in the burial rate from 60 Gt C/kyr in the late Permian to 24 Gt C/kyr in the early Triassic. During this time span, the rate of CO2 generation from the weathering of rock-bound kerogen on the continents is assumed to be constant at 60 Gt C/kyr (though it may have actually increased because of the exposure of coal-rich sediments from a drop in sea level; see ref. 42). To test the Broecker and Peacock hypothesis, we assume that the drop in burial rate, and consequent imbalance between organic burial and organic weathering, occurred essentially instantaneously at the P/Tr boundary and continued for a long time. Results are shown in Fig. 6. This situation results in a slow but persistent rise in atmospheric CO2 level and drop in δ13C until the carbon system returns to a new steady state. The reorganization hypothesis, thus, predicts the correct overall change in δ13C between the Permian and Triassic but fails to produce a rapid change. This result is due to the buffering capacity of the ocean plus the uptake of CO2 by continental weathering. However, if combined with the sudden drop in δ13C caused by mass mortality, as discussed above, the Broecker and Peacock idea predicts δ13C values closer to those observed (Fig. 1).
If there were a prolonged period whereby small land cover, such as bryophytes, replaced forests on the land surface, then the rate of consumption of atmospheric CO2 by the weathering of silicates could have been reduced by as much as a factor of 4 (19). With this assumption I examined the consequences of suddenly lowering the initial value for Ca–Mg silicate weathering (Fwsi; see Fig. 2) by a factor of 4 at the P/Tr boundary and letting it stay at this low value for 100,000 years, at which time reforestation of the land surface resulted in a rapid rise to the initial Permian Fwsi value. Results for this situation combined with the simultaneous imbalance in the burial and weathering of organic matter made no major difference in results from those for organic matter imbalance alone. The level of atmospheric CO2 attained a peak about 200 ppm higher than in the organic imbalance alone scenario, but with little difference in δ13C.
The effect of the reorganization of carbon burial can be seen in a broader context in terms of calculations of the burial rates of organic carbon and pyrite sulfur over the Phanerozoic. Results using the C and S isotope mass balance model are cited above (40), but results with the new late Permian high oceanic δ13C values of Grossman are shown in Fig. 7. The sudden drop in organic burial and rise in pyrite burial across the P/Tr boundary is apparent on the time scale of the entire Phanerozoic. This finding demonstrates the importance of the Permo–Triassic extinction as having a major effect on not only the global carbon cycle but also that of sulfur.
Figure 7.

Plots of the burial rate of sedimentary organic carbon (Fbg) and pyrite sulfur (Fbp) vs. time for the Phanerozoic computed by the means of the model of Berner (2001), amended to include new δ13C data for the late Permian (Mii et al., ref. 20; E. Grossman, personal communication)
The ratio of organic C burial to pyrite S burial over Phanerozoic time is shown in Fig. 8, along with calculated levels of atmospheric oxygen, which was done also by C–S isotope modeling (40). The P/Tr extinction is accompanied by a large drop in O2 and an increase in S/C ratio. The latter is just what would be expected for a shift from mainly terrestrial burial in low-S fresh water to mainly marine burial in high-S seawater. Accompanying the drop in O2, geocarb modeling (19) shows a concomitant rise in CO2 from the late Permian to the early Triassic (spanning about 20 million years) of 1,000–1,500 ppm.
Figure 8.

Plots of the ratio of the burial rates of pyrite S and organic C, and of atmospheric oxygen concentration vs time for the Phanerozoic based on the amended geocarb iii model (19) (see caption for Fig. 6 for details).
On a much shorter time scale (the Phanerozoic modeling of O2 and geocarb has a time resolution of only 10 million years), a rise in sedimentary pyrite burial across the P/Tr boundary is also shown by a rapid rise in oceanic δ34S (3, 15, 42). This is additional evidence of a rapid shift from burial of organic matter in low-S fresh continental waters to burial in sulfate-rich seawater, which provides sufficient sulfur for abundant pyrite formation (34). The finding of earliest Triassic pyrite-rich shales (37, 39) is in accord with the rapid rise in 34S at the boundary (3). Also, the S/C values found for the early Triassic are characteristic of an ocean with excessive pyrite burial as compared with that at present.
Combination of Processes.
It has been suggested that the P/Tr extinction was brought about by a synergistic combination of causes (1). With this idea in mind I have calculated a best fit to the observed δ13C data for at the P/Tr boundary by combining the hypotheses above. Here the carbon cycle reorganization is combined with sudden mass mortality of land plants and marine biota, Siberian volcanism, and rapid methane release from clathrates. Rates and durations of input and integrated amounts of CH4, volcanic CO2 (slow release), and biogenic CO2 are the same as those cited above. Results are shown in Fig. 9 and there is good overall agreement with observations, both on the short time scale and on the longer period time scale. The atmospheric CO2 values are the highest of all of the simulations as might be expected for an addition of all inputs. Of course, multiple δ13C pulses, such as those shown in Fig. 1, are not reproduced, but this could have been done by introducing a number of inputs at different times.
Figure 9.

Plots of δ13C and CO2 vs time for the combined inputs of carbon to the atmosphere from mass terrestrial mortality, CH4 hydrate decomposition, and volcanic CO2 degassing combined with an imbalance in the rates of burial and weathering of sedimentary organic matter.
Could this combined result be in accord with a reasonable scenario connecting all of the events? One suggestion is as follows: the impact of a bolide (or atmospheric disturbance) could have caused sudden mass mortality of marine and terrestrial organisms. The accompanying pressure wave could have triggered both the release of CH4 from stored hydrates and the initiation of Siberian volcanism. After the initial events of the first 10,000–30,000 years, the reduction of land plant productivity lasted for millions of years, leading to a drop in global organic carbon burial and a shift of the major site of burial from the terrestrial to the marine environments. This hypothesis helps to explain the very low level of Triassic coal deposition as compared with that during the Permian. Initial increased organic burial in the marine environment, caused by the sudden input of dead organisms, led to a higher frequency of euxinic basins and a greater burial of sedimentary pytite in the early Triassic. This is only one of several conceivable scenarios, but it shows that the Permian–Triassic extinction could have been brought about by a variety of causes.
Conclusion
Some of the theories for the mass biological extinction at the P/Tr boundary do not stand up to critical analysis with regards to their predictions concerning the global carbon cycle. The buildup of extremely high levels of dissolved CO2 in the subsurface ocean, followed by oceanic overturn, degassing, and mass CO2 poisoning, is unlikely if there were CaCO3 sediments present below the thermocline at that time. Neither volcanic degassing nor mass mortality of terrestrial organisms alone could have provided enough isotopically light carbon to bring about the drops in δ13C recorded at the P/Tr boundary. Release of large amounts of methane from stored methane hydrates to the carbon cycle best explains the δ13C measurements, but supplies little CO2 to the atmosphere. It is possible that a combination of mass mortality from an impact or radiation blast was combined with methane release and volcanic CO2 release to help account, at least partly, for the extinction. The long-lasting effect of this extinction was that the major locus of organic carbon deposition shifted from the continents to the oceans, which led to an overall lower global burial rate of organic matter, lower atmospheric O2, and higher atmospheric CO2 throughout the Triassic Period.
Acknowledgments
I thank D. L. Royer, D. J. Beerling, and L. J. Hickey for helpful comments on the manuscript. I also acknowledge fruitful discussions concerning the biological pump and the strangelove ocean with K. Caldeira and help in manuscript preparation by I. Miller and R. Wildman. Musical inspiration was provided by live performances of the piano concertos of S. Rachmaninoff. This research was supported by Department of Energy Grant DE-FG02-01ER15173 and National Science Foundation Grant EAR 0104797.
Abbreviations
- P/Tr
Permian–Triassic
- kyr
thousand years
References
- 1.Erwin D H. The Great Paleozoic Crisis. New York: Columbia Univ. Press; 1993. [Google Scholar]
- 2.Erwin D H. Nature (London) 1994;367:231–238. [Google Scholar]
- 3.Kaiho K, Kajiwara Y, Nakanao T, Miura Y, Kawahata H, Tazaki K, Ueshima M, Chen Z, Shi G R. Geology. 2001;29:815–818. [Google Scholar]
- 4.Becker L, Poreda R J, Hunt A G, Bunch T E, Rampino M. Science. 2001;291:1530–1533. doi: 10.1126/science.1057243. [DOI] [PubMed] [Google Scholar]
- 5.Detre C H, Toth I, Don G, Kiss A Z, Uzanyi I, Bodo P, Schleder Z. Meteorites Planet Sci. 1999;34:A125. [Google Scholar]
- 6.Twitchett R J, Looy C V, Morante R, Visscher H, Wignall P B. Geology. 2001;29:351–354. [Google Scholar]
- 7.Holser W T, Schonlaub H P, Attrep M, Boeckelmann K, Klein P, Magaritz M, Orth C J, Fenninger A, Jenny C, Kralik M, et al. Nature (London) 1989;337:39–44. [Google Scholar]
- 8.Baud A, Magaritz M, Holser W T. Geol Rundsch. 1989;78:649. [Google Scholar]
- 9.McLeod K G, Smith R M H, Koch P L, Ward P D. Geology. 2000;28:227–230. [Google Scholar]
- 10.Krull E S, Retallack G J. Geol Soc Am Bull. 2000;112:1459–1472. [Google Scholar]
- 11.Bowring S A, Erwin D H, Jin Y G, Martin M W, Davidek K, Wang W. Science. 1998;280:1039–1045. doi: 10.1126/science.280.5366.1039. [DOI] [PubMed] [Google Scholar]
- 12.Knoll A H, Bambach R K, Canfield D E, Grotzinger J P. Science. 1996;273:452–457. [PubMed] [Google Scholar]
- 13.Renne P R, Zhang Z C, Richards M A, Black M T, Basu A R. Science. 1995;269:1413–1416. doi: 10.1126/science.269.5229.1413. [DOI] [PubMed] [Google Scholar]
- 14.Campbell I H, Czamanske G K, Fedorenko V A, Hill R I, Stepanov V. Science. 1992;258:1760–1763. doi: 10.1126/science.258.5089.1760. [DOI] [PubMed] [Google Scholar]
- 15.Broecker W S, Peacock S. Global Biogeochem Cycles. 1999;13:1167–1172. [Google Scholar]
- 16.Berner R A. Am J Sci. 1987;287:177–196. [Google Scholar]
- 17.Berner R A, Lasaga A C, Garrels R M. Am J Sci. 1983;283:641–683. [Google Scholar]
- 18. Beerling, D. J. & Berner, R. A. (2002) Global Biogeochem. Cycles, in press.
- 19.Berner R A, Kothavala Z. Am J Sci. 2001;301:182–204. [Google Scholar]
- 20.Mii H-S, Grossman E L, Yancey T E. Geology. 1997;25:227–230. [Google Scholar]
- 21. Schrag, D. P., Berner, R. A., Hoffman, P. F. & Halvorsen, G. P. (2002) Geochem. Geophys. Geosys., in press.
- 22.Ekart D, Cerling T E, Montanez I P, Tabor N J. Am J Sci. 1999;299:805–827. [Google Scholar]
- 23.Walker L J, Wilkenson B H, Ivany L C. J Geology. 2002;110:75–87. [Google Scholar]
- 24.Rampino M R, Prokoph A, Adler A. Geology. 2000;28:643–646. [Google Scholar]
- 25.Dickens G R, O'Neil J R, Rea D K, Owen R M. Paleoceanography. 1995;10:965–971. [Google Scholar]
- 26.Visscher H, Brinkhuis H, Kuerschner W, Looy C, Van Konijnenburg-Van Cittert J H A. Eos Trans AGU. 2001;82:47–48. [Google Scholar]
- 27.Leavitt S W. Environ Geol. 1982;4:15–21. [Google Scholar]
- 28.Gerlach T M, Graeber E J. Nature (London) 1985;313:273–277. [Google Scholar]
- 29.Eshet Y, Rampino M R, Visscher H. Geology. 1995;23:967–970. [Google Scholar]
- 30.Visscher H, Brinkhuis H, Dilcher D L, Elsik W C, Eshet Y, Looy C V, Rampino M R, Traverse A. Proc Natl Acad Sci USA. 1996;93:2155–2158. doi: 10.1073/pnas.93.5.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Looy C V, Twitchett R J, Dilcher D L, Van Konijnenburg-Van Cittert J H A, Visscher H. Proc Natl Acad Sci USA. 2001;98:7879–7883. doi: 10.1073/pnas.131218098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.D'Hondt S, Donaghay P, Zachos J C, Luttenberg D, Lindinger M. Science. 1998;282:276–279. doi: 10.1126/science.282.5387.276. [DOI] [PubMed] [Google Scholar]
- 33.Sarmiento J L, Orr J C. Limnol Oceanogr. 1991;36:1928–1950. [Google Scholar]
- 34.Berner R A, Raiswell R. Geochim Cosmochim Acta. 1983;47:855–862. [Google Scholar]
- 35.Berner R A, Canfield D E. Am J Sci. 1989;289:333–381. doi: 10.2475/ajs.289.4.333. [DOI] [PubMed] [Google Scholar]
- 36.Retallack G J, Veevers J J, Morante R. Geol Soc Am Bull. 1996;108:195–207. [Google Scholar]
- 37.Wignall P B, Hallam A. Paleogeog Paleoclim Paleoecol. 1992;93:21–46. [Google Scholar]
- 38.Wignall P B, Twitchett R J. Science. 1996;272:155–1158. doi: 10.1126/science.272.5265.1155. [DOI] [PubMed] [Google Scholar]
- 39.Isozaki Y. Science. 1997;276:235–238. doi: 10.1126/science.276.5310.235. [DOI] [PubMed] [Google Scholar]
- 40.Berner R A. Geochim Cosmochim Acta. 2001;65:685–694. [Google Scholar]
- 41.Hayes J M, Strauss H, Kaufman A J. Chem Geol. 1999;161:103–125. [Google Scholar]
- 42.Holser W T, Magaritz M. Marine Geol. 1987;11:155–180. [Google Scholar]