

Abstract
Collagen consists of repetitive Gly–Xaa–Yaa tripeptide units with proline and hydroxyproline frequently found in the Xaa and Yaa position, respectively. This sequence motif allows the formation of a highly regular triple helix that is stabilized by steric (entropic) restrictions in the constituent polyproline-II-helices and backbone hydrogen bonds between the three strands. Concentration-dependent association reactions and slow prolyl isomerization steps have been identified as major rate-limiting processes during collagen folding. To gain information on the dynamics of triple-helix formation in the absence of these slow reactions, we performed stopped-flow double-jump experiments on cross-linked fragments derived from human type III collagen. This technique allowed us to measure concentration-independent folding kinetics starting from unfolded chains with all peptide bonds in the trans conformation. The results show that triple-helix formation occurs with a rate constant of 113 ± 20 s–1 at 3.7°C and is virtually independent of temperature, indicating a purely entropic barrier. Comparison of the effect of guanidinium chloride on folding kinetics and stability reveals that the rate-limiting step is represented by bringing 10 consecutive tripeptide units (3.3 per strand) into a triple-helical conformation. The following addition of tripeptide units occurs on a much faster time scale and cannot be observed experimentally. These results support an entropy-controlled zipper-like nucleation/growth mechanism for collagen triple-helix formation.
Keywords: collagen folding, nucleation mechanism, double jump, activation energy
Collagen folding is a complex process involving intermolecular and intramolecular interactions that lead to formation of the native triple helix. Folding and stability of collagen has been extensively studied over the last 25 years (for overview, see refs. 1–3). At low protein concentrations, the kinetics of triple-helix formation are limited by intermolecular association steps, whereas folding within a trimeric intermediate becomes rate-limiting at high protein concentrations. Compared with folding of globular proteins and of coiled-coil structures the concentration-independent folding steps of collagen are extremely slow (4). The triple-helical domains in collagens consist of Gly–Xaa–Yaa repeats with proline (Pro) and 4-hydroxyproline (Hyp) being the most frequent amino acids at positions Xaa and Yaa, respectively. In native collagen, all Gly–Pro and Xaa–Hyp peptide bonds are in the trans conformation, whereas in the unfolded state a significant fraction of cis isomers populates at each Gly–Pro and Xaa–Hyp peptide bond. cis to trans isomerization reactions at prolyl peptide bonds are the origin for the observed slow kinetics of triple-helix formation (3) as shown by their high activation energy [≈72 kJ/mol (5)] and on their acceleration by prolyl isomerases (6).
Another rate-determining step in collagen triple-helix formation is to bring the individual chains into correct register (7–9). During collagen folding in vitro, misalignment leads to concentration-dependent irreversible aggregation reactions. In vivo, this problem is circumvented by the presence of N- or C-terminal registration domains, which are usually cleaved off after formation of the correct triple helices (10–12). In type III and other collagens, the three chains are connected by disulfide bonds arranged in a knot-like structure (13). Introduction of the cysteine (Cys) knot into different collagen fragments and collagen model peptides resulted in monomeric molecules with disulfide-linked triple helices (14). All cross-linked structures showed reversible and concentration-independent kinetics of triple-helix formation. However, the folding kinetics in the cross-linked chains were complex and comprised fast processes occurring in the dead-time of the experiments (30 s) and of slow, prolyl-isomerization limited reactions (5). The fast process was interpreted as folding in regions devoid of cis residues. This process sets an upper limit for the rate of collagen folding, and its characterization would provide insight into the dynamics and molecular mechanism for triple-helix formation. It also would be interesting to compare the maximum rate of triple-helix formation with rates of related conformational transitions in proteins like α-helix formation or folding of two- or three-stranded α-helical coiled-coil structures. This information would be particularly valuable, because only little is known about the kinetics of formation of linear repetitive structures in proteins.
In the present work, we used trimeric fragments of various length derived from type III collagen containing the natural disulfide knot of this protein to eliminate concentration-dependent association steps. To study the fast process in the absence of prolyl isomerization reactions, we rapidly unfolded collagen at high guanidinium chloride (GdmCl) concentrations in a stopped-flow instrument and initiated refolding by diluting out denaturant before prolyl isomerization could occur (double-jump experiments). This technique allowed us to start refolding from collagen molecules with all Xaa–Pro peptide bonds in their native trans isomerization state and to characterize the process of triple-helix initiation.
Materials and Methods
Peptide Sequences. Peptides were either recombinantly expressed [(Col13Cys2)3 and (Col49Cys2)3] or chemically synthesized [(HypCol13Cys2)3]. The sequences of the peptides are as follows: (Col13Cys2)3, (GS GYP GPI GPP GPR GNR GER GSE GSP GHP GQP GPP GPP GAP GPCCGGG)3; (HypCol13Cys2)3, (GS GYO GPI GPO GPR GNR GER GSE GSO GHO GQO GPO GPO GAO GPCCGGG)3; and (Col49Cys2)3, (GS GYP GPP GPV GPA GKS GDR GES GPA GPA GAP GPA GSR GAP GPQ GPR GDK GET GER GAA GIK GHR GFP GNP GAP GSP GPA GQQ GAI GSP GPA GPR GPV GPS GPP GKD GTS GHP GPI GPP GPR GNR GER GSE GSP GHP GQP GPP GPP GAP GPC GGG)3. [O is 4(R)-hydroxyproline].
Recombinant Expression. The bacterial expression vector pHisMfCol13Cys2 (15) encoding the fusion protein consisting of the His-Tag sequence, the minifibritin domain, the thrombin cleavage site, and the collagen fragment Col13–Cys2 was used for the preparation of (Col13Cys2)3. The cDNA encoding the α1-chain of human type III collagen was a gift from Takako Sasaki (Max-Planck-Institut für Biochemie, Martinsried, Germany). The cDNA was used as a template to prepare the gene encoding the type III collagen fragment Col49Cys2, spanning residues GS-GYP-G874-G1023-G (numbering of mature human type III collagen) by PCR and cloned into the BamHI/EcoRI site of the bacterial expression vector pHisMf (15). Details of expression and purification are given in Supporting Text, which is published as supporting information on the PNAS web site.
Peptide Synthesis. HypCol13Cys2 was synthesized on an ABI433A peptide synthesizer with 0.25 mmol fluorenylmethoxycarbonyl (Fmoc)-Gly-PEG-PS resin, a 4-fold excess of Fmoc-amino acids and O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate as activating agent. Fmoc-Gly-Pro was used with the exception of the most C-terminal occurrence of this sequence. The Fmoc-amino acids carried the protection groups Cys-Trt, Hyp-t-Bu, Gln-Trt, His-Trt, Ser-t-Bu, Glu-O-t-Bu, Arg-Pbf, Asn-Trt, and Tyr-t-Bu. The peptide was cleaved off the resin and deprotected for 4 h at room temperature with 90% trifluoroacetic acid (TFA)/5% thioanisole/3% 1,2-ethanedithiol/2% anisole. The peptide was precipitated in cold ether, redissolved in H2O, and lyophilized. The reduced peptide was purified by RP-HPLC using a C18 column (Vydac, Hesperia, CA; 50 × 250 mm, 10- to 15-μm particle size, 300-Å pores) with an acetonitrile/water gradient and 0.1% TFA as ion-pairing agent.
Covalent Trimerization of the Peptides. Covalent trimerization of the chains of HypCol13Cys2 was achieved by controlled oxidation at 4°C following procedures described in ref. 16. Direct oxidation was not possible with Col13Cys2 and Col49Cys2 because of the low stability. Therefore, the trimerization domain minifibritin (bearing a His-tag at the N terminus) and a thrombin cleavage site were fused to the N terminus of the sequences. The minifibritin domain aligns and stabilizes the triple helix, which allows oxidation of the C-terminal Cys residues. The minifibritin domain was removed by thrombin cleavage after oxidation essentially as described in ref. 15. Col13Cys2 was extremely pure according to SDS/PAGE and mass spectroscopy, but Col49Cys2 was contaminated by noncovalently cross-linked and shortened fragments and therefore was further purified by hydroxyapatite chromatography. Purity and completeness of oxidation to trimers was tested in all peptides by SDS/PAGE under nonreducing conditions and by mass spectroscopy.
Equilibrium Measurements. Thermal unfolding of (HypCol13Cys2)3 was measured in a Cary 61 spectropolarimeter. Identical transitions were obtained upon heating and cooling with rates of 0.5°C/min. Because of slow equilibration, the nonhydroxylated fragments were measured by long-term incubation at given temperatures from both directions, i.e., heating and cooling (17). For GdmCl-induced unfolding transitions, the peptides were incubated at varying denaturant concentrations in 100 mM sodium acetate buffer (pH 4.75) until equilibration was reached. The data were fitted to a two-state model (18). The measurements were carried out in a DS62 spectropolarimeter (Aviv Associates, Lakewood, NJ).
Kinetic Measurements. All measurements were performed in 100 mM NaOAc (pH 4.75). Manual mixing experiments were performed in a DS62 spectropolarimeter (Aviv Associates). Stopped-flow single and double-mixing experiments were carried out in a PiStar spectropolarimeter (Applied Photophysics, Surrey, U.K.). In double-jump experiments at 3.7°C native protein was first unfolded for 30 s in 4.5 M GdmCl for (HypCol13Cys2)3, 1.5 M GdmCl for (Col13Cys2)3, or 3.6 M GdmCl for (Col49Cys2)3. This reaction leads to complete unfolding in all fragments. In a second mixing step, the protein was transferred to native conditions by diluting out the denaturant, and the resulting folding reactions was monitored by the change in far-UV CD. In double-jump experiments at higher temperatures, the unfolding time was reduced to 1 s at 25°C to compensate for faster prolyl isomerization.
The temperature dependence of the rate constants was analyzed according to the Arrhenius equation
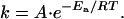 |
[1] |
Calculation of ΔASA. The difference in accessible surface area (ΔASA) was calculated by using the structure of a collagen model peptide (Protein Data Bank ID code 1CAG) (19) and the program molmol (20). For the unfolded state an extended conformation was assumed. The collagen model peptide contains mainly GPP units and thus differs from the sequence of type III collagen. However, because collagen structure is mainly stabilized by backbone interactions, the model peptide should allow an estimate of ΔASA occurring during folding of type III collagen.
Results and Discussion
Stability of the Collagen Fragments. We used three different model peptides to study fast processes during collagen folding. Col13Cys2 and Col49Cys2 are different-length peptides tailored after the sequence of human type III collagen (21). Col13Cys2 and Col49Cys2 were recombinantly expressed in Escherichia coli and consequently contain all Pro residues in a nonhydroxylated form. HypCol13Cys2 contains the same sequence as Col13Cys2 but has Hyp in all Yaa-position as in the natural sequence of type III collagen (21). All peptides contain the natural C-terminal disulfide knot sequence GPCCGGG, which produces monomeric molecules (Col13Cys2)3, (Col49Cys2)3, and (HypCol13Cys2)3 with covalently linked triple helices after oxidation (14).
The stability of the different fragments was compared by thermal unfolding transitions detected by the change in the characteristic positive CD-band at 222 nm (Fig. 1A). (Col13Cys2)3, is the least stable fragment with a midpoint of the transition (Tm) at 9.5 ± 0.1°C. (Col49Cys2)3 is significantly more stable (Tm = 18.1 ± 0.2°C) in accordance with its larger chain length. The highest thermal stability was found for (HypCol13Cys2)3, which can be attributed to the stabilizing effect of the Hyp residues. Its Tm of 35.0 ± 0.1°C is close to the Tm of ≈38°C for naturally occurring type III collagen. The increased stability of triple helices containing Hyp can be ascribed to an inductive effect of the OH group of hydroxyproline (22).
Fig. 1.

Effect of temperature and GdmCl on collagen stability. (A) Comparison of the thermal denaturation curves for (Col13Cys2)3 (red), (Col49Cys2)3 (blue), and (HypCol13Cys2)3 (black) (filled symbols, heating; open symbols, cooling). Tm = 9.5°C, 18.1°C, and 35.0°C, respectively. Analyzing the data with a two-state model gives values for ΔH0v.H. of 477 ± 5kJ/mol (12.9 kJ/mol), 834 ± 150 kJ/mol (5.75 kJ/mol), and 441 ± 10 kJ/mol (11.9 kJ/mol), respectively. In brackets are the values per interacting tripeptide unit (3). (B) GdmCl-induced unfolding transitions of (HypCol13Cys2)3 at 5°C (green), 10°C (blue), 15°C (violet), 20°C (red), and 25°C (orange). The transition curves were fitted with a global meq of 10.8 ± 0.8 (kJ/mol)·M–1. The resulting free energies extrapolated to zero denaturant [ΔG0(H2O)] show a linear temperature dependence (Inset) with an intersection at ΔG0 = 0of Tm = 35.3°C.
The stability of the (HypCol13Cys2)3 fragment against unfolding by denaturants was measured by GdmCl-induced unfolding transitions at different temperatures (Fig. 1B). The transitions could be fitted to a two-state model. The stability extrapolated to zero denaturant [ΔG0(H2O)] decreases linearly with temperature between 5 and 25°C (see Inset) indicating identical specific heat capacities (Cp) for the native and unfolded state (ΔCp ≈ 0). This finding is in agreement with results from other naturally occurring collagen sequences (23). Extrapolation to ΔG0(H2O) = 0 yields Tm of 35.3 ± 0.1°C (Fig. 1B Inset), which is virtually identical to the Tm value obtained from the thermal unfolding transition. The change in ΔG0 with [GdmCl] is temperature-independent and was fitted with a global meq value {meq = (∂ΔG0)/(∂[GdmCl])} of 10.8 ± 0.8 (kJ/mol)·M–1. For globular proteins, meq was found to correlate with the ΔASA between native and unfolded state (24). For unfolding of a collagen triple helix consisting of 37 interacting tripeptide units [(3 × 13) – 2], a ΔASA of ≈6,000 Å2 is expected (see Materials and Methods). Based on the correlations found in globular proteins a ΔASA of 6,000 Å2 should result in an meq of ≈9 (kJ/mol)·M–1 (24), which is similar to the value found for (HypCol13Cys2)3. This result is surprising because unfolding of (HypCol13Cys2)3 is associated with only little change in Cp. In globular proteins, a strong correlation between ΔCp and ΔASA and between ΔCp and meq was found (24). The difference in ASA between native and unfolded collagen is mainly due to exposure of the polypeptide backbone during unfolding, because only few side-chain interactions exist in collagen triple helices. Thus, our results suggest that GdmCl mainly binds to the polypeptide backbone, in agreement with recent conclusions from studies on the effect of denaturants on peptide dynamics (25). The change in heat capacity, in contrast, seems to be mainly based on the change in solvent exposure of the side chains, in particular of hydrophobic residues. In globular proteins backbone and side-chain exposure upon unfolding are correlated, whereas they are not correlated in collagen triple-helix unfolding.
Folding Kinetics of Disulfide-Linked Collagen. Because of the large number of prolyl residues in collagen, the majority of equilibrium-unfolded molecules contain one or more cis peptide bonds that have to undergo cis to trans isomerization reactions during refolding. When folding of the (HypCol13Cys2)3 fragment is started from equilibrium-unfolded protein, the majority of change in CD signal occurs in a slow reaction with a time constant (τ = 1/λ) of 1,070 ± 30sat3.7°C(λ = 9.3 × 10–4 s–1; Fig. 2A). This reaction is slightly slower than expected for isomerization reactions at single Gly–Pro and Pro–Pro peptide bonds, which have rate constants for cis → trans isomerization of 2.9 × 10–3 and 0.6 × 10–3 s–1, respectively, at 4°C (26). The slower kinetics are compatible with calculations showing that the apparent rate constant for protein folding decreases with the number of prolyl residues (4, 27). Stopped-flow CD measurements reveal that a fraction (20–30%) of the change in the CD signal occurs on a much faster time scale with two time constants of 53 ± 10 ms and 500 ± 110 ms (Fig. 2B). The amplitude of these reactions is much higher than the expected fraction of molecules with all-trans peptide bonds. The fast reactions therefore also may contain contributions from triple-helix formation in short regions that are devoid of cis-isomers.
Fig. 2.

Refolding kinetics of (HypCol13Cys2)3 at 25°C in the presence of 0.75 M GdmCl starting from equilibrium-unfolded protein. (A) Slow folding reaction [λ1 = 9.4 (±1.0) × 10–4 s–1] initiated by manual mixing. (B) Fast reaction measured in stopped-flow experiments. The kinetics are best described by a double-exponential function with rate constants of 19 ± 3 and 2.0 ± 0.6 s–1. The amplitudes of the two reactions cannot be compared directly because of different experimental setups required in the two experiments. The amplitudes of the fast reactions account for 20–30% of the total signal change.
Refolding Kinetics Starting from all-trans Chains. To investigate the fast processes during collagen folding in more detail, we started from unfolded chains with all peptide bonds in the native trans conformation by applying stopped-flow double-jump experiments (28). Native collagen was first unfolded at high concentrations of GdmCl until unfolding was complete. After this short unfolding pulse the majority of peptide bonds are still in the native trans conformation, because prolyl isomerization is slow compared with unfolding. Refolding then was initiated by a second mixing step to native conditions, and the folding reaction was monitored by the change in the CD signal at 231 nm. Fig. 3 shows folding starting from all-trans chains for the three model peptides. Triple-helix formation in all fragments occurs on the millisecond to seconds time scale. Folding of (HypCol13Cys2)3 is too fast to be resolved by stopped-flow mixing at GdmCl concentrations of <0.75 M. The kinetics at GdmCl concentrations of ≥0.75 M could be fitted by the sum of two exponentials. In the presence of 0.75 M GdmCl the fast process shows a time constant (τ = 1/λ) of 29 ms at 3.7°C and accounts for ≈90% of the measurable signal change (Fig. 3). This rate constant is similar to that of the fastest process measured in direct refolding (Fig. 2B). The remaining molecules fold on a much slower time scale with a time constant of 1,300 ± 250 s, which is identical to that observed for refolding starting from equilibrium unfolded protein. The amplitude of the slow reaction increases with increasing unfolding time on a time scale compatible with prolyl isomerization reactions (data not shown). This result shows that the slow reactions reflects folding of a small fraction of chains that have already undergone prolyl isomerization during the unfolding pulse. Because of the fast kinetics of this fragment at low denaturant concentrations, the extrapolation of the initial CD signal to t = 0is error-prone, and we cannot rule out the presence of a small fraction of a submillisecond burst phase reaction at GdmCl concentrations of <2 M.
Fig. 3.

Stopped-flow double-jump refolding kinetics starting from an unfolded state with all-trans peptide bonds. Folding of (HypCol13Cys2)3 is shown at 3.7°C and 25°C. At both temperatures a single kinetic phase is observed with rate constants of 28.0 and 35.2 s–1, respectively. Folding of (Col13Cys2)3 at 0.134 M GdmCl and (Col49Cys2)3 at 0.33 M GdmCl occurs in two kinetic phases with A1 = 0.11; λ1 = 0.47 s–1; A2 = 0.89, λ2 = 9.6 s–1; and A1 = 0.26; λ1 = 0.15 s–1; A2 = 0.74; λ2 = 4.7 s–1, respectively.
Triple-helix formation in the nonhydroxylated peptides (Fig. 3) is significantly slower compared with (HypCol13Cys2)3. The fast events in the refolding kinetics of (Col13Cys2)3 and (Col49Cys2)3 are best described by the sum of two exponentials with the major signal change occurring in the fastest process. For (Col13Cys2)3 time constants of 70 ± 4 ms (83% amplitude) and 630 ± 70 ms (17% amplitude) were obtained in the presence of 0.13 M GdmCl. The kinetics of (Col49Cys2)3 shows time constants of 200 ± 6 ms (75% amplitude) and 5.6 ± 0.7 s (25% amplitude) in the presence of 0.33 M GdmCl. Both fast reactions are at least 3–4 orders of magnitude faster than folding of equilibrium unfolded protein (cf. Fig. 2).
Denaturant-Dependence of Triple-Helix Formation. To determine the folding rate constant in the absence of denaturant and gain information on the structure of the transition state for triple-helix formation, we measured the GdmCl-dependence of the folding kinetics for the three fragments at 3.7°C (Fig. 4). The kinetics of the (Col13Cys2)3 peptide could only be measured at a very low denaturant concentration due to the low stability of this fragment (see Fig. 1A). The two rate constants observed for (Col13Cys2)3 folding at 0.13 M GdmCl are displayed for comparison. Analysis and interpretation of the results is most straightforward for (HypCol13Cys2)3 due to the observation of a single fast rate constant (λ) indicating a two-state folding reaction. The logarithm of the rate constant for this reaction increases linearly with decreasing denaturant concentration. Because the native state of (HypCol13Cys2)3 is significantly more stable than the unfolded state under the applied experimental conditions, the measured apparent rate constant λ = kf + ku nearly exclusively represents the rate constant for folding (kf). Thus, the experimentally determined rate constant allows us to determine kf in the absence of denaturant [kf(H2O)] by extrapolation to zero denaturant, which gives a value of kf(H2O) = 113 ± 20 s–1. Comparing the effect of denaturants on kf (mf value)
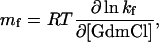 |
[2] |
with the corresponding effect on Keq (meq value)
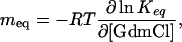 |
[3] |
allows a structural characterization of the transition state according to the Leffler relationship (29, 30).
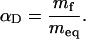 |
[4] |
Because protein folding m values were found to correlate with the change in ASA (24), αD is a measure for the relative change in ASA between unfolded and transition states (30). Comparing mf of 2.9 ± 0.3 (kJ/mol)·M–1 (Fig. 3) with the meq of 10.8 ± 0.7 (kJ/mol)·M–1 (Fig. 1B) results in an αD of 0.27. This result suggests that 27% of the total change in ASA between native and unfolded collagen has already occurred in the transition state for triple-helix formation. Collagens form a highly repetitive and regular structure, and the only major changes in ASA during folding occur at the polypeptide backbone. Thus, the αD value can be used to estimate the degree of triple-helix formation in the transition state. The observed αD of 0.27 for (HypCol13Cys2)3 suggests that the rate-limiting step in (HypCol13Cys2)3 folding is represented by bringing 10 consecutive tripeptide units (3.3 in each chain) into a triple-helical conformation. The following addition of tripeptide units must be much faster because no additional kinetic phases are observed in this fragment. This model is based on the single sequence approximation, which assumes that a triple helix will grow at a nucleated site rather than form a second nucleus.
Fig. 4.

Effect of GdmCl and temperature on the rate constants for triple-helix formation. (A) Denaturant dependence of the apparent rate constants (λ) for folding of the different collagen fragments starting from the all-trans conformation. Linear extrapolation of ln(λ) to zero denaturant concentration yields values for kf(H2O) of 113 ± 20 and 13 ± 2 s–1 for the fastest observable rate constants for (HypCol13Cys2)3 (•) and (Col49Cys2)3 (▴) at 3.7°C, respectively, with mf of 2.9 ± 0.3 (kJ/mol)·M–1 for (HypCol13Cys2)3 and 6.4 ± 0.2 (kJ/mol)·M–1 for (Col49Cys2)3. For comparison, the fastest kinetic reaction for folding of (Col13Cys2)3 at 0.13 M GdmCl (▪) is displayed. Additionally, the minor slow kinetic phases observed for (Col13Cys2)3 (□) and (Col49Cys2)3 (▵) peptides are shown. This reaction is virtually independent of GdmCl concentration in the (Col49Cys2)3 peptide. (B) Temperature dependence of (HypCol13Cys2)3 folding starting from the unfolded state with all-trans peptide bonds. A fit to the Arrhenius equation (Eq. 1) yields an activation energy (Ea) for the fast refolding reaction (•) of 8.8 ± 6.4 kJ/mol and an preexponential factor (A) of 1,700 ± 1,000 s–1. Additionally, the rate constant of the minor slow phase observed for (HypCol13Cys2)3 folding in double-jump experiments is shown (□) and compared with the slow Proisomerization limited reaction starting from equilibrium-unfolded protein (○). The results indicate that the two reactions are identical and have an activation energy of 71.7 ± 0.3 kJ/mol [A = 2.6 (± 0.4) × 1010 s–1].
In contrast to (HypCol13Cys2)3, refolding of (Col13Cys2)3 and (Col49Cys2)3 shows two fast kinetic phases. The GdmCl-dependence of the faster phase in (Col49Cys2)3 has an mf of 6.4 ± 0.2 (kJ/mol)·M–1 and kf(H2O) of 13 ± 2 s–1. The mf value is significantly larger than that of 2.9 ± 0.3 (kJ/mol)·M–1 in (HypCol13Cys2)3. Assuming that formation of triple-helical segments leads to the same ΔASA in hydroxylated and nonhydroxylated peptides, the observed mf of 6.4 ± 0.2 (kJ/mol)·M–1 for the (Col49Cys2)3 peptide corresponds to 22 tripeptide units (7.3 per chain) in a triple-helical conformation in the transition state for folding of (Col49Cys2)3.
Comparison of the kinetics for (Col13Cys2)3 with the extrapolated value for (Col49Cys2)3 shows that the rate constant of the faster kinetic phase is virtually independent of chain length. This finding argues for the same rate-limiting step for folding of the different-length peptides and supports a nucleation mechanism. The amplitude and the rate constant of the minor kinetic phase of the (Col49Cys2)3 fragment are independent of GdmCl concentration, which suggests that this process is not associated with a significant change in ASA. This behavior is typical for amide bond isomerization reactions in globular proteins (31). The observed time constant of 5.6 s at 3.7°C is much faster than for Pro isomerization reactions but is compatible with non-Pro isomerization reactions (31, 32). This model is supported by the increased amplitude of this reaction in the longer peptide (25% vs. 11%). However, this reaction is ≈8 times faster in the shorter (Col13Cys2)3 peptide compared with the (Col49Cys2)3 fragment, which also may indicate that it is due to local structural rearrangements in the folded triple helix.
Temperature Dependence of Triple-Helix Formation. To obtain information on the barriers for triple-helix formation, we measured folding of the (HypCol13Cys2)3 fragment in stopped-flow double-jump experiments at different temperatures between 3.7 and 25°C. The resulting Arrhenius plot is shown in Fig. 4B. The fast reaction, which represents triple-helix formation starting from all-trans chains is virtually independent of temperature (see also Fig. 3) with an Arrhenius activation energy of 8.8 ± 6.4 kJ/mol (≈3 RT) and an Arrhenius preexponential factor (A) of 1,700 ± 1,000 s–1. This result suggests that triple-helix formation in the cross-linked peptide is limited nearly exclusively by an entropic barrier. For comparison, measurements of the temperature dependence of folding of the same peptides starting from equilibrium-unfolded chains yields an activation energy of 72.8 ± 0.3 kJ/mol and a preexponential factor (A) of 2.6 (± 0.4) × 1010 s–1 (Fig. 4), which are typical values for Pro isomerization reactions (33). Similar values have been observed for many collagens or collagen model peptides (3).
Properties of the Transition State for Triple-Helix Formation. Collagen folding is a sterically demanding reaction even in cross-linked peptides. It requires the formation of the correct hydrogen bonds between three peptide chains containing Gly–Xaa–Yaa repeats (3). Folding of collagen with all peptide bonds in the native trans isomerization state was proposed to proceed by means of a zipper-like mechanism in which nucleation of the triple helix is the slow rate-limiting step, which is followed by fast growth steps (4). The use of stopped-flow double-jump experiments in cross-linked collagen fragments allowed us to characterize the process of triple-helix formation starting from all-trans chains. Folding of the hydroxylated collagen fragment (HypCol13Cys2)3 shows single-exponential kinetics, which are virtually independent of temperature. This result is in agreement with a nucleation process that is limited by entropic search for a critical size of triple-helical segments that form a sufficient number of interactions to compensate for the loss in conformational entropy. The results from the effect of GdmCl on folding and stability indicate that the nucleus for triple-helix formation in (HypCol13Cys2)3 consists of 10 consecutive tripeptide units (3.3 in each chain) in a triple-helical conformation. The nucleation step has a time constant of 8.5 ms at zero denaturant concentration. The following addition of further triple-helical segments must be much faster than the nucleation reaction because no second kinetic phase is observed. It is very likely that nucleation takes place near the disulfide knot because the local concentration of interacting tripeptide segments is highest in this region. It was estimated to be in the millimolar range (14). The Arrhenius preexponential factor for triple-helix nucleation is 1,700 ± 1,000 s–1 (Fig. 4B). It contains contributions from the maximum rate constant for chain dynamics and from entropic barriers encountered during triple-helix nucleation. Recent results showed that trans-Pro residues slow chain dynamics compared with other amino acids, but Pro-containing peptides can still explore conformational space on the 50–100 ns time scale (34). The low preexponential factor most likely originates in major entropic costs for formation of a large nucleus, in which 30 aa (10 tripeptide units) have to adopt a specific backbone conformation, which will lead to a major loss in conformational entropy.
Folding of the nonhydroxylated collagen fragments is significantly slower and more complex compared with the hydroxylated peptide. The fastest process in these peptides is independent of chain length, which supports the idea that this reaction represents a nucleation reaction. The nucleus in the nonhydroxylated peptide is larger than in the hydroxylated peptide (22 vs. 10 tripeptide units). This result is compatible with the lower stability of a tripeptide unit in the nonhydroxylated peptides, which requires formation of more interactions during nucleation to compensate for the loss in conformational entropy. This observation is in agreement with Hammond behavior, which, applied to protein folding, postulates that the transition state of a folding reaction becomes more structured if the native state is destabilized (30). The origin of the second fast phase observed in folding of the nonhydroxylated peptides remains unclear. The insensitivity of this reaction toward GdmCl shows that it is not associated with major changes in backbone exposure and argues for local structural changes occurring during this process. The rate constant of 0.2–1 s–1 for this reaction at 3.7°C may indicate a non-Pro isomerization reaction.
Comparison with Other Fast Processes in Protein Folding. The rate constant for folding of (HypCol13Cys2)3 is comparable with the fastest folding reactions reported for globular proteins of similar size. It would be interesting to compare our results with nucleation reactions in other regular protein structures. For α-helix formation a zipper model has been proposed similar to the model for formation of the collagen triple helix (35, 36). However, the kinetics of α-helix formation starting from an all-coil state have not been measured. Studies on helix–coil dynamics applied various relaxation methods on unfolding of model helices (37–39). For helix–coil transitions in long homo-polypeptides relaxation times in the range of 10 ns to 1 μs were observed, depending on the nature of the side chains and the solvent. From these values growth rate constants of 108 to 1010 s–1 were calculated (40). Similar results were obtained in temperature-jump studies on short Ala-based model peptides (41). However, relaxation experiments do not allow a model-free determination of nucleation rate constants (42). Estimated time constants for helix formation range from 100 ns to 1 μs, which would be 4–5 orders of magnitude faster than the nucleation step measured for triple-helix formation in (HypCol13Cys2)3. The rather slow triple-helix nucleation in collagen might partly be associated with the slower chain dynamics around Pro residues (34), which reduces the Arrhenius preexponential factor. The major contributions, however, most likely result from the need to bring three adjacent strands in the correct backbone conformation and the significantly larger nucleus for triple-helix formation compared with nucleation of an α-helix, for which formation of a single turn was proposed to be rate-limiting.
Based on theoretical considerations it was proposed that the nucleation barrier for α-helix formation contains enthalpic contributions due to unfavorable dipole–dipole interactions in the polypeptide backbone (43). Our results show that collagen triple-helix nucleation represents nearly exclusively an entropic barrier. This finding might be due to the favorable dipole–dipole interactions (43, 44) and solvation energy (45) in the extended chain conformations required for triple-helix initiation.
The α-helical coiled-coil motif represents another well-studied linear and regular structural element in proteins. Folding of the dimeric GCN4 fragment was studied in a disulfide-bonded monomeric variant to eliminate the concentration-dependent association reactions. It showed a rate constant for folding at ≈200 s–1 in the presence of 2.5 M GdmCl at 20°C (46), which is slightly faster than folding of (HypCol13Cys2)3. However, folding of GCN4 only requires the interaction of two chains. It is unknown whether folding of GCN4 represents a nucleation-limited zipper-like process or whether it encounters additional barriers during folding.
Implications for the Mechanism of Triple-Helix Formation. Folding of collagen starting from equilibrium unfolded chains is limited by slow prolyl isomerization reactions. For type III collagen, which contains ≈20% Xaa–Pro and Yaa–Hyp bonds, the average interval between two peptide bonds in cis configuration was estimated to be 30 tripeptide units (4). Because hydroxylated peptides as short as 15 aa per strand were shown to form stable triple helices, formation of triple-helical stretches could occur rapidly in the regions between two cis-peptide bonds. Fast structure formation in regions with all-trans peptide bonds is compatible with the observation of a minor fast folding reaction during refolding of equilibrium-unfolded (HypCol13Cys2)3 fragment (Fig. 2). Our results show that this fast process occurs with a rate constant of 113 s–1 and requires 10 consecutive tripeptide units to adopt a triple-helical structure in hydroxylated collagen sequences. The triple helix grows until a cis bond is encountered in either of the three chains. The final slow steps in triple-helix formation are limited by slow prolyl isomerization reactions until all peptide bonds are in the native trans isomerization state. The slow isomerization reactions are ≈3–4 orders of magnitude slower than triple-helix formation in regions devoid of cis-prolyl bonds.
Supplementary Material
Acknowledgments
We thank Dr. R. L. Baldwin for discussion and comments on the manuscript. This work was funded by grants from the Swiss National Science Foundation and the University of Basel.
Author contributions: A.B., J.E., and H.P.B. designed research; A.B., S.B., and H.P.B. performed research; S.B. and H.P.B. contributed new reagents/analytic tools; and A.B. and T.K. analyzed data and wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: Hyp, 4-hydroxyprolines; GdmCl, guanidinium chloride.
References
- 1.Brodsky, B. & Persikov, A. V. (2005) Adv. Protein Chem. 70, 301–339. [DOI] [PubMed] [Google Scholar]
- 2.Engel, J. & Bächinger, H. P. (2005) Top. Curr. Chem. 247, 7–34. [Google Scholar]
- 3.Bächinger, H. P. & Engel, J. (2005) in Handbook of Protein Folding, eds. Buchner, J. & Kiefhaber, T. (Wiley-VCH, Weinheim, Germany), Vol. 2, pp. 1059–1110. [Google Scholar]
- 4.Bächinger, H. P., Bruckner, P., Timpl, R., Prockop, D. J. & Engel, J. (1980) Eur. J. Biochem. 106, 619–632. [DOI] [PubMed] [Google Scholar]
- 5.Bächinger, H. P., Bruckner, P., Timpl, R. & Engel, J. (1978) Eur. J. Biochem. 90, 605–613. [DOI] [PubMed] [Google Scholar]
- 6.Bächinger, H. P. (1987) J. Biol. Chem. 262, 17144–17148. [PubMed] [Google Scholar]
- 7.Engel, J. & Prockop, D. J. (1991) Annu. Rev. Biophys. Biophys. Chem. 20, 137–152. [DOI] [PubMed] [Google Scholar]
- 8.Boudko, S. P., Frank, S., Kammerer, R. A., Stetefeld, J., Schulthess, T., Landwehr, R., Lustig, A., Bächinger, H. P. & Engel, J. (2002) J. Mol. Biol. 317, 459–470. [DOI] [PubMed] [Google Scholar]
- 9.Baum, J. & Brodsky, B. (1999) Curr. Opin. Struct. Biol. 9, 122–128. [DOI] [PubMed] [Google Scholar]
- 10.Prockop, D. J., Kadler, K. E., Hojima, Y., Constantinou, C. D., Dombrowski, K. E., Kuivaniemi, H., Tromp, G. & Vogel, B. (1988) Ciba Found. Symp. 136, 142–160. [DOI] [PubMed] [Google Scholar]
- 11.Fessler, J. H. & Fessler, L. I. (1978) Annu. Rev. Biochem. 47, 129–162. [DOI] [PubMed] [Google Scholar]
- 12.McAlinden, A., Crouch, E. C., Bann, J. G., Zhang, P. & Sandell, L. J. (2002) J. Biol. Chem. 277, 41274–41281. [DOI] [PubMed] [Google Scholar]
- 13.Bruckner, P., Bächinger, H. P., Timpl, R. & Engel, J. (1978) Eur. J. Biochem. 90, 595–603. [DOI] [PubMed] [Google Scholar]
- 14.Frank, S., Kammerer, R. A., Mechling, D., Schulthess, T., Landwehr, R., Bann, J., Guo, Y., Lustig, A., Bächinger, H. P. & Engel, J. (2001) J. Mol. Biol. 308, 1081–1089. [DOI] [PubMed] [Google Scholar]
- 15.Boudko, S. P. & Engel, J. (2004) J. Mol. Biol. 335, 1289–1297. [DOI] [PubMed] [Google Scholar]
- 16.Mann, K., Mechling, D. E., Bachinger, H. P., Eckerskorn, C., Gaill, F. & Timpl, R. (1996) J. Mol. Biol. 261, pp. 255–266. [DOI] [PubMed] [Google Scholar]
- 17.Persikov, A. V., Xu, Y. & Brodsky, B. (2004) Protein Sci. 13, 893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santoro, M. M. & Bolen, D. W. (1988) Biochemistry 27, 8063–8068. [DOI] [PubMed] [Google Scholar]
- 19.Bella, J., Eaton, M., Brodsky, B. & Berman, H. M. (1994) Science 266, 75–81. [DOI] [PubMed] [Google Scholar]
- 20.Koradi, R., Billeter, M. & Wüthrich, K. (1996) J. Mol. Graphics 14, 51–55. [DOI] [PubMed] [Google Scholar]
- 21.Glanville, R. W., Allmann, H. & Fietzek, P. P. (1976) Hoppe-Seylers Z. Physiol. Chem. 357, 1663–1665. [PubMed] [Google Scholar]
- 22.Jenkins, C. L. & Raines, R. T. (2002) Nat. Prod. Rep. 19, 49–59. [DOI] [PubMed] [Google Scholar]
- 23.Privalov, P. L. (1982) Adv. Protein Chem. 35, 1–104. [PubMed] [Google Scholar]
- 24.Myers, J. K., Pace, C. N. & Scholtz, J. M. (1995) Protein Sci. 4, 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Möglich, A., Krieger, F. & Kiefhaber, T. (2004) J. Mol. Biol. 345, 153–162. [DOI] [PubMed] [Google Scholar]
- 26.Reimer, U., Scherer, G., Drewello, M., Kruber, S., Schutkowski, M. & Fischer, G. (1998) J. Mol. Biol. 279, 449–460. [DOI] [PubMed] [Google Scholar]
- 27.Kiefhaber, T., Kohler, H. H. & Schmid, F. X. (1992) J. Mol. Biol. 224, 217–229. [DOI] [PubMed] [Google Scholar]
- 28.Brandts, J. F., Halvorson, H. R. & Brennan, M. (1975) Biochemistry 14, 4953–4963. [DOI] [PubMed] [Google Scholar]
- 29.Leffler, J. E. (1953) Science 117, 340–341. [DOI] [PubMed] [Google Scholar]
- 30.Kiefhaber, T., Sánchez, I. E. & Bachmann, A. (2005) in Protein Folding Handbook, eds. Buchner, J. & Kiefhaber, T. (Wiley/VCH, Weinheim, Germany), Vol. 1, pp. 411–453. [Google Scholar]
- 31.Pappenberger, G., Aygün, H., Engels, J. W., Reimer, U., Fischer, G. & Kiefhaber, T. (2001) Nat. Struct. Biol. 8, 452–458. [DOI] [PubMed] [Google Scholar]
- 32.Scherer, G., Kramer, M. L., Schutkowski, M., Reimer, U. & Fischer, G. (1998) J. Am. Chem. Soc. 120, 5568–5574. [Google Scholar]
- 33.Harrison, R. K. & Stein, R. L. (1992) J. Am. Chem. Soc. 114, 3464–3471. [Google Scholar]
- 34.Krieger, F., Möglich, A. & Kiefhaber, T. (2005) J. Am. Chem. Soc. 127, 3346–3352. [DOI] [PubMed] [Google Scholar]
- 35.Qian, H. & Schellman, J. A. (1992) J. Phys. Chem. 96, 3978–3994. [Google Scholar]
- 36.Zimm, B. H. & Bragg, J. K. (1959) J. Chem. Phys. 31, 526–535. [Google Scholar]
- 37.Schwarz, G. & Seelig, J. (1968) Biopolymers 6, 1263–1277. [DOI] [PubMed] [Google Scholar]
- 38.Cummings, A. L. & Eyring, E. M. (1975) Biopolymers 14, 2107–2114. [Google Scholar]
- 39.Wada, A., Tanaka, T. & Kihara, H. (1972) Biopolymers 11, 587–605. [DOI] [PubMed] [Google Scholar]
- 40.Gruenewald, B., Nicola, C. U., Lustig, A., Schwarz, G. & Klump, H. (1979) Biophys. Chem. 9, 137–147. [DOI] [PubMed] [Google Scholar]
- 41.Thompson, P., Eaton, W. & Hofrichter, J. (1997) Biochemistry 36, 9200–9210. [DOI] [PubMed] [Google Scholar]
- 42.Schwarz, G. (1965) J. Mol. Biol. 11, 64–77. [DOI] [PubMed] [Google Scholar]
- 43.Brant, D. A. & Flory, P. J. (1965) J. Am. Chem. Soc. 87, 663–664. [Google Scholar]
- 44.Brant, D. A. & Flory, P. J. (1965) J. Am. Chem. Soc. 87, 2791–2800. [Google Scholar]
- 45.Baldwin, R. L. (2005) in Protein Folding Handbook, eds. Buchner, J. & Kiefhaber, T. (Wiley/VCH, Weinheim, Germany), Vol. 1, pp. 127–162. [Google Scholar]
- 46.Moran, L. B., Schneider, J. P., Kentsis, A., Reddy, G. A. & Sosnick, T. R. (1999) Proc. Natl. Acad. Sci. USA 96, 10699–10704. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.