

Abstract
Purpose
Malaria, caused by the parasite Plasmodium falciparum, remains a critical health challenge. Its enzyme Dihydrofolate Reductase (PfDHFR) is vital for the parasite’s survival and a key target for antimalarial drugs. Mutations in PfDHFR are primary cause of drug resistance in malaria parasites, particularly to antifolate drugs, like pyrimethamine. Herein, a comprehensive structure-based virtual screening (SBVS) benchmarking analysis of three generic docking tools against both wild-type (WT) and quadruple-mutant (Q) PfDHFR variants were investigated. Furthermore, re-scoring of the docking outcome via two popular pretrained machine learning scoring functions (ML SFs) were explored. The study provides valuable recommendations into enhancing the SBVS performance against both the WT and the resistant Q PfDHFR variants.
Methods
Three generic docking tools (AutoDock Vina, PLANTS, and FRED) were evaluated using the DEKOIS 2.0 benchmark set against both WT and Q PfDHFR variants. Furthermore, we analyzed the re-scoring performance of two pretrained ML SFs, namely CNN-Score and RF-Score-VS v2. In depth analysis of the screening performance and enrichment behavior using pROC-AUC, pROC-Chemotype plots and EF 1% were deliberated.
Results
Overall, eighteen docking and re-scoring outcomes for both variants were conducted. For the WT PfDHFR, PLANTS demonstrated the best enrichment when combined with CNN re-scoring reflecting an EF 1% value of 28. Re-scoring with RF and CNN significantly improved AutoDock Vina’s screening performance from worse-than-random to better-than-random. For the Q variant, FRED exhibited the best enrichment when combined with the CNN re-scoring scheme, exhibiting the maximum value of EF 1% (ie, EF 1% = 31). pROC-Chemotype plots analysis revealed that these re-scoring combinations effectively retrieved diverse and high-affinity actives at early enrichment.
Conclusion
The findings demonstrate that re-scoring with CNN-Score consistently augments the SBVS performance and enriches diverse and high-affinity binders for both PfDHFR variants, offering important endorsements for improving malaria drug discovery, especially against the highly resistant Q variant.
Keywords: DEKOIS 2.0, MLSFs, docking, malaria
Graphical Abstract

Introduction
The number of cases of malaria remains dramatic and estimates for the year 2023 indicated 263 million cases which is a significant rise compared to previous years. Similarly, the higher cases reported during this period can be explained by the COVID-19 outbreak and other factors such as the changes in environmental features.1 Plasmodium falciparum is a type of protozoan parasite that causes malaria in humans and it is the most dangerous species among the genus Plasmodium.2 It causes a vast majority of severe and most life-threatening malaria cases.2
The enzyme Dihydrofolate Reductase-Thymidylate Synthase in Plasmodium falciparum (PfDHFR-TS) is very crucial in the lifecycle of the parasite.2 This bifunctional enzyme plays a critical role in the parasite’s reproduction as part of the folate pathway as it is involved in an integral pathway for making purines and pyrimidines, which are the building blocks of RNA and DNA. It catalyzes the reduction of dihydrofolate to tetrahydrofolate using nicotinamide adenine dinucleotide phosphate (NADPH) as its cofactor, while TS (thymidylate synthase) converts deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP) using tetrahydrofolate.3 This makes it a very attractive target in the development of antimalarial drugs. For instance, proguanil and pyrimethamine are among the drugs used in the treatment and prevention of malaria as PfDHFR inhibitors.4 Mutations of the PfDHFR binding site, especially the quadruple-mutant (Q) PfDHFR variant (N51I/C59R/S108N/I164L) represent a common resistance mechanism which can alter the efficacy of the drug-DHFR interactions by making therapies impossible.5 In sequence, this represents a major problem in the control of malaria, reaffirming the need for alternative approaches to the development of therapies and drugs that act at different stages of the life cycle of the pathogen.4,6
In the context of SBVS, benchmarking serves as an assessment method to evaluate the performance of docking tools.7–9 Benchmark datasets typically include bioactive molecules and structurally similar inactive molecules, referred to as “decoys”, for a specific protein target.7–9 The effectiveness of a docking tool in SBVS is determined by its ability to prioritize known bioactive molecules over decoys.7–9 Consequently, benchmarking is a crucial approach for identifying optimal virtual screening pipelines that enhance predictive accuracy.10–13
Recent studies have reported the application of the DEKOIS 2.0 benchmark set to rigorously evaluate and elucidate the SBVS performance across a wide array of clinically relevant targets that extend beyond the original sets of 81 protein targets encompassed by the standard DEKOIS 2.0 collection.7,10–13 Notably, an in-depth benchmarking analysis was conducted against the fascin protein, a target of interest in cancer therapy.11 Additionally, a cross-benchmarking study utilized the SARS-CoV papain-like protease (PLpro) benchmark set against SARS-CoV-2 PLpro, dedicated to COVID-19 research.13 Similarly, cross-benchmarking investigations evaluated the SARS-CoV-2 RNA-dependent RNA polymerase (RdRp) palm subdomain using the DEKOIS 2.0 benchmark set originally generated for the hepatitis C virus (HCV) NS5B palm subdomain, owing to the high sequence homology between these two proteins.12 Furthermore, in response to emerging COVID-19 resistant variants, comprehensive benchmarking analyses have been carried out for both the wild-type and Omicron SARS-CoV-2 main protease (Mpro), utilizing newly compiled DEKOIS 2.0 benchmark sets specific to each protein.10 However, in the context of benchmarking, the dual focus on both wild-type (WT) and drug-resistant Q variants of PfDHFR remain underrepresented in the literature, and enhanced efforts are essential to combat malaria resistance.
Recently, with the emergence of machine learning scoring functions (ML SFs),14 numerous studies have shown that these methods significantly outperform traditional scoring functions in predicting binding affinities for protein-ligand complexes based on their X-ray crystal structures. Consequently, ML SFs are now considered the leading approach for this docking application.14,15 For instance, an ML SF designed for virtual screening, known as “RF-Score-VS”,16 which is based on a random forest algorithm, has achieved an average hit rate that is more than three times higher than that of the classical scoring function DOCK3.717 at the top 1% of ranked molecules. Similar improvements have been noted with convolutional neural network-based scoring functions, eg, CNN-Score18 which showed hit rates three times greater than those of traditional scoring functions like Smina/Vina19 at the top 1%.
While classical docking tools and ML SFs have shown promise in drug discovery, their combined use for complex and clinically relevant targets, like PfDHFR, is still a less explored but a very promising area in literature. Traditional docking methods often struggle to accurately predict binding strengths for different chemical structures or in case of binding site mutations due to resistance. Using ML re-scoring can help refining the initial docking poses and better distinguish between active compounds and decoys.20
Our current benchmarking study conducted this combined strategy to both WT and Q PfDHFR (as depicted in Figure 1). By evaluating this integrated workflow, we aim to create a solid framework for speeding up the discovery of new antimalarial agents that can work better against resistant PfDHFR strains. To achieve this, a diverse and potent dataset of non-covalent, non-peptidomimetic inhibitors was compiled from literature and BindingDB to create a high-quality DEKOIS 2.0 benchmark set for both variants. The study evaluated the performance of three docking tools—AutoDock Vina,21 FRED,22 and PLANTS23—using protein structures of both PfDHFR variants to identify the most suitable tool for virtual screening against these targets. Additionally, the ligand poses generated by these docking tools were re-scored using two ML SFs, RF-Score-VS v2 and CNN-Score, resulting in eighteen combined docking and scoring outcomes for both variants. The screening performance analysis and the chemotype enrichment behavior provided valuable insights to enhance the success rate of virtual screening in malaria drug discovery.
Figure 1.

The logical flow of the study.
Methods
Preparation of the Data Sets
Preparation of Protein Structures
Crystal structures (PDB ID. 6A2M) and (PDB ID. 6KP2) for PfDHFR for WT and Q structures, respectively, were downloaded from the Protein Data Bank (PDB). Protein preparation was carried out using “Make Receptor” (version 4.3.2.0) GUI of OpenEye at default settings due to its broader applicability in VS campaigns. The water molecules, unnecessary ions, redundant chains, crystallization molecules (if any) were removed. Hydrogen atoms were added and optimized. After preparation, the protein structures were saved as OEDU file and converted to PDB file format for the docking steps afterwards.
Preparation of Small Molecules of DEKOIS 2.0 Benchmark Set
The DEKOIS 2.0 protocol was employed on the collected and curated 40 bioactive molecules for each WT and Q PfDHFR to create 1200 challenging decoys (1:30 ratio) for both variants.7 After that, preparation of all molecules was performed using Omega24 to generate multiple conformations for each ligand for FRED25 docking, while a single conformer per ligand was retained for subsequent docking in both PLANTS and AutoDock Vina. The prepared compounds were saved as SDF files which were transformed and split into PDBQT files using OpenBabel26 for AutoDock Vina27 docking experiments. For PLANTS28 docking experiments, the SDF files were converted into mol2 files, and the types of correct atoms were performed using SPORES29 software.
Benchmarking
Docking Experiments
For AutoDock Vina (1.5.7 version)27 docking, the protein files were converted to PDBQT files via prepare_receptor4.py script from the MGLTools package (version 1.5.7).30 The dimensions of the docking grid box for WT PfDHFR (PDB ID: 6A2M) were 21.33Å × 25.00Å × 19.00Å, and for the Q PfDHFR (PDB ID: 6KP2) were 21.00Å × 21.33Å × 19.00Å with a 1 Å grid spacing to ensure that all docked compound geometries were covered. The docking method’s search efficiency was retained at its default setting.
Regarding docking via PLANTS (1.2 version),28 the SDF files were converted into mol2 format and the correct atom types were set via SPORES software. “ChemPLP”, was the employed scoring function with selecting “screen” mode. Within 5 Å of the co-crystal ligand coordinates, the binding site was identified.28
For FRED docking22 (OEDocking v4.1.1.0) was used at default settings. MakeReceptor GUI of OpenEye was utilized to describe the binding site as a search box in the vicinity of the co-crystal ligand with dimensions for WT PfDHFR (PDB ID: 6A2M) 21.33Å × 25.00Å × 19.00Å, and Q PfDHFR (PDB ID: 6KP2) were 21.00Å × 21.33Å × 19.00Å with a 1 Å grid spacing. These nearly identical dimensions of grid boxes for both variants are due to high level of conservation in the binding site’s shape and size for both WT and Q PfDHFR proteins.
pROC and pROC-Chemotype Calculations
The score-based docking rank was employed in the calculation of pROC-AUC (semi-logarithmic receiver operating characteristic area under the curve) utilizing the KNIME “R-Snippet” component according to the following equation:31,32
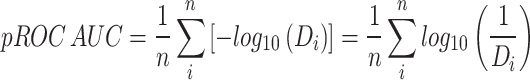 |
Di is the decoy fraction that is ordered higher than the bioactive that was detected, and n is the bioactives number where i corresponds to the rank’s bioactive number.
The “pROC-Chemotype” plot is an automated tool that integrates ligand chemotype and scaffold characteristics with potency classifications, allowing for their simultaneous visualization within pROC profiles generated from docking or re-scoring workflows.31 Generation of the “pROC-Chemotype” plot can be accomplished using a specific KNIME node as reported and provided in its original study.31 The docking scores are represented as fitness values (fitness = docking score multiplied by −1) to illustrate the bioactive distributions.
The enrichment factor (EF) was calculated using the following formula to evaluate the docking tool’s capacity to identify true positives from the active set in the score-ordered list as opposed to the random collection:31,32
 |
Re-Scoring ML SFs
The docking outcome from AutoDock Vina, FRED and PLANTS were re-scored by the pretrained ML SFs of CNN-Score and RF-Score-VS v2.33 The CNN-Score (v1.0.1) is a deep learning framework for molecular docking.18 It consists of an ensemble of five CNN models, each featuring a deep learning architecture with 7 to 20 hidden layers, which strikes a balance between pose prediction quality, VS performance, and execution time.18,33 These models were trained using data from two primary sources: true active compounds and property-matched decoys derived from the DUD-E dataset,34 as well as experimental data obtained from the PDBbind database.18,33 It uses the gnina function of GNINA v1.0.35 The output of the “CNNScore” by the SF for each docked pose is used for re-scoring.35
RF-Score-VS v2 is a pretrained scoring function built using the Random Forest algorithm.15 It is trained on the complete DUD-E dataset, which includes over 15,000 active molecules and approximately 900,000 property-matched decoys across 102 targets, all successfully docked to their respective DUD-E targets.15,16
The docked poses were simultaneously re-scored using RF-Score-VS and converted into SDF files, and relevant fields were extracted for pROC-AUC and EF 1% calculations.
Nevertheless, both CNN-Score and RF-Score-VS were trained on DUD-E datasets, which, despite its extensive use, contains decoys designed to be physically dissimilar from actives but may not fully represent the chemical space of true non-binders.34 This can lead to optimistic performance estimates when evaluated on similar benchmark sets. Furthermore, both models have been independently validated on external diverse datasets beyond DUD-E to assess their generalization capability. Nonetheless, performance can be influenced by such biases. Within the context of our study, these pretrained models were employed as established scoring functions to provide comparative insights rather than as definitive predictors. Therefore, we acknowledge these limitations to ensure balanced interpretation and address generalizability questions of our study.
Results
Selection of PfDHFR Bioactive Molecules
As an initial step, all available PfDHFR inhibitors for the WT, and the Q variant were collected from the literature and BindingDB, then the bioactive molecules were manually curated. Many compounds were reported to be in a nanomolar range of activity. Therefore, we decided to include them all, and only exclude the ones with no determined affinity/activity or the ones having Ki values >260 nM. We also excluded all stereo-active molecules that exist as R/S. This ended up collecting 93 bioactive molecules for the WT and 56 actives for Q variant. Then we clustered the actives using DataWarrior software36 where the structure similarity falls below 0.6 based on Tanimoto similarity index. Consequently, 40 actives per WT, and 40 actives per Q variant were selected to represent the chemotype diversity. The compiled actives represent 12 chemical scaffolds in the WT (Table S1, in the Supplementary Material - SM) and 8 chemical scaffolds in the Q variant (PDB ID: 6KP2) (Table S2 in SM). This diversity would reflect the available chemical space for malarial PfDHFR inhibition and provide unbiased insights into the screening performance of the docking tools. The Ki values range from 0.011 nM to 1.17 nM for the WT, and from 0.04 nM to 254 nM for the Q variant. For decoys generation, the bioactive molecules were subjected to the DEKOIS 2.0 protocol8 which creates 30 structurally related decoys per compound. Eventually, we compiled a challenging decoy set of 1200 compounds for each WT and Q variant. The whole set of bioactives and decoys was used to assess the performance of the three docking tools investigated and the re-scoring employing two ML SFs.
Selection of Representative PDB Structure(s) for PfDHFR
The PfDHFR-TS polypeptide is usually in a dimeric assembly containing 608 amino acids, with the 231 first amino acids being the residues of the PfDHFR domain. In addition, it consists of 89 residues known as the junction region, while the remaining 288 residues form the TS domain 3 (Figure 2). The active site of the PfDHFR domain is lined with the amino acids Asp54, Ile164, Ile14, Phe116, and Phe58. These residues are often involved in the binding of small molecules.5
Figure 2.

Structural illustration of PfDHFR-TS. (A) WT PfDHFR-TS dimeric assembly (PDB ID: 6A2M). The TS domain is shown in yellow, the junction region in green, and the DHFR domain in grey. The DHFR domain is complexed with nicotinamide adenine dinucleotide phosphate (NADPH), colored in blue, and the co-crystal binder in red. (B) Superposition of WT PfDHFR (PDB ID: 6A2M) in grey and Q PfDHFR (PDB ID: 6KP2) in magenta. The key resistance-associated mutations—C59R, N51I, S108N, and I164L—are highlighted in sphere representation to emphasize their spatial positions within the protein structures.
In the study, we downloaded all PfDHFR structures with resolution better than 2Å from the Protein Data Bank (PDB). We focused especially on the PfDHFR structures co-crystallized with ligands in their binding site to account for the structural changes occurring during the ligand-protein binding.37 We did not observe major differences in the binding site conformations between the PfDHFR structures. Consequently, based on our analysis, we selected (PDB ID: 6A2M) as a WT PfDHFR structure,38 and (PDB ID: 6KP2) as a Q variant (N51I/C59R/S108N/I164L).5
Benchmarking
Generic Docking Tools
One of the aims of this study is to evaluate the performance of diverse docking via benchmarking, eg, AutoDock Vina,21 PLANTS,23 and FRED.22 These docking tools represent different architectures in the development of their optimization/search algorithms and scoring functions. For instance, AutoDock Vina (1.5.7 version)21 is based on the Broyden–Fletcher–Goldfarb–Shanno (BFGS) method for local optimization and uses its own Vina scoring function, while PLANTS (1.2 version)23 employs the Protein-Ligand ANT System algorithm and PLANTSCHEMPLP scoring function. While FRED (v4.1.1.0)22 Developed by OpenEye Scientific Software, uses chemgauss4 which is the default scoring function, it is a Gaussian-based function that evaluates the fit of the ligand in the binding pocket. Afterward, we investigate whether re-scoring with ML SFs, such as, RF-Score-VS and CNN-Score, will outperform the classical scoring functions or not.
The benchmarking results against WT PfDHFR (PDB ID: 6A2M), and Q PfDHFR variant (PDB ID: 6KP2) revealed that PLANTS and FRED showed better-than-random performance in both structures using the pROC-AUC metric,32 ie, pROC-AUC value >0.43, compared to AutoDock Vina. Higher pROC-AUC reflects higher actives recognition at the early enrichment compared to the decoys. Interestingly, FRED showed the best screening performance against both structures. The WT structure yielded pROC-AUC values of 0.85 for FRED compared to 0.33 (worse-than-random) for AutoDock Vina, and 0.75 for PLANTS, as shown in Figure 3. The poor performance of AutoDock Vina in this case reflects the target-specific nature of its virtual screening performance. This fact is augmented by various benchmarking studies where AutoDock Vina exhibited pROC-AUC values >0.43 with DEKOIS 2.0 benchmark data sets against other protein targets.7,10–13 In addition, the Q structure yielded pROC-AUC values of 1.65 for FRED compared to 0.60 and 1.07 for AutoDock Vina and PLANTS, respectively (Figure 3).
Figure 3.

pROC plots illustrating the screening performance of AutoDock Vina, FRED and PLANTS. The left panel is for the WT PfDHFR (PDB ID: 6A2M). The right panel is for Q PfDHFR (PDB ID: 6KP2).
To provide an in-depth analysis, we examined the chemotype/scaffold enrichment with the “pROC-Chemotype”31 Plot for the best-performing tool (FRED docking) against both WT and Q PfDHFR mutant variants.
The pROC-Chemotype plot for WT displayed that FRED could not retrieve any active molecule at the early enrichment (eg, library cutoff 1%), as shown in (Figure 4). In fact, the first active molecule recognized is at docking rank of 16 onwards, reflecting library cutoff of 1.3%. Therefore, re-scoring approaches are highly recommended to improve the screening performance (details in the re-scoring section). As seen in Figure 4, the bioactivity data are symbolized by the level of activity (LOA) extending from 10−6 to 10−11 for the active molecules for WT PfDHFR, reported as Ki as a type of data (TOD). The Box plot chart (Figure 4) demonstrates the docking fitness distribution of the bioactive molecules. A scaffold represented by cluster 2 molecules lies in a superior region of fitness (ie, fitness >13), while the docking scores of actives vary from −13.34 (best score) to – 8.72 (worst score) and are presented as fitness values of 13.34 to 8.72.
Figure 4.

pROC-Chemotype plot for the benchmarking using FRED against WT PfDHFR. (A) The pROC-Chemotype plot where the docking information matches the chemotype described by the cluster number and the bioactivity information. (B) Box plot of the fitness vs chemotype clusters.
For the Q mutant PfDHFR, the pROC-Chemotype plot exhibited that FRED is able recognize high-affinity binders at early enrichment, as seen in (Figure 5). The bioactivity data of the collected actives in the plot are symbolized by the level of activity (LOA) extending from 10−7 to 10−11 and reported as Ki as a type of data (TOD). Like in Figure 4, the Box plot chart in (Figure 5) demonstrates the docking fitness distribution. A scaffold represented by cluster 1 molecules lies in a superior region of fitness (ie, fitness >13) among fitness values of 13.07 to 8.13 of the bioactive molecules. Interestingly, an EF 1% = 10 indicates a promising predictive power of FRED since it can recognize active molecules 10 times more than the random performance at early enrichment (eg, library cutoff 1%). The first two ranked active molecules with docking ranks 3 and 8 (Figure 5) have bioactivity ranks 35 and 16 reflecting Ki values of 5.40 nM and 1.16 nM, respectively. This highlights the ability of FRED to enrich high-affinity binders at the early enrichment.
Figure 5.

pROC-Chemotype plot for the benchmarking using FRED against Q PfDHFR. (A) The pROC-Chemotype plot where the docking information matches the chemotype described by the cluster number and the bioactivity information. (B) Box plot of the fitness vs chemotype clusters.
Re-Scoring via ML SFs
The Bar chart in Figure 6A shows the re-scoring impact of RF-Score-VS and CNN-Score as pretrained ML SFs on the three docking tools. Generally, the screening performance is improved in all cases when employing CNN re-scoring for all docking tools for both WT and Q variants. Likewise, the screening performance is enhanced when recruiting RF re-scoring for all cases, except for a slight deterioration in performance for the FRED docking on the Q variant.
Figure 6.

The screening performance and early enrichment behavior of the three docking tools and the re-scoring outcome via the ML SFs of CNN-Score and RF-Score-VS (A) The screening performance is depicted as pROC-AUC, while in (B) the early enrichment behavior is depicted as EF 1%.
Regarding the re-scoring performance for the WT PfDHFR, the best performance is observed for the combination of PLANTS docking with CNN and RF re-scoring, with pROC-AUC values of 1.85 and 1.38, respectively. Both re-scoring schemes were able to enhance the performance of all generic docking tools (FRED, PLANTS, and AutoDock Vina), as shown in Figure 6A. Interestingly, the RF and CNN re-scoring were able to dramatically improve the screening performance of AutoDock Vina from worse-than-random (pROC-AUC <0.43) into better-than-random performance, with pROC-AUC values of 0.98 and 1.19 for CNN and RF, respectively.
For the Q mutant PfDHFR, it is remarkable that the CNN re-scoring scheme produced the best performance when combined with any of the three generic docking tools as shown in (Figure 6A). Its performance reaches pROC-AUC values of 1.82, 1.86, and 1.78 when combined with FRED, PLANTS, and AutoDock Vina, respectively.
Collectively, for the WT PfDHFR structure, PLANTS with CNN re-scoring shows the highest pROC-AUC, indicating a strong performance. Likewise, for the Q mutant structure, all the three tools profited from the CNN re-scoring intensely, with PLANTS and FRED showing nearly identical high scores, slightly outperforming AutoDock Vina. Re-scoring with CNN benefits the screening performance across FRED and PLANTS docking tools in all protein types while re-scoring with RF improved the docking with AutoDock Vina, especially in for the WT structure. This recommends the re-scoring procedure with ML SFs post-docking re-scoring for enhancing the screening performance in VS against PfDHFR. This improvement can be attributed to the target-specific performance of docking and post-docking re-scoring in VS. Evaluating the screening enrichment at a 1% library cutoff (EF1%) for the WT PfDHFR indicates that the docking with PLANTS combined with a re-scoring with CNN is the best scoring scheme, as demonstrated in (Figure 6B). This aligns with the overall screening performance highlighted with pROC-AUC, in Figure 6A. The EF1% value of PLANTS combined with CNN re-scoring is 28.41 which is superior to FRED and AutoDock Vina combined with RF or CNN, as shown in (Figure 6B).
For the enrichment performance of the Q PfDHFR, as observed from the overall screening performance pROC-AUC values, CNN re-scoring enhanced the enrichment when combined with any of the three docking tools, as shown in Figure 6B. It is worth mentioning that the best EF1% value is observed for the docking with FRED combined with a re-scoring with CNN (EF1% = 31.02). This enrichment slightly outperforms the EF1% values of PLANTS and AutoDock Vina combined with CNN, as seen in Figure 6B. Nonetheless, the re-scoring with RF fails to enrich actives for FRED and PLANTS at this enrichment threshold, while a good enrichment can be observed with AutoDock Vina (Figure 6B).
Visualizing the best re-scoring combinations the CNN re-scoring for WT PfDHFR via pROC-Chemotype plots (Figure 7A), revealed that CNN re-scoring was able to enrich the most potent active compound (Bioactivity rank = 1, Ki = 0.04 nM) as the best-ranked docking pose, highlighting its superior predictive capability in ranking highly bioactive compounds, ultimately in early enrichment. This result suggests the model’s efficiency in correlating docking scores with actual biological activity, ensuring reliable prioritization of high affinity candidates. Similarly, the pROC-Chemotype plot for CNN re-scoring of FRED for Q PfDHFR exhibited potent and diverse clusters of active molecules as shown in Figure 7B, with no decoys detected at all at 1% library cutoff, reflected with a maximum value of EF 1% (ie EF 1% = 31). Again, this suggests an outstanding performance of CNN re-scoring when prioritizing actives over the challenging decoys at the early enrichment.
Figure 7.

pROC-Chemotype plots for CNN-Score re-scoring performance. (A) Re-scoring of PLANTS docking against WT PfDHFR. (B) Re-scoring of FRED docking against Q PfDHFR.
Re-scoring with CNN-Score consistently outperforms RF-Score-VS, which can be attributed to the model being trained using convolutional neural networks that effectively capture complex spatial and physicochemical interactions within protein-ligand complexes.18 Besides, it automatically learns the key features of protein–ligand interactions that correlate with binding.18 This enables CNN-Score to generate richer and more hierarchical representations compared to the more traditional ensemble-based methods employed by RF.16,18 However, it is important to note that the superior performance of CNN-Score in re-scoring can also be influenced by target-specific factors. Therefore, broader conclusions regarding its generalizability require further validation across a large and diverse set of targets.
Conclusion
The parasite’s PfDHFR enzyme—a critical folate pathway component for DNA synthesis—remains a key antimalarial target despite widespread resistance to drugs like pyrimethamine caused by PfDHFR mutations. Mutations, especially in the Q PfDHFR variant, have caused widespread resistance to key antimalarial drugs like pyrimethamine. This makes these treatments ineffective in many areas. The growing drug resistance presents a serious threat to global efforts to control and eliminate malaria. We urgently need to focus on finding new strategies for developing new antimalarial agents that can bypass these resistance mechanisms. As part of these strategies, this study assessed virtual screening approaches for identifying PfDHFR inhibitors against both WT and drug-resistant Q variants. Three docking tools (AutoDock Vina, FRED and PLANTS) were evaluated using the DEKOIS 2.0 benchmark set, and re-scored utilizing two ML SFs via pretrained models of CNN-Score and RF-Score-VS
The results highlight that PLANTS combined with CNN re-scoring achieved the highest early enrichment (EF1% = 28) for the WT PfDHFR. Moreover, the performance of AutoDock Vina was improved from sub-random to better-than-random performance after both RF-Score-VS and CNN-Score re-scoring. It is important to emphasize that enhanced performance of ML re-scoring may be affected by target-specific variables. Consequently, broader conclusions about its generalizability warrant additional validation using a large and diverse range of targets. For the Q PfDHFR, the combination of FRED docking with CNN re-scoring showed superior enrichment with a maximum EF 1% value (ie, EF1% = 31), outperforming other combinations. Furthermore, examining the chemotype behavior, pROC-Chemotype plots confirmed that re-scoring methods enhanced early retrieval of diverse and high-affinity binders for both variants. Importantly, this study offers a practical guideline for virtual screening campaigns aimed at targeting a resistant Q variant of PfDHFR, recommending the use of FRED docking as a preliminary step followed by re-scoring with CNN-Score.
Moreover, these results demonstrate that ML-based re-scoring can significantly boost virtual screening efficacy for antimalarial drug discovery, particularly against resistant strains. Importantly, this study offers a practical guideline for virtual screening campaigns aimed at targeting a resistant Q variant of PfDHFR, recommending the use of FRED docking as a preliminary step followed by re-scoring with CNN-Score. The approach addresses challenges posed by PfDHFR mutations that undermine traditional antifolate therapies, offering a pathway to identify next-generation inhibitors less prone to resistance.
The WT and Q PfDHFR DEKOIS 2.0 sets will be made available on www.dekois.com.
Acknowledgments
TMI acknowledges the Alexander von Humboldt Foundation for partially supporting this project via the Return Fellowship Program. The authors gratefully acknowledge the support of OpenEye Scientific Software Inc. for offering a free academic license.
Abbreviations
SBVS, structure-based virtual screening; PfDHFR, Plasmodium falciparum Dihydrofolate Reductase; NADPH, nicotinamide adenine dinucleotide phosphate; dUMP, Deoxyuridine monophosphate; dTMP, deoxythymidine monophosphate; pROC-AUC, semi-logarithmic receiver operating characteristic area under the curve; EF, enrichment factor; FRED, Fast Rigid Exhaustive Docking; PLANTS, Protein-Ligand ANT System; OEDU, OpenEye Digital Unit; PDB, Protein Data Bank.
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Venkatesan P. WHO world malaria report 2024. Lancet Microbe. 2025;6(4):101073. doi: 10.1016/j.lanmic.2025.101073 [DOI] [PubMed] [Google Scholar]
- 2.Sato S. Correction to: plasmodium-a brief introduction to the parasites causing human malaria and their basic biology. J Physiol Anthropol. 2021;40(1):3. doi: 10.1186/s40101-021-00254-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raimondi M, Randazzo O, la Franca M, et al. DHFR inhibitors: reading the past for discovering novel anticancer agents. Molecules. 2019;24:1140. doi: 10.3390/molecules24061140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yuvaniyama J, Chitnumsub P, Kamchonwongpaisan S, et al. Insights into antifolate resistance from malarial DHFR-TS structures. Nat Struct Biol. 2003;10(5):357–365. doi: 10.1038/nsb921 [DOI] [PubMed] [Google Scholar]
- 5.Saepua S, Sadorn K, Vanichtanankul J, et al. 6-Hydrophobic aromatic substituent pyrimethamine analogues as potential antimalarials for pyrimethamine-resistant Plasmodium falciparum. Bioorg Med Chem. 2019;27(24):115158. doi: 10.1016/j.bmc.2019.115158 [DOI] [PubMed] [Google Scholar]
- 6.Vanichtanankul J, Taweechai S, Uttamapinant C, et al. Combined spatial limitation around residues 16 and 108 of Plasmodium falciparum dihydrofolate reductase explains resistance to cycloguanil. Antimicrob Agents Chemother. 2012;56(7):3928–3935. doi: 10.1128/aac.00301-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bauer M, Ibrahim T, Vogel S, Boeckler F. Evaluation and optimization of virtual screening workflows with DEKOIS 2.0-A public library of challenging docking benchmark sets. J Chem Inf Model. 2013;53(6):1447–1462. doi: 10.1021/ci400115b [DOI] [PubMed] [Google Scholar]
- 8.Ibrahim TM, Bauer MR, Boeckler FM. Applying DEKOIS 2.0 in structure-based virtual screening to probe the impact of preparation procedures and score normalization. J Cheminform. 2015;7(1):21. doi: 10.1186/s13321-015-0074-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vogel SM, Bauer MR, Boeckler FM. DEKOIS: demanding evaluation kits for objective in silico screening — a versatile tool for benchmarking docking programs and scoring functions. J Chem Inf Model. 2011;51(10):2650–2665. doi: 10.1021/ci2001549 [DOI] [PubMed] [Google Scholar]
- 10.Galal N, Beshay B, Soliman O, et al. Evaluating the structure-based virtual screening performance of SARS-CoV-2 main protease: a benchmarking approach and a multistage screening example against the wild-type and Omicron variants. PLoS One. 2025;20(2):e0318712. doi: 10.1371/journal.pone.0318712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hassan HHA, Ismail MI, Abourehab MAS, Boeckler FM, Ibrahim TM, Arafa RK. In silico targeting of fascin protein for cancer therapy: benchmarking, virtual screening and molecular dynamics approaches. Molecules. 2023;28(3):1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elghoneimy LK, Ismail MI, Boeckler FM, Azzazy HME, Ibrahim TM. Facilitating SARS CoV-2 RNA-Dependent RNA polymerase (RdRp) drug discovery by the aid of HCV NS5B palm subdomain binders: in silico approaches and benchmarking. Comput Biol Med. 2021;134:104468. doi: 10.1016/j.compbiomed.2021.104468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ibrahim TM, Ismail MI, Bauer MR, Bekhit AA, Boeckler FM. Supporting SARS-CoV-2 papain-like protease drug discovery: in silico methods and benchmarking. Front Chem. 2020;8:592289. doi: 10.3389/fchem.2020.592289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ballester PJ, Mitchell JB. A machine learning approach to predicting protein-ligand binding affinity with applications to molecular docking. Bioinformatics. 2010;26(9):1169–1175. doi: 10.1093/bioinformatics/btq112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fresnais L, Ballester PJ. The impact of compound library size on the performance of scoring functions for structure-based virtual screening. Brief Bioinform. 2021;22(3):bbaa095. doi: 10.1093/bib/bbaa095 [DOI] [PubMed] [Google Scholar]
- 16.Wojcikowski M, Ballester PJ, Siedlecki P. Performance of machine-learning scoring functions in structure-based virtual screening. Sci Rep. 2017;7:46710. doi: 10.1038/srep46710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coleman RG, Carchia M, Sterling T, Irwin JJ, Shoichet BK. Ligand pose and orientational sampling in molecular docking. PLoS One. 2013;8(10):e75992. doi: 10.1371/journal.pone.0075992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ragoza M, Hochuli J, Idrobo E, Sunseri J, Koes DR. Protein-ligand scoring with convolutional neural networks. J Chem Inf Model. 2017;57(4):942–957. doi: 10.1021/acs.jcim.6b00740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koes DR, Baumgartner MP, Camacho CJ. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J Chem Inf Model. 2013;53(8):1893–1904. doi: 10.1021/ci300604z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zayed AOH. Optimizing protein-ligand docking through machine learning: algorithm selection with AutoDock Vina. Discov Chem. 2025;2(1):164. doi: 10.1007/s44371-025-00246-4 [DOI] [Google Scholar]
- 21.Eberhardt J, Santos-Martins D, Tillack AF, Forli S. AutoDock Vina 1.2.0: new docking methods, expanded force field, and python bindings. J Chem Inf Model. 2021;61(8):3891–3898. doi: 10.1021/acs.jcim.1c00203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McGann M. FRED pose prediction and virtual screening accuracy. J Chem Inf Model. 2011;51(3):578–596. doi: 10.1021/ci100436p [DOI] [PubMed] [Google Scholar]
- 23.Korb O, Stützle T, Exner TE. PLANTS: application of ant colony optimization to structure-based drug design. In: International Workshop on Ant Colony Optimization and Swarm Intelligence. Springer Berlin Heidelberg; 2006:247–258. [Google Scholar]
- 24.Hawkins PC, Skillman AG, Warren GL, Ellingson BA, Stahl MT. Conformer generation with OMEGA: algorithm and validation using high quality structures from the protein databank and Cambridge structural database. J Chem Inf Model. 2010;50(4):572–584. doi: 10.1021/ci100031x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McGann M. FRED and HYBRID docking performance on standardized datasets. J Comput Aided Mol Des. 2012;26(8):897–906. doi: 10.1007/s10822-012-9584-8 [DOI] [PubMed] [Google Scholar]
- 26.O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open chemical toolbox. J Cheminform. 2011;3(1):33. doi: 10.1186/1758-2946-3-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korb O, Stützle T, Exner TE. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J Chem Inf Model. 2009;49(1):84–96. doi: 10.1021/ci800298z [DOI] [PubMed] [Google Scholar]
- 29.ten Brink T, Exner TE. Influence of protonation, tautomeric, and stereoisomeric states on protein−ligand docking results. J Chem Inf Model. 2009;49(6):1535–1546. doi: 10.1021/ci800420z [DOI] [PubMed] [Google Scholar]
- 30.Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model. 1999;17(1):57–61. [PubMed] [Google Scholar]
- 31.Ibrahim TM, Bauer MR, Dörr A, Veyisoglu E, Boeckler FM. pROC-chemotype plots enhance the interpretability of benchmarking results in structure-based virtual screening. J Chem Inf Model. 2015;55(11):2297–2307. doi: 10.1021/acs.jcim.5b00475 [DOI] [PubMed] [Google Scholar]
- 32.Clark RD, Webster-Clark DJ. Managing bias in ROC curves. J Comput Aided Mol Des. 2008;22(3–4):141–146. doi: 10.1007/s10822-008-9181-z [DOI] [PubMed] [Google Scholar]
- 33.Tran-Nguyen V-K, Junaid M, Simeon S, Ballester PJ. A practical guide to machine-learning scoring for structure-based virtual screening. Nat Protoc. 2023;18(11):3460–3511. doi: 10.1038/s41596-023-00885-w [DOI] [PubMed] [Google Scholar]
- 34.Mysinger MM, Carchia M, Irwin JJ, Shoichet BK. Directory of Useful Decoys, Enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem. 2012;55(14):6582–6594. doi: 10.1021/jm300687e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McNutt AT, Francoeur P, Aggarwal R, et al. GNINA 1.0: molecular docking with deep learning. J Cheminform. 2021;13(1):43. doi: 10.1186/s13321-021-00522-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sander T, Freyss J, von Korff M, Rufener C. DataWarrior: an open-source program for chemistry aware data visualization and analysis. J Chem Inf Model. 2015;55(2):460–473. doi: 10.1021/ci500588j [DOI] [PubMed] [Google Scholar]
- 37.Shamshad H, Bakri R, Mirza AZ. Dihydrofolate reductase, thymidylate synthase, and serine hydroxy methyltransferase: successful targets against some infectious diseases. Mol Biol Rep. 2022;49(7):6659–6691. doi: 10.1007/s11033-022-07266-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tarnchompoo B, Chitnumsub P, Jaruwat A, et al. Hybrid inhibitors of malarial dihydrofolate reductase with dual binding modes that can forestall resistance. ACS Med Chem Lett. 2018;9(12):1235–1240. doi: 10.1021/acsmedchemlett.8b00389 [DOI] [PMC free article] [PubMed] [Google Scholar]