

Abstract
Cyanide oxygenase (CNO) from Pseudomonas fluorescens NCIMB 11764 catalyzes the pterin-dependent oxygenolytic cleavage of cyanide (CN) to formic acid and ammonia. CNO was resolved into four protein components (P1 to P4), each of which along with a source of pterin cofactor was obligately required for CNO activity. Component P1 was characterized as a multimeric 230-kDa flavoprotein exhibiting the properties of a peroxide-forming NADH oxidase (oxidoreductase) (Nox). P2 consisted of a 49.7-kDa homodimer that showed 100% amino acid identity at its N terminus to NADH peroxidase (Npx) from Enterococcus faecalis. Enzyme assays further confirmed the identities of both Nox and Npx enzymes (specific activity, 1 U/mg). P3 was characterized as a large oligomeric protein (∼300 kDa) that exhibited cyanide dihydratase (CynD) activity (specific activity, 100 U/mg). Two polypeptides of 38 kDa and 43 kDa were each detected in the isolated enzyme, the former believed to confer catalytic activity based on its similar size to other CynD enzymes. The amino acid sequence of an internal peptide of the 43-kDa protein was 100% identical to bacterial elongation factor Tu, suggesting a role as a possible chaperone in the assembly of CynD or a multienzyme CNO complex. The remaining P4 component consisted of a 28.9-kDa homodimer and was identified as carbonic anhydrase (specific activity, 2,000 U/mg). While the function of participating pterin and the roles of Nox, Npx, CynD, and CA in the CNO-catalyzed scavenging of CN remain to be determined, this is the first report describing the collective involvement of these four enzymes in the metabolic detoxification and utilization of CN as a bacterial nitrogenous growth substrate.
From its role as an important prebiotic reactant to its notorious reputation as a genocidal and homicidal agent, the significance of cyanide (referred to herein as CN and indicating the free form, which exists as either CN− or HCN) to biology has a long history. However, gaps in our understanding of how this toxic substance is tolerated and metabolized in nature remain. Effective means of detoxifying cyanide have presumably evolved given the preponderance of cyanogenic organisms in nature. For example, a large number of plant species (∼2,000) as well as some fungi and bacteria are known to be cyanogenic (25, 28). Bacteria exhibit a range of natural resistance to cyanide related in part to their ability to synthesize cyanide-detoxifying enzymes. The added ability to assimilate metabolites derived therefrom accounts further for the unusual capacity of some bacteria to grow on CN as a provisionary nitrogen source (14). Pseudomonas fluorescens NCIMB 11764 is one such organism that grows at concentrations of CN equivalent to ≤300 μM, above which growth is inhibited (10, 16). CN utilization requires that the substrate be enzymatically converted to ammonia, which is then assimilated by established pathways (9, 17). In P. fluorescens NCIMB 11764, this occurs by a novel enzymatic mechanism in which CN is cleaved oxygenatively to give formate and ammonia via reactions involving formamide as an inferred intermediate (equations 1 and 2) (6). The responsible enzyme, variously described as cyanide oxygenase (CNO), is located in the cytosolic fraction of cells induced with CN and requires both reduced pyridine nucleotide (NADH) and a source of reduced pterin as a cofactor (6, 15).
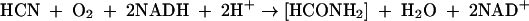 |
(1) |
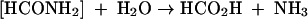 |
(2) |
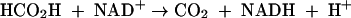 |
(3) |
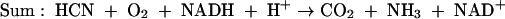 |
(4) |
Formate is further oxidized by a soluble formate dehydrogenase (FDH) simultaneously elevated in CN-induced cells (equation 3). Together, these two enzymes accomplish the complete oxidation of CN to carbon dioxide and ammonia, in the process consuming one molecule each of NADH and oxygen (equation 4). CNO represents the first enzyme of its kind to utilize molecular oxygen to catalyze the enzymatic cleavage of CN. It should be noted that while there is no net change between the oxidation state of the carbon atom residing in the reactants and products (+2 for both CN and formamide) (equations 1 and 2), a more oxidized substance is presumably transiently formed (e.g., isocyanate [HNCO], in which carbon is +4), since substrate conversion proceeds by a monooxygenative mechanism (6, 29). That CNO is absolutely required for growth is supported by studies showing that mutants devoid of enzyme activity cannot grow (R. Fernandez, H. Wessler, R. Benjamin, and D. Kunz, Abstr. 101st Gen. Meet. Am. Soc. Microbiol., abstr. K106, 2001). This report extends work reported previously (6) on the partial purification of the enzyme. We now show that CNO is comprised of four separate metabolic enzymes having functions related to oxidative stress and CN degradation. Together these enzymes facilitate the rapid scavenging and adventitious utilization of CN as a nitrogenous bacterial growth substrate.
MATERIALS AND METHODS
Chemicals.
KCN (97%) was obtained from Sigma/Aldrich. K14CN (1.9 GBq/mmol) was purchased from Perkin Elmer Life Sciences. CN solutions were prepared immediately before use and handled according to Environmental Protection Agency and the Texas Commission on Environmental Quality standards. 7,8-Dihydro-l-biopterin (dihydrobiopterin) and other pterin compounds were purchased from Schirck's Laboratory (Jona, Switzerland). All other chemicals were obtained commercially and used without further purification.
Growth of bacteria and preparation of cell extracts.
The bacterium used in this study was Pseudomonas fluorescens strain NCIMB 11764 (6, 10, 17). Cells were grown in minimal glucose-ammonia medium and induced for enzyme activity with 0.1 mM KCN as earlier described (6). Cells were harvested and washed in 50 mM Na2HPO4-KH2PO4 (Na-K phosphate buffer [pH 7.0]) and frozen at −80°C. Frozen cells were broken in a chilled (4°C) French press (20,000 lb/in2), and enzyme activity was recovered in the cytosol.
Separation of cyanide oxygenase into four separate enzyme fractions.
All purification steps were performed at 9 to 12°C with ÄKTA (Amersham-Pharmacia) fast protein liquid chromatography. In step 1, concentrated cell extracts (330 mg protein) obtained by passage of high-speed supernatants at 150,000 × g through a Centriprep-10 ultraconcentrator (nominal molecular mass cutoff, 10 kDa; Amicon) were applied to a Source 30Q (Amersham-Pharmacia) anion-exchange column (10 by 24 cm) equilibrated with 50 mM piperazine buffer, pH 10.0. A linear gradient of 0 to 0.5 M Na2SO4 was applied, and fractions (6 ml) were assayed for CNO activity by measuring the extent of radiolabeled K14CN conversion to 14CO2 in reactions mixtures supplemented with FDH (see enzyme assays). Active fractions were pooled, desalted in Na-K buffer, and concentrated with 10-kDa-cutoff ultraconcentrators. In step 2, proteins recovered in step 1 (100 μl containing 1 mg protein) were loaded on a Superdex 200 (Amersham-Pharmacia) gel filtration column (30 cm by 10 mm) equilibrated with 20 mM Na2HPO4 phosphate buffer (pH 7.0). Proteins were eluted at a flow rate of 0.5 ml min−1, and fractions (1 ml) were screened for CNO activity.
Enzyme assays. (i) Cyanide oxygenase (CNO).
The fixed-time measurement of CNO activity was determined by measuring the extent of K14CN conversion to 14CO2 in the presence of commercial formate dehydrogenase (FDH) (Sigma) as described previously (6). The time-dependent determination of CNO activity was determined by measuring either the rate of CN disappearance and/or ammonia (or formate) accumulation in reaction mixtures or CN-dependent O2 and/or NADH consumption as described previously (6).
(ii) NADH oxidase (Nox).
Nox activity was determined by measuring O2 consumption at 30°C with an oxygen electrode (Clark-type; Rank Bros., Cambridge, United Kingdom). Reactions were initiated by the addition of NADH (0.2 mM) to 0.5 ml of air-saturated (0.224 mM O2) Na-K phosphate buffer (pH 7.0) containing enzyme (10 to 40 μg protein).
(iii) NADH peroxidase (Npx).
Npx activity was measured spectrophotometrically by measuring the H2O2-dependent decrease in A340. Reaction cuvettes (0.5 ml) contained 0.4 mM NADH and protein (10 to 40 μg) in 0.5 ml Na-K phosphate buffer (pH 7.0), and reactions were initiated by the addition of 1 mM H2O2. Rates were corrected for the spontaneous breakdown of NADH.
(iv) Cyanide dihydratase (CynD).
CynD activity was determined by measuring the rate of CN consumption or ammonia accumulation in sealed vials at 30°C. In some cases reaction vials were made anaerobic by flushing with argon to establish that reactions occurred independent of molecular oxygen. The standard reaction mixture (0.25 ml) contained 0.5 to 70 μg protein and 50 mM Na-K phosphate buffer (final pH, 8.2). Samples were withdrawn at periodic intervals, and the amount of remaining cyanide or ammonia was quantified as earlier described (6, 17).
(v) Carbonic anhydrase (CA).
CA activity was measured essentially as described by Alber and Ferry (1). Reaction mixtures contained 0.25 to 1 μg protein in 0.5 ml 26 mM HEPES buffer, pH 7.9, containing 0.113 mM phenol red, the latter providing a means of visually observing CA-mediated pH changes. Following the addition of 0.5 ml of CO2-saturated water, the time required for the pH to decrease from 7.9 to approximately 7.1 was recorded. The time for a similar pH decrease in a chemical control in which no enzyme or boiled enzyme was supplied was also recorded and subtracted from that observed when enzyme was present. Units of activity were calculated as (t − to)/t, where to is the time required for pH change in the absence of enzyme or boiled enzyme.
Molecular mass determination.
Native protein molecular mass determination was determined by gel filtration chromatography on a Superdex 200 column. The column was calibrated with urease (545- and 272-kDa hexamer and trimer, respectively), bovine serum albumin (132- and 66-kDa dimer and monomer, respectively), ovalbumin (42 kDa), carbonic anhydrase (29 kDa), and α-lactalbumin (14 kDa), all obtained from Sigma.
Gel electrophoresis and electroblotting.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (16% total gel; 4% cross-linking) was carried out using Mini-Protean (Bio-Rad) acrylamide gels according to the procedure of Laemmli (18). The protein solubilizing buffer contained 150 mM dithiothreitol, 150 mM Trizma (pH 6.8), 21% glycerol, 6% lithium dodecyl sulfate, and 0.003% bromphenol blue. Electrophoresis was conducted in SDS-denaturing buffer (25 mM Trizma, 192 mM glycine, and 0.1% sodium dodecyl sulfate [SDS], pH 8.3) at 200 V for 55 min at 4°C. Low-molecular-mass protein standards (97 to 14 kDa) used for calibrating gels were purchased from Bio-Rad. Native PAGE (16%) analysis was conducted similarly at 4°C using the same buffers but without added SDS at constant power (2 W) for 1.5 h. Proteins were electroblotted onto polyvinylidene difluoride membranes at 4°C by overnight electroelution in a Bio-Rad transfer cell held at 30 V. The transfer buffer contained 25 mM Trizma, pH 8.0, 192 mM glycine, and 10% methanol. Bands of interest were excised using dust-free scissors after membranes were allowed to dry at room temperature.
Matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) and amino acid sequence analysis.
All analyses were performed at the Protein Chemistry Laboratory, University of Texas Medical Branch, Galveston. An Applied Biosystems Voyager DE STR Biospectrometer was used for MALDI-TOF. N-terminal amino acid sequencing was performed on an Applied Biosystems Procise 494HT Sequencer. Tryptic digests, where performed, were resolved using an Applied Biosystems 173A MicroBlotter. Sequence comparisons were performed using BLAST (2).
RESULTS AND DISCUSSION
CNO activity requires four (P1 to P4) separate protein components.
As described previously (6), CNO activity, defined as the oxygen-, reduced pterin-, and NADH-dependent conversion of cyanide to formate and ammonia was enriched ∼71-fold in one step by anion-exchange chromatography at pH 10 (Table 1). A complex molecular nature for the responsible enzyme was inferred from the nonlinear relationship observed when enzyme activity was measured as a function of protein concentration (data not shown). Proteins required for CNO activity were further resolved by gel filtration chromatography into four major components (P1 to P4) as shown in Fig. 1A. Only when all four resolved components were combined and a source of reduced pterin and NADH were each provided was CNO activity detected (Fig. 1B). Formate and ammonia were each formed quantitatively from CN, with one and two molar equivalents, respectively, of O2 and NADH being consumed in the process. This agrees with values for the reaction stoichiometry reported previously also for the partially purified enzyme (6). When FDH was also provided, formate was oxidized to CO2, thus reconstituting the complete enzymatic system for CN oxidation. The specific activity for the purified CNO enzyme determined either by measuring the rate of CN disappearance and ammonia appearance as well as O2 or NADH consumption by the combined P1 to P4 fractions was ∼1 U/mg. This represents approximately a 112-fold enrichment of the activity detected in crude extracts (Table 1, fraction H). P1 to P4 components were recovered in essentially a pure or nearly homogenous state after only two purification steps, inferring that the four may exist in cells as a multienzyme complex.
TABLE 1.
Activity-based purification of cyanide oxygenase from P. fluorescens NCIMB 11764
| Purifi- cation stepa | Total protein (mg) | Volume (ml) | Total activityb (mU) | Sp act (mU mg−1) | Purifi- cation (fold) | Recovery (%) |
|---|---|---|---|---|---|---|
| Fraction H | 330 | 8 | 2,640 | 8 | 1 | 100 |
| Source 30Q | 1 | 1 | 500 | 500 | 71 | 18 |
| Superdex 200 | 0.98 | 0.5 | 450 | 900 | 112 | 17 |
Fraction H, high-molecular weight retentate recovered after passing 150,000 × g high-speed supernatant through a 10-kDa mwco ultraconcentrator; Source 30Q, fraction recovered after anion-exchange chromatography; Superdex 200, combined P1 to P4 fractions recovered after gel filtration chromatography.
CNO activity was determined by measuring O2 or NADH consumption at 30°C in reaction mixtures (0.5 ml) supplied with 0.1 mM KCN, 0.2 mM NADH, and 0.5 μM dihydrobiopterin in 50 mM Na-K phosphate buffer (pH 7.0). The amounts of protein were as follows: 0.5 to 1 mg ml−1 for Fraction H and Source 30Q and 50 μg ml−1 and for Superdex 200 fractions equally proportioned between P1, P2, P3, and P4.
FIG. 1.

Identification of proteins required for cyanide oxygenase activity in P. fluorescens. (A) Resolution of CNO into four active protein fractions (P1 to P4) by gel filtration chromatography. Proteins (1 mg) were loaded onto a Superdex 200 column (30 cm by 10 mm) and eluted with 20 mM Na2HPO4 buffer (pH 7.0) at 0.5 ml per min. (B) The CNO activity of reconstituted protein components recovered as described for panel A was measured either as the relative amount of radioactive K14CN converted to 14CO2 when supplemented with formate dehydrogenase (+FDH) or the amount of unlabelled CN converted to ammonia (or formate) in its absence (−FDH). In each case, reactions (0.25 ml) were conducted in sealed vials at 30oC and supplied with ∼24 μg protein (equal component proportions), 0.1 mM KCN, 0.2 mM NADH, and 0.5 μM dihydrobiopterin (H2B) in 50 mM Na-K phosphate buffer (pH 7.0). Reactions mixtures supplied with FDH (2 mU) contained 1 μCi K14CN (48 KBq, ∼100 μM) and were incubated for 1 h before 14CO2 was quantified versus those from which FDH was omitted, in which case ammonia (or formate) was determined after 10 min. (C) SDS-PAGE (12%) analysis of CNO components. Lanes: Std., molecular size standards (5 μg); H, crude extract (5 μg); B, source 30Q fraction (5 μg); and P1 to P4 (10 μg each).
P1 is NADH oxidase (oxidoreductase) (Nox).
P1 was yellow in color, displayed UV maxima at 375 and 454 nm (data not shown), and consumed oxygen independent of the other three protein fractions when NADH was provided (specific activity, 1 U/mg protein) (Fig. 2). Flavin adenine dinucleotide (50 μM) stimulated the O2-consuming activity above that which was observed in controls containing no protein and was unaffected by the addition of 1 mM KCN. The activity, however, was unstable and declined by about 50% after 1 to 2 days at 4°C. In the absence of KCN, catalase (5,000 U/ml) caused a significant reduction in the rate of O2 consumption, implying that H2O2 was generated as a reaction product (data not shown). Collectively, these properties are consistent with P1 being a flavoprotein NADH oxidase (Nox) (3, 21). The stimulatory effect of flavin adenine dinucleotide further implies that the enzyme has flavin reductase activity (20). On denaturing gels, two weakly staining polypeptides of 46 and 31 kDa were observed along with a dense-staining polypeptide of ∼76 kDa (Fig. 1C). Quantitative N-terminal amino acid sequencing is expected to provide further information on whether the 76-kDa protein is composed of two smaller 46- and 31-kDa components, studies which are currently in progress. On a gel filtration column, P1 migrated as a single peak of 230 kDa, which, if the enzyme is composed of two components, could imply an α3β3 native structure (Table 2). The detection of a predominant protein of 76 kDa for the enzyme gives cause to believe that the enzyme more closely resembles group II versus group I and III Nox homologues for which molecular masses of ∼76, ∼49, and 20 kDa, respectively, have been reported (13).
FIG. 2.

Individual activities of P1 to P4 protein components of CNO. Enzymes were assayed as described in Materials and Methods. P3-containing reaction mixtures were supplied 17.5 μg of protein. Data sets are those from at least two independent experiments.
TABLE 2.
Components of cyanide oxygenase from P. fluorescens NCIMB 11764
| Analysis | P1 | P2 | P3 | P4 |
|---|---|---|---|---|
| Enzyme | NADH oxidase (Nox) | NADH peroxidase (Npx) | Cyanide dehydratase (CynD) | Carbonic anhydrase (CA) |
| E.C. no. | 1.6.99.3 | 1.11.1 | 3.5.5.- | 4.2.1.1 |
| Sp act (U/mg)a | 1 | 1 | 100 | 2000 |
| Molecular mass (kDa) | ||||
| SDS-PAGE | 76b (46, 31) | 47 | 38 (43c) | 29 |
| Mass spectrometry | NDe | 49.7 | ND | 28.9 |
| Gel filtrationd | 230 | 100 | ≥300 | 58 |
| Suggested composition | α3β3 | α2 | Unknown | α2 |
| N-terminal sequence | ND | MKVIVLGSSHGGYEAVEELLNLHPD | ND | N-blocked |
| Tryptic peptide sequence | ND | ND | ND | LVQFHFHWGS YAAELHL |
Defined as 1 μmol of substrate converted per min.
Possible dimer of 46- and 31-kDa species.
Internal peptide from trypic digest shows 100% amino acid sequence homology (VQDPLEIVGLR) to elongation factor Tu from P. putida.
Determined at pH 7.0, except for CynD, which was performed at pH 8.0.
ND, not determined.
P2 is NADH peroxidase.
MALDI-TOF analysis confirmed the presence of a single protein of 49,725 Da in P2, which agrees with the size of a single polypeptide (47 kDa) detected on denaturing gels (Fig. 1C). Gel filtration chromatography and native PAGE each confirmed a native mass of 100 kDa, indicating a homodimeric structure. N-terminal amino acid sequence analysis by automated Edman degradation revealed that the first 25 amino acids of the protein were 100% identical to those found in NADH peroxidase (Npx) from Enterococcus faecalis (Table 2), and enzyme assays further confirmed the Npx activity of the purified enzyme (specific activity, 1 U/mg) (Fig. 2).
P3 is cyanide dihydratase (CynD).
When P3 was analyzed by SDS-PAGE a number of polypeptides were detected; however, of these a band at ∼40 kDa was consistently the most abundant (Fig. 1C). The size of this protein, parenthetically, coincided also with the ∼35-kDa size for P3 based on its elution from a gel filtration column (Fig. 1A). By comparison, native gels gave evidence of several large (∼200 to 500 kDa) protein species. These observations, taken together with reports (11, 12, 26, 30) that cyanide dihydratase (CynD) from bacteria is composed of a 38-kDa protein that oligomerizes into a large nonglobular structure, led us to hypothesize that the discrepancy between the size of P3 based on its elution from a gel filtration column and that observed on native gels might be due to the presence of a related enzyme. We assayed P3 for CynD activity and found that, indeed, the isolated protein had high activity (specific activity, 100 U/mg protein). Figure 2 shows that P3 rapidly consumed CN provided at 20 mM and converted it quantitatively into ammonia and formate. Additional kinetic studies have shown that like other CynD enzymes, CynDPf11764 has a low affinity for CN (Km = 8.3 mM) (R. Fernandez, E. Dolghih, and D. Kunz, Abstr. 104th Gen. Meet. Am. Soc. Microbiol., abstr. K165, 2004). Moreover, the activity, as expected, was completely independent of molecular oxygen, reduced pterin, or NADH. This contrasts sharply with the enzymatic properties recognized for CNO for which the affinity for CN is approximately 2,300-fold higher (Km app = 3.5 μM), and substrate turnover absolutely requires oxygen, reduced pterin, and NADH. Hence, the possibility that CynD could somehow masquerade as CNO seems remote.
Consistent with CynD enzymes having been described from Bacillus pumilus (CynDBp) (12) and Pseudomonas stutzeri (CynDPs) (26), the oligomerization of CynDPf11764 was favored at alkaline pH. This was evidenced by the detection of a major protein peak with a mass value of ≥300 kDa when P3 was subjected to gel filtration chromatography at pH 8.0 (Fig. 3) (as opposed to pH 7.0, in which case P3 eluted with a mass of ∼35 kDa). Smaller amounts of two additional proteins having masses of 43 and 38 kDa were also detected at pH 8.0, the latter presumably being the same protein as that detected at pH 7.0. On a native gel several large proteins (∼200 to 500 kDa) and the 38- and 43-kDa species were each detected (Fig. 3, inset). We interpreted these results as indicating that either one or both of the latter were components of CynDPf11764 and that the native size of the enzyme could vary depending on the extent of oligomerization. To further identify the 38- and 43-kDa proteins, we attempted to electroelute each from a native gel and determine the amino acid sequence after trypsin digestion. Unfortunately, no amino acid sequence could be obtained for the 38-kDa species due to peptide decomposition. This reinforces our interpretation that many of the peptides observed when P3 was subjected to SDS-PAGE (Fig. 1C) can also be attributed to protein degradation. The 43-kDa species, in contrast, yielded a peptide for which the amino acid sequence (VQDPLEIVGLR) was shown to be 100% identical to elongation factor Tu (Ef-Tu) from P. putida (Table 2). The occurrence of this protein with CynD was interpreted as follows. (i) Ef-Tu is a structural component of the native P. fluorescens NCIMB 11764 enzyme; (ii) Ef-Tu is not a structural component, but its presence is necessary for formation of the enzyme's native state; or (iii) Ef-Tu is a coincidental impurity. Further studies are needed to distinguish between these possibilities; however, Ef-Tu proteins are known to have chaperone activity (4, 5). It therefore seems reasonable, given the complex oligomeric structure of CynD, to think that a chaperone might be required in its assembly. Furthermore, other enzymes in the nitrilase superfamily (22), of which CynD is a member, have been reported also to copurify with chaperonins, providing further reason to think that the occurrence of Ef-Tu is more than just a coincidence. Finally, the possibility that CNO may exist in cells as a large protein complex whose assembly might also be expected to be chaperone dependent may further help explain the occurrence of Ef-Tu.
FIG. 3.

Gel filtration chromatography of P3 (CynDPf11764) at pH 8.0. P3 (20 μg protein) was applied to a Superdex 200 column, and the molecular mass of eluted proteins was determined against calibrated molecular mass standards. Inset, native PAGE analysis of CynDPf11764 (peak at 300 kDa) recovered from gel filtration and molecular mass standards (Std).
P4 is carbonic anhydrase (CA).
When P4 was subjected to MALDI-TOF, a single polypeptide of 28,992 Da was detected. This is close to that estimated (29 kDa) by SDS-PAGE and gel filtration (Fig. 1C). When analyzed on a native gel, the purified protein had an intense band at 58 kDa and a lighter staining band at 29 kDa, implying that its native structure was that of a homodimer. N-terminal amino acid sequencing could not be performed because of a blocked N terminus; however, a BLAST search of amino acid sequences determined for two internal peptides generated by trypsin digestion gave a 100% match to mammalian carbonic anhydrase (CA) (Table 2). The native and subunit structures were further consistent with those of a CA, the activity of which also was confirmed by enzyme assays (specific activity, 2,000 U/mg) (Fig. 2).
It is noteworthy that of the four enzymes found to comprise CNO, three are known to have oxidative stress functions. These include Nox, Npx, and CA, the latter reported to protect against oxyradicals in both yeast and humans (7, 24, 31). The significance of this is not entirely clear, but it does parallel reports of the general effect of CN to induce oxidative stress in higher organisms (8, 23). The remaining component, CynD, might be expected to be a key player in substrate turnover. However, there are also reports of the ability of peroxidase enzymes, including NADH peroxidase, to act on CN (19, 27), making it possible that substrate destabilization via an Npx-initiated process could also occur. Further studies on the catalytic roles of each component are expected to provide important new information on the mechanism of substrate decomposition.
While conceptually related to the more established hydrolytic route of CN conversion to formate and ammonia catalyzed by CynD enzymes,
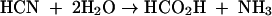 |
(5) |
the utilization of molecular oxygen by CNO for this purpose is, as far as we are aware, unknown in biochemistry. Our evidence indicates that CynD is not able to carry out the oxygen-dependent turnover of CN on its own. This makes it reasonable to inquire as to whether there are potential advantages to be gained physiologically from utilizing one or the other enzyme. We suggest that because the affinity for CN by CNO (Km app = 3.5 μM) (15) is orders of magnitude (2,300 times) higher than that for CynD, CNO provides for a much more effective means of scavenging CN. Another reason may be related to the large thermodynamic displacement to be achieved from coupling the oxygenolytic step catalyzed by CNO with the oxidation of formate by FDH (see equations 1 to 4). The calculated free energy change (ΔGo′), for example, for CN hydrolysis under standard conditions is −38.63 kJ mol−1. This compares with −466.31 kJ mol−1 when CN is completely oxidized to CO2, implying that bacteria would much prefer to evade the toxic consequences accompanying the adventitious utilization of CN as a growth substrate by accomplishing its complete oxidation.
Acknowledgments
We are grateful to Rebecca Dickstein, Paul Weimer, Avinash Srivastava, and two anonymous reviewers for critiquing the manuscript.
This work was supported by National Science Foundation grant MCB 0136280.
REFERENCES
- 1.Alber, B. G., and J. G. Ferry. 1994. A carbonic anhydrase from the archaeon Methanosarcina thermophila. Proc. Natl. Acad. Sci. USA 91:6903-6913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altshul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipmann. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Argurou, A., and J. S. Blanchard. 2004. Flavoprotein disulfide reductases: advances in chemistry and function. Prog. Nucleic Acids Res. Mol. Biol. 78:89-142. [DOI] [PubMed] [Google Scholar]
- 4.Caldas, T., S. Laalami, and G. Richarme. 2000. Chaperone properties of bacterial elongation factor EF-G and initiation factor IF2. J. Biol. Chem. 275:855-860. [DOI] [PubMed] [Google Scholar]
- 5.Caldas, T. D., A. El Yaagoubi, and G. Richarme. 1998. Chaperone properties of bacterial elongation factor EF-Tu. J. Biol. Chem. 273:11478-11482. [DOI] [PubMed] [Google Scholar]
- 6.Fernandez, R., E. Dolghih, and D. A. Kunz. 2004. Enzymatic assimilation of cyanide via pterin-dependent oxygenolytic cleavage to ammonia and formate in Pseudomonas fluorescens NCIMB 11764. Appl. Environ. Microbiol. 70:121-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Götz, R., A. Gnann, and F. K. Zimmerman. 1999. Deletion of carbonic anhydrase-like gene NEC103 of the yeast Saccharomyces cerevisiae causes an oxygen-sensitive growth defect. Yeast 15:855-864. [DOI] [PubMed] [Google Scholar]
- 8.Gunsekara, P. G., J. L. Borowitz, and G. E. Isom. 1998. Cyanide-induced generation of oxidative species: involvement of nitric oxide synthase and cyclooxygenase-2. J. Pharmacol. Exp. Ther. 285:236-241. [PubMed] [Google Scholar]
- 9.Harris, R., and C. J. Knowles. 1983. The conversion of cyanide to ammonia by extracts of a strain of Pseudomonas fluorescens that utilizes cyanide as a source of nitrogen for growth. FEMS Microbiol. Lett. 20:337-341. [Google Scholar]
- 10.Harris, R., and C. J. Knowles. 1983. Isolation and growth of a Pseudomonas species that utilizes cyanide as a source of nitrogen. J. Gen. Microbiol. 129:1005-1011. [DOI] [PubMed] [Google Scholar]
- 11.Ingvorsen, K., B. Højer-Pedersen, and S. E. Godtfredsen. 1991. Novel cyanide-hydrolyzing enzyme from Alcaligenes xylosoxidans subsp. denitrificans. Appl. Environ. Microbiol. 57:1783-1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jandhyala, D., M. Berman, P. R. Meyers, B. T. Sewell, R. C. Wilson, and M. J. Benedik. 2003. CynD, the cyanide dihydratase from Bacillus pumilus: gene cloning and structural studies. Appl. Environ. Microbiol. 69:4794-4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kengen, S. W. M., J. van der Oost, and W. M. de Vos. 2003. Molecular characterization of H2O2-forming NADH oxidases from Archaeglobus fulgidus. Eur. J. Biochem. 270:2885-2894. [DOI] [PubMed] [Google Scholar]
- 14.Knowles, C. J. 1988. Cyanide utilization and degradation by microorganisms, p. 3-15. In D. Evered and S. Harnett (ed.), Cyanide compounds in biology, vol. 140. John Wiley & Sons Ltd., Chicester, United Kingdom. [DOI] [PubMed] [Google Scholar]
- 15.Kunz, D., A. Fernandez, and P. Parab. 2001. Evidence that bacterial cyanide oxygenase is a pterin-dependent hydroxylase. Biochem. Biophys. Res. Commun. 287:514-518. [DOI] [PubMed] [Google Scholar]
- 16.Kunz, D. A., O. Nagappan, J. Silva-Avalos, and G. T. Delong. 1992. Utilization of cyanide as a nitrogenous substrate by Pseudomonas fluorescens NCIMB 11764: evidence for multiple pathways of metabolic conversion. Appl. Environ. Microbiol. 58:2022-2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kunz, D. A., C.-S. Wang, and J.-L. Chen. 1994. Alternative routes of enzymic cyanide metabolism in Pseudomonas fluorescens NCIMB 112764. Microbiology 140:1705-1712. [DOI] [PubMed] [Google Scholar]
- 18.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 19.Moreno, S. N. J., K. Stolze, E. G. Janzen, and R. P. Mason. 1988. Oxidation of cyanide to the cyanyl radical by peroxidase/H2O2 systems as determined by spin trapping. Arch. Biochem. Biophys. 265:267-271. [DOI] [PubMed] [Google Scholar]
- 20.Nimura, Y., Y. Nishiyama, D. Saito, H. Tsuji, M. Hidaka, T. Miyaji, T. Watanabe, and V. Massey. 2000. A hydrogen-peroxide-forming NADH oxidase that functions as an alkyl hydroperoxide reductase in Amphibacillus xylanus. J. Bacteriol. 182:5046-5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishiyama, N., V. Massey, K. Takeda, S. Kawasaki, J. Sato, T. Watanabe, and Y. Nimura. 2001. Hydrogen peroxide-forming NADH oxidase belonging to the peroxiredoxin oxidoreductase family: existence and physiological role in bacteria. J. Bacteriol. 183:2431-2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Reilly, C., and P. D. Turner. 2003. The nitrilase family of CN hydrolyzing enzymes -a comparative study. J. Appl. Microbiol. 95:1161-1174. [DOI] [PubMed] [Google Scholar]
- 23.Prabhakaran, K., L. Li, E. M. Mills, J. L. Borowitz, and G. E. Isom. 2005. Up-regulation of Ucp-2 by cyanide is linked with cytotoxicity in mesencephalic cells. J. Pharmacol. Exp. Ther. doi: 10.1124/jpet.105.088625. [DOI] [PubMed]
- 24.Räisänen, S. R., P. Lehenkari, M. Tasanen, P. Rahkila, P. L. Härkönen, and H. K. Väänänen. 1999. Carbonic anhydrase III protects cells from hydrogen peroxide-induced apoptosis. FASEB J. 13:513-522. [DOI] [PubMed] [Google Scholar]
- 25.Reed, R. E. 1988. Cyanide compounds in plants and their effects on animals, p. 47-50. In D. Van Zyl (ed.), Cyanide and the environment, vol. 1. Geotechnical engineering program. Colorado State University, Fort Collins, Colo. [Google Scholar]
- 26.Sewell, B. T., M. N. Berman, P. R. Meyers, D. Jandhyala, and M. J. Benedik. 2003. The cyanide degrading nitrilase from Pseudomonas stutzeri AK61 is a two-fold symmetric, 14-subunit spiral. Structure (Cambridge) 11:1413-1422. [DOI] [PubMed] [Google Scholar]
- 27.Stolze, K., S. N. Moreno, and R. P. Mason. 1989. Free radical intermediates formed during the oxidation of cyanide by horseradish peroxidase/H2O2 as detected with nitroso spin traps. J. Inorg. Chem. 37:45-53. [DOI] [PubMed] [Google Scholar]
- 28.Taylor, C. A., and M. H. Ralphs. 1988. The importance of poisonous plants as forages in the prairies and Southwest, p. 363-375. In M. H. R. L. F. James and D. B. Nielsen (ed.), The ecology and economic impact of poisonous plants on livestock production. Westview Press, Boulder, Colo.
- 29.Wang, C.-S., D. A. Kunz, and B. J. Venables. 1996. Incorporation of molecular oxygen and water during enzymatic oxidation of cyanide by Pseudomonas fluorescens NCIMB 11764. Appl. Environ. Microbiol. 62:2195-2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watanabe, A., K. Yano, K. Ikebukuro, and I. Karube. 1998. Cyanide hydrolysis in a cyanide-degrading bacterium, Pseudomonas stutzeri AK61, by cyanidase. Microbiology 144:1677-1682. [DOI] [PubMed] [Google Scholar]
- 31.Zimmerman, U., P. Wang, X. Zhang, S. Bogdanovich, and R. E. Forster. 2004. Anti-oxidative response of carbonic anhydrase III in skeletal muscle. IUBMB Life 56:343-347. [DOI] [PubMed] [Google Scholar]