

Abstract
Numerous sophisticated systems have been described that protect bacteria from increased levels of reactive oxygen species. Although indispensable during prolonged oxidative stress, these response systems depend on newly synthesized proteins, and are hence both time and energy consuming. Here, we describe an “express” cytoprotective system in Bacillus subtilis which depends on nitric oxide (NO). We show that NO immediately protects bacterial cells from reactive oxygen species by two independent mechanisms. NO transiently suppresses the enzymatic reduction of free cysteine that fuels the damaging Fenton reaction. In addition, NO directly reactivates catalase, a major antioxidant enzyme that has been inhibited in vivo by endogenous cysteine. Our data also reveal a critical role for bacterial NO-synthase in adaptation to oxidative stress associated with fast metabolic changes, and suggest a possible role for NO in defending pathogens against immune oxidative attack.
Keywords: Fenton reaction, nitric oxide, thiols
NO has many of the properties of a prototypical signaling molecule. It is small, freely diffusible, short-lived, and highly reactive in biological systems. NO is synthesized by NO synthases (NOS) in a wide variety of cells and is involved in numerous physiological and pathological processes in mammals (1–4). In contrast, bacterial-derived NO has been known only as an intermediate in the process of anaerobic respiration. However, some recent evidence suggests that NO and/or its equivalents [S-nitrosothiols (SNO)] may also be involved in signaling in bacteria. Several bacterial proteins have been shown to change their properties upon interaction with NO. For example, the transcription factors OxyR, SoxR, NorR, and Fur (5–7) in Escherichia coli and ResDE in Bacillus subtilis (8) activate corresponding regulons upon reaction with NO. Furthermore, several Gram-positive bacteria, including B. subtilis, possess an enzyme orthologous to eukaryotic NOS (9–13). The ability of B. subtilis NOS to synthesize NO from arginine has been confirmed in vitro (9, 10), although its physiological role remains obscure.
NO bioactivity depends on its target (2, 3, 14). In mammals, NO/SNO influence ranges from cytoprotection to cytotoxicity (5, 15–17). NO has been shown to protect various types of eukaryotic cells from H2O2 and organic peroxide-mediated toxicity (18–24), although the molecular mechanism of NO mediated cytoprotection has not been elucidated.
In bacteria, H2O2 toxicity is attributable primarily to DNA damage (25–27). Upon interaction with free cellular iron, H2O2 forms hydroxyl radicals (OH·) (reaction 1) that react at diffusion-limited rates with DNA bases and sugar moieties causing modifications and strand breaks (25–27).
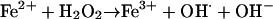 |
[1(Fenton reaction)] |
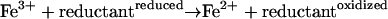 |
[2] |
Significantly, free reduced iron, which is required for the Fenton reaction, is scarce in vivo and would be depleted almost instantaneously upon H2O2 challenge (25). Thus, to persistently drive the Fenton reaction, ferric iron must be continuously rereduced to the ferrous state by cellular reductants (reaction 2). It has been shown that rereduction of ferric ion by cellular reducing equivalents (RE) such as FADH2 and cysteine sustain the Fenton reaction, ultimately leading to cellular death (25, 28). Here, we demonstrate that B. subtilis utilizes endogenous and exogenous NO for rapid protection from oxidative damage. NO suppresses the Fenton reaction by transiently inhibiting cysteine reduction. Independently, NO specifically activates catalase to detoxify excess H2O2. We explain how these two components of NO-mediated cytoprotection function in bacteria, and propose that this dual mechanism may be universal.
Experimental Procedures
Reagents, Strains, and Plasmids. 3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)2H-tetrazolium inner salt (MTS) was purchased from Promega. All other reagents, thioredoxin (Trx), and Trx reductase (TrxRed) from E. coli were from Sigma. NO solution was prepared in an airtight device by bubbling NO gas (Aldrich) that had been purified from higher oxides by passing it through a 1 M solution of KOH in to water, until the concentration of dissolved NO reached ≈300 μM. Water (Milli-Q grade) was deaerated by boiling and then cooling under argon (Praxair, Danbury, CT). Immediately before the reaction, the NO concentration was measured by using an ISO-NO Mark II electrode (WPI Instruments, Waltham, MA). B. subtilis IS75 (his leu met) was used as a parent strain. Plasmids were constructed by using standard methods and amplified in E. coli BL21 (Novagen). All PCR fragments were amplified from B. subtilis IS75 chromosomal DNA by using Pwo DNA polymerase (Roche). Oligonucleotide primers were purchased from Integrated DNA Technologies (Coralville, IA). To construct pUSNO1, two 400-bp fragments upstream and downstream of nos (yflM) were amplified by PCR and cloned into pUS19 (a gift from D. Dubnau, Public Health Research Institute, Newark, NJ). The resulting plasmid (pUSNO1) carries the spectinomycin (spc) resistance gene flanked with these fragments. IS75 was transformed with pUSNO1 to obtain the Δnos strain. Spectinomycin-resistant colonies were selected, and double cross-over recombination events were confirmed by PCR. The same procedure was used to construct ΔkatA and ΔcitB deletion strains. The pMutin2 plasmid was used for the complementation test (29) (Figs. 7 and 8, which are published as supporting information on the PNAS web site). Preparation of B. subtilis competent cells was carried out by the Spizizen method (30). Antibiotics were used at the following concentrations: chloramphenicol (Cm), 5 μg/ml; erythromycin, 1 μg/ml; and spectinomycin, 100 μg/ml.
General Methods. B. subtilis IS75 and Staphylococcus aureus wt strain RN6734 (a gift from R. Novick, New York University Medical Center, New York) overnight cultures grown in liquid Luria–Bertani (LB) media were diluted 1:100 in fresh LB and grown at 37°C with aeration until OD600 ∼ 0.5, unless indicated otherwise. To determine H2O2 resistance, B. subtilis cells were exposed to 1 or 10 mM H2O2 for 30 min. S. aureus was challenged with 370 mM H2O2. The number of viable cells was determined by colony formation on LB agar. Colony-forming units (CFU) were counted the following day, and the percentage of survival was calculated. To prepare bacterial cell extracts, B. subtilis cells were harvested, dissolved in lysis buffer (20 mM Tris·HCl, pH 7.9/150 mM NaCl) containing 125 μg/ml lysozyme (Sigma), incubated for 5 min at 37°C, sonicated, and clarified by centrifugation. Protein concentration was determined by using the Bio-Rad protein assay kit. Nitrite was measured in clarified cell culture supernatants by using the fluorimetric nitrite assay kit (Cayman Chemical, Ann Arbor, MI).
Catalase Activity Assay. Degradation of H2O2 was monitored in real time by spectroscopy, detected as a decrease in absorbance at 240 nm (31). Total H2O2 degrading activity was measured as the decrease of H2O2 concentration per mg of total protein per sec. OD240 was converted to the concentration of H2O2 according to the calibration curve (10 mM H2O2 = 0.36 OD240).
Quantification of Reduced Thiols in Vitro in Vivo. Cys and other thiols react with mBB to form a fluorescent dye (32). To determine Trx/TrxRed activity in the reconstituted system, the amount of reduced Cys was measured by reaction with mBB. The reaction mixture contained TrxRed (0.05 units per 100 μl) and Trx (2 μM) from E. coli and 1 mM cystine dissolved in 0.15 M NaCl/0.2 M Hepes (pH 7.6). Reaction was initiated by addition of 0.5 mM NADPH at 25°C. Twenty-microliter aliquots were withdrawn and mixed with 80 μl of 0.4 mM mBB in 0.15 M NaCl/0.2 M Hepes (pH 7.6). Reactions were incubated for 10 min in the dark, and the fluorescence was measured by using a PerkinElmer (LC55) fluorometer (λem = 479 nm, λex = 390 nm). Addition of NO after thiol reaction with mBB did not affect the fluorescent yield. Fluorescence was converted to the concentration of thiols according to the Cys standard curve. TrxRed activity in B. subtilis extracts was measured spectrophotometrically by reduction of 5,5′-dithio-bis(2-nitrobenzoic acid) in the presence of NADPH (33). The concentration of reduced thiols in vivo was measured by using mBB (32). B. subtilis cells (≈40 ml) were grown to the mid-log phase (OD ∼ 0.6–0.8), collected by centrifugation, and resuspended in 1 ml of fresh LB with 180 μg/ml Cm. Cells were treated with lysozyme (100 μg/ml) for 2 min at 37°C to remove cell walls. At indicated time points, 20-μl aliquots were withdrawn and mixed with 80 μlofstop solution (1 mM mBB/6 M guanidine·HCl/0.2 M Tris·HCl, pH 7.9). Reactions were vigorously shaken to facilitate cell lysis and kept in the dark for 10 min before measuring fluorescence.
Measurement of DNA Damage by Quantitative PCR. Total genomic DNA was isolated from 10 ml of culture and quantified by using a PicoGreen dsDNA quantitation reagent (Molecular Probes) and lambda DNA as a standard. Approximately 10-kb fragments near zwf regions were used for quantitative PCR. Primer sequences were as follows: 5′-GGATGCCTGTCTCGGTACAACACACG (forward primer) and 5′-GACCAGCCGGTTGTAGCTGTTACACC (reverse primer). PCR was performed by using Phusion DNA polymerase (Finnzymes). The 50-μl PCR mixture contained 0.05–0.5 ng of genomic DNA as a template, 1.5 μM primers, 200 μM dNTPs (Fermentas), 5× Phusion GC PCR buffer, and 0.5 μl of DNA polymerase. DNA was subjected to 30 cycles of PCR (98°C for 30 sec, 58°C for 30 sec, and 72°C for 9 min). PCR products were separated by 1% agarose gel electrophoresis, stained with ethidium bromide, scanned, and quantified with an AlphaImager (Imgen Technologies).
Determination of the Rate of RE Rereduction. IS75 cells were grown to mid-log phase in LB, and aliquots were treated with 1 mM diamide, 50 μM chloro(triethylphosphine)gold(I) (TepAu), 30 μM NO, or water. MTS (0.1 mg/ml) was added 5 sec after NO or H2O and 30 sec after diamide and TepAu. Tubes were incubated at 37°C, 1-ml aliquots were withdrawn every 2 min and separated from cells, and an absorbance was measured at 490 nM (34, 35). All reactions were preformed without phenazine methosulfate, because under this condition MTS is reduced by Cys 10 times more efficiently than by NADPH.
Results and Discussion
NO Rapidly Protects B. subtilis from Oxidative Stress. To examine the possibility that cytoprotection is induced by NO, we first investigated the effect of a harmless, single dose of NO (30 μM) on the survival of B. subtilis exposed to oxidative stress. As shown in Fig. 1A, within 5 sec of NO administration, resistance to H2O2 increased ≈100-fold (lane 4). The addition of NO simultaneously with or after H2O2 had no protective effect (lanes 5 and 6) apparently because of NO scavenging by radicals generated by H2O2 (26, 36). Also, no cytoprotection was observed if oxidized NO or nitrite were added instead of NO (lanes 7 and 8). Notably, pretreatment with the same low concentration of H2O2 (30 μM) could not protect cells from a lethal doze (10 mM) of H2O2 (lane 9), indicating that the protective effect of NO was highly specific. The above-mentioned controls (lanes 5 and 6) and large excess (>300 times) of H2O2 over NO rule out the possibility that the protective effect of NO was due to direct reaction with H2O2 or its derivatives. A similar level of protection from H2O2 was achieved with the NO-donors SNAP and MAHMA NONOATE (unpublished observation). Moreover, NO also protected B. subtilis against organic peroxides such as t-butyl hydroperoxide and cumene hydroperoxide (see Fig. 9, which is published as supporting information on the PNAS web site).
Fig. 1.

Cytoprotection by exogenous NO. (A) Effect of exogenous NO on H2O2 toxicity. The final concentration of NO after a bolus application was 30 μM. The concentrations of H2O2 and NaNO2 were 10 mM and 30 μM, respectively. Reagents were added as indicated to aerobically grown wt cells at OD600 ∼ 0.5 (in LB at 37°C). Incubation with H2O2 was for 30 min. In lanes 4, 7, and 8, H2O2 was added 5 sec after NO, NaNO2, or oxidized NO, respectively. In lane 6, NO was mixed with H2O2 before addition to cells. In lane 8, NO was oxidized before addition to cells by bubbling air into an aqueous solution of NO for 2 h. In lane 9, 10 mM H2O2 was added after a 5-sec incubation with 30 μMH2O2. The percentage of surviving cells was determined by colony formation and is shown as the mean ± SD from five experiments. (B) Time course of NO-mediated cytoprotection. At the times shown after the addition of NO, aliquots of culture were removed and challenged with 10 mM H2O2 for 30 min. Cm (200 μg/ml) was added for 5 min before NO/H2O2 treatment. Values shown are the means ± SD from three experiments.
NO has been shown to activate various genes in E. coli and B. subtilis to protect cells from oxidative and nitrosative stress (7, 8, 37, 38). However, in our experiments the full protective effect of NO was established within 5 sec of NO administration, eliminating the necessity of gene activation for cytoprotection. We consistently found that inhibition of protein synthesis by Cm did not compromise NO-mediated cytoprotection (Fig. 1B). Notably, the effect of a bolus of NO was transient, with a maximum attained within 5 sec of application (Fig. 1B). This finding is consistent with the short life time of NO in physiological solutions (3) and argues for a rapid reversibility of the process. Taken together, these data suggest that NO directly and reversibly activates some latent, readily available oxidative stress defense system(s) in B. subtilis.
Catalase Activation by NO: The First Component of NO-Mediated Cytoprotection. NO could rapidly protect cells from H2O2 by boosting the activity of a preexisting H2O2 scavenging enzyme(s). B. subtilis vegetative catalase KatA is the major antioxidant enzyme. It is an iron–heme protein, and thus a natural target for NO. To test whether NO activates KatA, we measured the rate of H2O2 decomposition in crude cell extracts (31). Challenging a B. subtilis extract with our standard NO dose boosted H2O2 degradation by 75% (Fig. 2A) but failed to do so in an extract from ΔkatA cells (Fig. 2A), demonstrating that NO indeed potentiates the activity of KatA but not that of other enzymes of related function. Our in vitro data indicate that free Cys partially inhibits KatA (see Supporting Text and Fig. 10, which are published as supporting information on the PNAS web site). NO relieves this inhibition by disrupting the KatA–Cys complex apparently via an S-nitrosylation mechanism.
Fig. 2.

Activation of catalase and inhibition of the Fenton reaction, two components of NO-mediated cytoprotection. (A) Stimulating effect of NO on H2O2 degrading activity in crude extracts of wt and ΔkatA cells. Total H2O2 degrading activity was measured as described in Experimental Procedures (31). Where indicated, extracts were incubated with 45 μM NO for 5 sec. 100% catalase activity = 30 mM H2O2 min–1·mg–1. Values shown are the means ± SE from six experiments. (B) Transient protection of Δkat cells from oxidative stress by NO. The graph shows the time course of H2O2-mediated toxicity. Ten millimolar H2O2 was added at t = 0. Where indicated, 30 μM NO was added 5 sec before H2O2. Values shown are the means ± SD from three experiments. (C) Protection of wt cells from oxidative stress by the iron chelator dipyridyl and thiol oxidizer diamide. After 5 min of incubation with Cm (50 μg/ml), aerobically grown wt cells (OD600 ∼ 0.5) were treated with dipyridyl (1 mM for 10 min) or diamide (200 μM for 3 min) and/or NO (30 μM for 5 sec), followed by the addition of 10 mM H2O2 for 5 min. Values shown are the means ± SE from four experiments. (D) Chromosomal DNA damage from the Fenton reaction. A representative agarose gel shows a 10-kb PCR fragment amplified from B. subtilis chromosome. Chromosomal DNA was isolated from cells treated with diamide, dipyridyl, or NO and H2O2 as described in Figs. 1 A and 2C. M, 1-kb DNA ladder. The relative intensity of the full size DNA band is indicated at the bottom. Values shown are the means ± SD from three experiments.
Inhibition of the Fenton Reaction: A Second Component of NO-Mediated Cytoprotection. The above results suggested that KatA deletion would compromise NO-mediated protection from reactive oxygen species in vivo. Indeed, we found that NO failed to elicit any significant protection of ΔkatA cells from H2O2 after 15 min of H2O2 application (Fig. 2B). However, NO-treated ΔkatA cells still retained most of their resistance to H2O2 during the first 10 min of treatment (Fig. 2B), indicating that another mechanism unrelated to KatA activation was responsible for initial, transient cytoprotection by NO.
In E. coli, DNA damage from hydroxyl radicals generated by the Fenton reaction is a primary mechanism of H2O2 cytotoxicity (25, 26). If the same mechanism operates in B. subtilis, an early cytoprotective effect of NO must be in suppressing the Fenton reaction and DNA damage. To test this hypothesis, we examined the effect of a cell permeable iron chelator (dipyridyl) on H2O2 toxicity and NO cytoprotection. Dipyridyl protected cells from a lethal doze of H2O2 (Fig. 2C), whereas NO failed to further protect dipyridyl-treated cells. This result shows that H2O2 toxicity in B. subtilis (like in E. coli) is attributed to DNA damage induced by Fenton chemistry.
To further support this conclusion, chromosomal DNA damage was measured by quantitative PCR (28). We found that the yield of full-length PCR fragments (10 kb) decreased considerably after challenging cells with 10 mM H2O2, with DNA fragments of a smaller size becoming apparent (Fig. 2D, lane 4). These smaller fragments reflect multiple DNA lesions that prematurely interrupted the PCR reaction. The iron chelator and inhibitor of the Fenton reaction dipyridyl (39) eliminated all DNA damage (Fig. 2D, lane 1). NO also restored the yield of the full-length fragment and eliminated smaller bands (Fig. 2D, lane 5), implying that NO protects cellular DNA from the destructive Fenton reaction.
As mentioned in the Introduction, ferric iron must be repeatedly rereduced to maintain the Fenton reaction. In E. coli, Cys reduces cellular iron making the Fenton reaction processive (28, 36). To verify whether Cys or other free thiols are capable of supporting the Fenton reaction in B. subtilis, we used diamide, a specific thiol-oxidizing reagent (40). Diamide eliminated lesions in chromosomal DNA (Fig. 2D, lane 2) and protected cells from H2O2 toxicity (Fig. 2C). NO failed to further protect diamide-treated cells from H2O2 (Fig. 2C), indicating that NO protection occurs via the inhibition of Fenton chemistry.
Taken together, these results explain the biphasic NO protection pattern shown in Fig. 2B and imply that NO inhibition of the Fenton reaction along with NO activation of catalase constitute the mechanism of NO cytoprotection.
Mechanism of NO-Mediated Inhibition of the Fenton Reaction. Because the amount of NO used in our experiments was insufficient to eliminate OH· directly (NO:H2O2 = 1:300), we assumed that NO inhibited the Fenton reaction by either scavenging cellular iron or preventing its rereduction. First, we examined the effect of NO on the Fenton reaction in vitro. Hydrogen peroxide alone did not produce any strand brakes in pBR322 DNA (Fig. 3A, lane 6). Addition of Fe3+ caused only a slight increase in strand breaks (Fig. 3A, lane 2), whereas addition of Cys [a major free thiol in B. subtilis (41)] along with Fe3+ dramatically accelerated DNA damage (Fig. 3A, lane 3). Addition of excess NO (NO:Fe3+ = 3:2) did not inhibit DNA damage (Fig. 3A, lane 4). These results show that NO cannot interfere with Fenton chemistry by scavenging cellular iron. Indeed, the NO–iron complex is highly unstable in biological solutions [Koff ∼ 24–660 sec–1 (42)] and dissociates rapidly in the presence of H2O2 in vitro [t1/2 = 15 sec (data not shown)].
Fig. 3.

Inhibition of Cys reduction is a mechanism of NO cytoprotection. (A) Effect of Cys and NO on Fenton-mediated DNA damage in vitro. As indicated, the supercoiled pBR322 plasmid (0.5 μg) was treated with 30 μM FeCl3, 10 mM Cys, 45 μM NO, or 10 mM H2O2 in 20 mM Tris·HCl buffer (pH 7.9). After a 10-min incubation at room temperature, the reaction was stopped and separated in a 1% agarose gel. RF, relaxed form; SF, supercoiled form. (B) NO-mediated suppression of cellular RE rereduction. The graph shows the negative effect of NO on the rate of formazan dye formation in the culture of wt cells grown in LB. Where indicated, cells were treated with 30 μM NO for 5 sec before the addition of 0.1 mg/ml MTS. Values shown are the means ± SE from three experiments. (C) Effects of NO, diamide, or TepAu on the rate of formazan dye accumulation. Conditions are as in B. TepAu and diamide were added 30 sec before MTS. Values shown are the means ± SD from three experiments. (D) Effect of carbon availability on oxidative stress survival and NO protection. Cells in mid-log phase were resuspended in M9 minimal medium containing 200 μg/ml Cm and incubated with or without glucose (50 mM for 15 min at 37°C). NO (30 μM) was added for 5 sec, followed by H2O2 (10 mM). Values shown are the means ± SD from three experiments.
An alternative mechanism by which NO could suppress the Fenton reaction in vivo is to inhibit enzymes that generate RE [RE reduce cellular iron, thus driving the Fenton reaction (see Introduction)] (25). Indeed, NO readily binds iron–heme and iron–sulfur centers of various proteins [Kd ∼ 10–6 to 10–12 M (42)] and can also inhibit enzymes via S-nitrosylation (43).
We first examined the possibility that NO inhibits aconitase, a Fe-S enzyme that metabolizes citrate. Citrate is a natural iron chelator. We therefore reasoned that its accumulation due to aconitase inhibition should interfere with Fenton chemistry. However, cells bearing a chromosomal deletion of the aconitase gene (citB) were protected by NO to the same extent as wt cells (data not shown), thus excluding aconitase from the mechanism of NO cytoprotection.
We then examined whether NO suppresses major catabolic pathways (glycolysis, the pentose phosphate pathway and TCA cycle) and as a result depletes cells of RE [Cys, FADH2, and NAD(P)H]. To address this issue, we took advantage of the MTS reagent, which forms a colored dye upon reduction by cellular RE (34, 35). The rate of MTS dye accumulation is directly correlated with the rate of RE rereduction in vivo. As shown in Fig. 3B, exponentially growing B. subtilis cells accumulate RE at a steady rate as represented by the linearity of the dye formation curve; note that the slope of the line reflects the cumulative activity of reducing enzymes. Remarkably, after bolus NO application, the total cellular concentration of RE was diminished. More importantly, the rate of RE rereduction also decreased >2-fold, as reflected by the lower slope of the line (k1/k2 = 2.2) (Fig. 3B). The slower rate of RE rereduction would result in a slower rate of cellular iron reduction; hence, the inefficient Fenton reaction. Significantly, the ≈10-min delay in RE accumulation correlated well with the period (≈10 min) of NO-mediated protection from H2O2 in ΔkatA cells (Fig. 2B).
MTS is reduced by Cys 10 times more efficiently than by NADPH (see Experimental Procedures and Fig. 11, which is published as supporting information on the PNAS web site), suggesting that dye accumulation in Fig. 3B is mostly attributed to MTS reaction with Cys. To confirm this hypothesis, we monitored the dye accumulation in the presence of thiol-depleting reagents diamide or TepAu (Fig. 3C). TepAu is a specific inhibitor of TrxRed (44). Trx/TrxRed is the only and essential system in B. subtilis dedicated to thiol reduction (45). Inhibition of this system would quickly deplete cells of reduced thiols. Indeed, treating cells with either diamide or TepAu leads to a significant decrease in the rate and amount of MTS dye formation (Fig. 3C), i.e., thiol reduction. These results indicate that NO inhibits the rate of thiols reduction and thus inhibits Fenton chemistry (Fig. 3C).
The proposed mechanism of NO cytoprotection implies that the extent of H2O2 toxicity is directly proportional to the rate of thiols reduction, i.e., to the activity of glycolysis, the pentose phosphate pathway, and Trx/TrxRed. Consistently, we noticed that B. subtilis cells deprived of a carbon source acquired resistance to H2O2 (Fig. 3D). A similar observation has been made with E. coli (25, 46). Moreover, the addition of glucose sensitized B. subtilis cells to H2O2, whereas NO reversed this effect (Fig. 3D).
We next proceeded to determine which RE [FADH2, NAD(P)H, or Cys] serves as a major driving force of the Fenton reaction in B. subtilis. Cys is the major reducing agent that drives the Fenton reaction in E. coli (28). NAD(P)H does not support the Fenton reaction as well (25), whereas free FADH2 is scarce in the cell. The amount of free FADH2 to be sufficient to drive the Fenton reaction could be achieved only upon inhibition of respiration (25). Thus, Cys, the only abundant small thiol in B. subtilis (41), is the primary candidate for iron reduction.
NO could either deplete free cellular Cys or prevent cystine rereduction. Because the concentration of free intracellular Cys was not affected by NO (data not shown), we proposed that NO inhibited cystine reduction. Because Trx/TrxRed is the only (and essential) system in B. subtilis that reduces thiols (45), and NO can inhibit eukaryotic Trx (47), we examined the effect of NO on bacterial Trx/TrxRed. Reconstituted Trx/TrxRed plus NADPH efficiently reduce cystine (Fig. 4A). Forty-five and 100 μM NO decreased the reduction rate 4- and 40-fold, respectively (Fig. 4A). NO also inhibited Trx/TrxRed in a cell extract (data not shown). These results demonstrate that Trx/TrxRed is a target for NO inhibition. Consistently, a specific inhibitor of TrxRed (TepAu) protected cells against Fenton-mediated toxicity (unpublished observations).
Fig. 4.

Inhibition of Cys reduction by NO in vitro and in vivo.(A) Inhibition of Trx/TrxRed-mediated cystine reduction by NO in the reconstituted system (see Experimental Procedures). NO donor MAHMA (45 and 100 μM) was added before initiation of the reaction with NADPH. (B) NO transiently inhibits Cys reduction in vivo. Cells were treated with 1 mM diamide and/or the indicated amount of NO donor MAHMA. At the indicated time points, aliquots were withdrawn, and thiols were quantified as described in Experimental Procedures. NO added to the reaction after it was stopped did not affect the fluorescence yield (data not shown). Values shown are the means ± SD from three experiments.
To confirm that NO inhibits thiol reduction in vivo, we oxidized cellular thiols with diamide and then monitored the rate of their rereduction. It took ≈6 min for B. subtilis to rereduce its thiols after diamide challenge (Fig. 4B, open squares). Two hundred and 500 μM NO delayed thiol reduction by 3 (Fig. 4B, open circles) and 6 (Fig. 4B, open triangles) min, respectively. We used higher concentrations of NO and diamide because cells were concentrated 40-fold to allow for thiols detection. NO on its own could not oxidize thiols efficiently (Fig. 4B, filled circles and filled triangles). The gradual decrease of the thiol concentration in this case reflects spontaneous thiol oxidation apparently due to Trx/TrxRed inhibition by NO.
Taken together, our results explain the mechanism of instantaneous NO protection from oxidative stress (Fig. 5). H2O2 oxidizes cellular iron to yield highly toxic hydroxyl radicals. Cys reduces iron back again by forming cystine. Trx/TrxRed then reduces cystine back to Cys at the expense of NADPH. To keep the Fenton reaction going, oxidized iron and cystine must be continuously rereduced. This mechanism explains why cells depleted of a carbon source acquire resistance to H2O2. Such cells are restricted to a “single-round” Fenton reaction because all free Cys becomes rapidly oxidized in the presence of H2O2. In contrast, cells grown on glucose are able to regenerate Cys, thus enabling a “multi-round” Fenton reaction. NO transiently interrupts cystine reduction by inhibiting Trx/TrxRed, thus rendering the Fenton reaction non-processive. Simultaneously, NO directly activates catalase, which detoxifies excess H2O2 (Fig. 5).
Fig. 5.

Mechanism of NO-mediated protection from oxidative stress in B. subtilis. NO instantly protects cells from H2O2 toxicity by a dual mechanism. In the schematic shown, NO transiently interrupts the production of damaging hydroxyl radicals from the Fenton reaction by suppressing Cys reduction by Trx/TrxRed. In parallel, NO activates catalase (KatA).
NOS Protects B. subtilis from Metabolically Linked Oxidative Stress. Many organisms use glutathione to maintain their internal reducing environment. It has been shown that, unlike Cys, glutathione does not support the Fenton reaction (28). Because B. subtilis uses Cys instead of glutathione, it is reasonable to speculate that it has developed a dedicated system to control the rate of Cys reduction so as to keep the Fenton reaction at bay.
As noted in the Introduction, B. subtilis possesses NOS that is capable of generating NO from l-arginine in vitro (9). One can therefore speculate that endogenous NO renders cells more resistant to oxidants via both catalase activation and diminishing the rate of Cys reduction. To examine a possible cytoprotective role of endogenous NO (produced by bacterial NOS), we compared the ability of wild-type (wt) and nos (yflM) deletion strains to withstand oxidative stress. Because wt and Δnos had the same sensitivity to peroxide under steady growth conditions, we hypothesized that changes in thiol availability may activate NOS. To simulate this situation, we grew cells to late exponential phase and then diluted them with fresh LB medium. The intracellular level of RE (mostly Cys) increased 2-fold in Δnos mutant upon dilution as compared with 1.5-fold in wt (Table 1, which is published as supporting information on the PNAS web site). Such nutrient shift sensitized both wt and nos cells to H2O2. However, Δnos cells were ≈25 times more sensitive to oxidative stress than wt (Fig. 6A). Such a dramatic difference between the two strains became obvious within the first 2 min after dilution, arguing against any contribution from de novo protein synthesis. This situation is reminiscent of the instant anti-oxidant effect of exogenous NO (Fig. 1).
Fig. 6.

NOS-mediated protection from oxidative stress in B. subtilis. (A) Effect of nos deletion on H2O2 sensitivity. wt and Δnos cells were grown aerobically in LB to late log phase (OD600 ∼ 0.8–0.9) at 30°C. An aliquot from each culture was diluted with an equal amount of fresh prewarmed LB for 2 min (diluted). Both diluted and undiluted aliquots were treated with 1 mM or 10 mM H2O2 for 30 min. The percentage of surviving cells was determined by colony formation. Values shown are the means and SD (error bars) from four independent experiments. (B) Exogenous Cys sensitizes B. subtilis to oxidative stress and induces NOS-mediated protection. Cells were grown as in A and diluted with an equal volume of saline or saline plus Cys (100 μM). Arg (100 μM) was added to all samples. After 2 min of incubation, cells were treated with 10 mM H2O2 for 30 min. Values shown are the means ± SD from three experiments. (C) Effect of fresh medium dilution on nitrite levels in wt and Δnos cell cultures. Conditions were as in A. Samples for nitrite measurements were taken 5 min after dilution. LB, nitrite level in LB. Values shown are the means ± SD from three experiments. (D) Effect of dilution on KatA activity. Conditions were as in A. Cells were collected 1 min after dilution and lysed immediately, and catalase activity was measured as described in Experimental Procedures. Values shown are the means ± SE from three experiments.
To prove that exogenous Cys was responsible for increased H2O2 sensitivity, we reproduced the above experiment using saline (with or without Cys) instead of LB for dilution. Cys sensitized Δnos cells to H2O2 >20 times more than wt (Fig. 6B). Notably, dilution per se could not render cells sensitive to H2O2 (Fig. 6B). Moreover, addition of glucose or various amino acids also did not sensitize cells to H2O2 (data not shown). These results strongly suggest that a rapidly increased concentration of Cys triggers (directly or indirectly) NOS-mediated NO production to protect cells against oxidative stress.
To test this hypothesis, we measured the concentration of nitrite in the medium before and after dilution. Nitrite is a direct product of NO oxidation serving as a standard quantitative marker of NO. Fig. 6C shows that the level of nitrite was higher in the wt culture than Δnos, reflecting the basal level of NOS activity. More importantly, upon dilution, the concentration of nitrite went down in the Δnos culture but increased in wt. These results demonstrate that NOS is indeed activated upon dilution thus protecting cells from oxidative stress.
To determine whether endogenous NO activates catalase, we measured KatA activity upon dilution. As noted above, NO reactivates catalase that has been inhibited by thiols (Fig. 10). Cellular level of reduced Cys was increased upon dilution (Table 1), and catalase activity was inhibited by 20% in Δnos cells (Fig. 6D). In contrast, dilution of wt cells did not affect catalase activity. These results indicate that NOS-derived NO reactivates catalase that has been inhibited by endogenous Cys (Fig. 6D).
These findings directly implicate NOS in the delicate regulation of cellular redox homeostasis and adaptation of B. subtilis to oxidative stress during rapid metabolic changes. This function of NOS may be crucial under conditions of competitive growth in natural environments. Note that dilution exacerbates an otherwise subtle difference in growth rates between wt and Δnos strains (Fig. 12, which is published as supporting information on the PNAS web site).
As mentioned above, the dilution experiment simulates the natural situation when the demand for biosynthesis temporarily stops, leading to transient accumulation of reduced Cys, a major thiol in B. subtilis (41). The excess of Cys would not only drive the Fenton reaction (28) but also suppress catalase activity and thus elevate the level of H2O2 (Fig. 5). In this regard, the protective effect of NO is twofold. It immediately and transiently suppresses Cys reduction, thus suppressing the Fenton reaction. At the same time, NO activates a preexisting antioxidant enzyme, catalase, which quickly detoxifies the excess H2O2.
NO-Mediated Cytoprotection May Be General for NOS-Containing Bacteria and Contribute to Pathogenesis. NOS has been found only in Gram-positive bacteria, suggesting that Gram-negative bacteria do not use NO for adaptation to oxidative stress. Indeed, treating E. coli with NO does not provide any immediate protection from H2O2 toxicity (data not shown). In light of the mechanism presented above, there are at least three explanations for such a difference between bacterial species. First, in contrast to B. subtilis KatA, major catalase from E. coli is inhibited by NO (48). Second, Cys does not inhibit catalase from E. coli to the same extent as B. subtilis KatA (49). Third, Cys is a major low-molecular-weight thiol in many Gram-positive including Bacilli and S. aureus but not in E. coli (28, 41). The most abundant small thiol in E. coli, glutathione (GSH), has not been found in B. subtilis (41). Unlike Cys, GSH does not support the Fenton reaction and acts as an antioxidant rather than a prooxidant in vivo (28).
The “express” mechanism of NO-mediated cytoprotection reported here could play an important role in the adaptation of pathogens to oxidative stress imposed by the immune system. Macrophages and other phagocytes produce 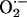 and NO in large quantities to combat infecting bacteria (4, 50–52). The ability of pathogens to survive the immunological “respiratory burst” critically depends on the state of their oxidative stress defense system. Macrophages start to produce high levels of NO after they encounter bacteria (53). Whereas superoxide anion stays inside the phagocyte vacuole, exceptionally pervasive NO escapes from immune cells. Thus, NO should be able to reach the bacterial interior even outside the macrophage, serving as a forewarning of the impending full-scale onslaught of the host. This event would postpone the damaging Fenton reaction and activate preexisting catalase to neutralize excess H2O2, giving bacteria critical time for de novo synthesis of their major stress response components. In support of this hypothesis, we provide evidence that the notorious Gram-positive pathogen S. aureus employs the same NO-mediated cytoprotection system as B. subtilis (see Fig. 13, which is published as supporting information on the PNAS web site).
and NO in large quantities to combat infecting bacteria (4, 50–52). The ability of pathogens to survive the immunological “respiratory burst” critically depends on the state of their oxidative stress defense system. Macrophages start to produce high levels of NO after they encounter bacteria (53). Whereas superoxide anion stays inside the phagocyte vacuole, exceptionally pervasive NO escapes from immune cells. Thus, NO should be able to reach the bacterial interior even outside the macrophage, serving as a forewarning of the impending full-scale onslaught of the host. This event would postpone the damaging Fenton reaction and activate preexisting catalase to neutralize excess H2O2, giving bacteria critical time for de novo synthesis of their major stress response components. In support of this hypothesis, we provide evidence that the notorious Gram-positive pathogen S. aureus employs the same NO-mediated cytoprotection system as B. subtilis (see Fig. 13, which is published as supporting information on the PNAS web site).
Supplementary Material
Acknowledgments
We thank Ruslan Rafikov and Nick Cowan for comments. This work was supported by National Institutes of Health Grant AI060762.
Author contributions: E.N. designed research; I.G. performed research; I.G. and E.N. analyzed data; and E.N. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: Cm, chloramphenicol; mBB, monobromobimane; MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)2H-tetrazolium inner salt; NOS, NO synthase; RE, reducing equivalents; TepAu, chloro(triethylphosphine)gold(I); Trx, thioredoxin; TrxRed, Trx reductase.
References
- 1.Ignarro, L. J. (2002) J. Physiol. Pharmacol. 53, 503–514. [PubMed] [Google Scholar]
- 2.Moncada, S., Palmer, R. M. & Higgs, E. A. (1991) Pharmacol. Rev. 43, 109–142. [PubMed] [Google Scholar]
- 3.Kerwin, J. F., Jr., Lancaster, J. R., Jr., & Feldman, P. L. (1995) J. Med. Chem. 38, 4343–4362. [DOI] [PubMed] [Google Scholar]
- 4.Nathan, C. & Shiloh, M. U. (2000) Proc. Natl. Acad. Sci. USA 97, 8841–8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Demple, B. (1999) Braz. J. Med. Biol. Res. 32, 1417–1427. [DOI] [PubMed] [Google Scholar]
- 6.Marshall, H. E., Merchant, K. & Stamler, J. S. (2000) FASEB J. 14, 1889–1900. [DOI] [PubMed] [Google Scholar]
- 7.Mukhopadhyay, P., Zheng, M., Bedzyk, L. A., LaRossa, R. A. & Storz, G. (2004) Proc. Natl. Acad. Sci. USA 101, 745–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakano, M. M. (2002) J. Bacteriol. 184, 1783–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adak, S., Aulak, K. S. & Stuehr, D. J. (2002) J. Biol. Chem. 277, 16167–16171. [DOI] [PubMed] [Google Scholar]
- 10.Adak, S., Bilwes, A. M., Panda, K., Hosfield, D., Aulak, K. S., McDonald, J. F., Tainer, J. A., Getzoff, E. D., Crane, B. R. & Stuehr, D. J. (2002) Proc. Natl. Acad. Sci. USA 99, 107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi, W. S., Chang, M. S., Han, J. W., Hong, S. Y. & Lee, H. W. (1997) Biochem. Biophys. Res. Commun. 237, 554–558. [DOI] [PubMed] [Google Scholar]
- 12.Chen, Y. & Rosazza, J. P. (1995) J. Bacteriol. 177, 5122–5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen, Y. & Rosazza, J. P. (1994) Biochem. Biophys. Res. Commun. 203, 1251–1258. [DOI] [PubMed] [Google Scholar]
- 14.Moncada, S. & Higgs, E. A. (1991) Eur. J. Clin. Invest. 21, 361–374. [DOI] [PubMed] [Google Scholar]
- 15.Schopfer, F. J., Baker, P. R. & Freeman, B. A. (2003) Trends Biochem. Sci. 28, 646–654. [DOI] [PubMed] [Google Scholar]
- 16.Radi, R. (2004) Proc. Natl. Acad. Sci. USA 101, 4003–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thomas, D. D., Espey, M. G., Vitek, M. P., Miranda, K. M. & Wink, D. A. (2002) Proc. Natl. Acad. Sci. USA 99, 12691–12696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wink, D. A., Hanbauer, I., Krishna, M. C., DeGraff, W., Gamson, J. & Mitchell, J. B. (1993) Proc. Natl. Acad. Sci. USA 90, 9813–9817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wink, D. A., Cook, J. A., Krishna, M. C., Hanbauer, I., DeGraff, W., Gamson, J. & Mitchell, J. B. (1995) Arch. Biochem. Biophys. 319, 402–407. [DOI] [PubMed] [Google Scholar]
- 20.Robb, S. J. & Connor, J. R. (2002) Ann. N.Y. Acad. Sci. 962, 93–102. [DOI] [PubMed] [Google Scholar]
- 21.Monastyrskaya, E., Folarin, N., Malyshev, I., Green, C. & Andreeva, L. (2002) Nitric Oxide 7, 127–131. [DOI] [PubMed] [Google Scholar]
- 22.Degnim, A. C., Morrow, S. E., Ku, J., Zar, H. A. & Nakayama, D. K. (1998) J. Surg. Res. 75, 127–134. [DOI] [PubMed] [Google Scholar]
- 23.Gorbunov, N. V., Yalowich, J. C., Gaddam, A., Thampatty, P., Ritov, V. B., Kisin, E. R., Elsayed, N. M. & Kagan, V. E. (1997) J. Biol. Chem. 272, 12328–12341. [DOI] [PubMed] [Google Scholar]
- 24.Joshi, M. S., Ponthier, J. L. & Lancaster, J. R., Jr. (1999) Free Radical Biol. Med. 27, 1357–1366. [DOI] [PubMed] [Google Scholar]
- 25.Woodmansee, A. N. & Imlay, J. A. (2002) J. Biol. Chem. 277, 34055–34066. [DOI] [PubMed] [Google Scholar]
- 26.Aruoma, O. I., Halliwell, B., Gajewski, E. & Dizdaroglu, M. (1989) J. Biol. Chem. 264, 20509–20512. [PubMed] [Google Scholar]
- 27.Liochev, S. I. & Fridovich, I. (1999) IUBMB Life 48, 157–161. [DOI] [PubMed] [Google Scholar]
- 28.Park, S. & Imlay, J. A. (2003) J. Bacteriol. 185, 1942–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vagner, V., Dervyn, E. & Ehrlich, S. D. (1998) Microbiology 144 (Pt 11), 3097–3104. [DOI] [PubMed] [Google Scholar]
- 30.Anagnostopoulos, C. & Spizizen, J. (1961) J. Bacteriol. 81, 741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen, L., Keramati, L. & Helmann, J. D. (1995) Proc. Natl. Acad. Sci. USA 92, 8190–8194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Newton, G. L., Dorian, R. & Fahey, R. C. (1981) Anal. Biochem. 114, 383–387. [DOI] [PubMed] [Google Scholar]
- 33.Holmgren, A. & Bjornstedt, M. (1995) Methods Enzymol. 252, 199–208. [DOI] [PubMed] [Google Scholar]
- 34.Cory, A. H., Owen, T. C., Barltrop, J. A. & Cory, J. G. (1991) Cancer Commun. 3, 207–212. [DOI] [PubMed] [Google Scholar]
- 35.Debnam, P. M. & Shearer, G. (1997) Anal. Biochem. 250, 253–255. [DOI] [PubMed] [Google Scholar]
- 36.Imlay, J. A. (2003) Annu. Rev. Microbiol. 57, 395–418. [DOI] [PubMed] [Google Scholar]
- 37.Nunoshiba, T., DeRojas-Walker, T., Wishnok, J. S., Tannenbaum, S. R. & Demple, B. (1993) Proc. Natl. Acad. Sci. USA 90, 9993–9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hausladen, A., Privalle, C. T., Keng, T., DeAngelo, J. & Stamler, J. S. (1996) Cell 86, 719–729. [DOI] [PubMed] [Google Scholar]
- 39.Asad, N. R. & Leitao, A. C. (1991) J. Bacteriol. 173, 2562–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leichert, L. I., Scharf, C. & Hecker, M. (2003) J. Bacteriol. 185, 1967–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Newton, G. L., Arnold, K., Price, M. S., Sherrill, C., Delcardayre, S. B., Aharonowitz, Y., Cohen, G., Davies, J., Fahey, R. C. & Davis, C. (1996) J. Bacteriol. 178, 1990–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cooper, C. E. (1999) Biochim. Biophys. Acta 1411, 290–309. [DOI] [PubMed] [Google Scholar]
- 43.Mohr, S., Stamler, J. S. & Brune, B. (1996) J. Biol. Chem. 271, 4209–4214. [DOI] [PubMed] [Google Scholar]
- 44.Pia Rigobello, M., Messori, L., Marcon, G., Agostina Cinellu, M., Bragadin, M., Folda, A., Scutari, G. & Bindoli, A. (2004) J. Inorg. Biochem. 98, 1634–1641. [DOI] [PubMed] [Google Scholar]
- 45.Scharf, C., Riethdorf, S., Ernst, H., Engelmann, S., Volker, U. & Hecker, M. (1998) J. Bacteriol. 180, 1869–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Imlay, J. A. & Linn, S. (1986) J. Bacteriol. 166, 519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nikitovic, D. & Holmgren, A. (1996) J. Biol. Chem. 271, 19180–19185. [DOI] [PubMed] [Google Scholar]
- 48.Brunelli, L., Yermilov, V. & Beckman, J. S. (2001) Free Radical Biol. Med. 30, 709–714. [DOI] [PubMed] [Google Scholar]
- 49.Switala, J. & Loewen, P. C. (2002) Arch. Biochem. Biophys. 401, 145–154. [DOI] [PubMed] [Google Scholar]
- 50.Hampton, M. B., Kettle, A. J. & Winterbourn, C. C. (1998) Blood 92, 3007–3017. [PubMed] [Google Scholar]
- 51.MacMicking, J., Xie, Q. W. & Nathan, C. (1997) Annu. Rev. Immunol. 15, 323–350. [DOI] [PubMed] [Google Scholar]
- 52.Fang, F. C. (1999) Nitric Oxide and Infection (Kluwer Academic/Plenum, New York).
- 53.Vazquez-Torres, A., Xu, Y., Jones-Carson, J., Holden, D. W., Lucia, S. M., Dinauer, M. C., Mastroeni, P. & Fang, F. C. (2000) Science 287, 1655–1658. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.