

Abstract
Sequencing 16S rRNA genes (SSU) cloned from Aeromonas strains revealed that strains contained up to six copies differing by ≤1.5%. The SSU copies from Aeromonas veronii LMG13695 clustered with sequences from four Aeromonas species. These results demonstrate intragenomic heterogeneity of SSU and suggest caution when using SSU to identify aeromonads.
Differences in virulence of Aeromonas species, ranging from environmental isolates and digestive-tract symbionts of blood-feeding animals to human pathogens (11, 12, 16, 24, 25), led to the increased use of 16S rRNA gene (SSU) sequences to identify Aeromonas species (4, 9, 10). A potentially complicating factor is that bacteria contain up to 15 copies of the ribosomal operon (17). Evidence suggests that concerted evolution does not always effectively homogenize rRNA operons in bacteria and that intragenomic heterogeneity exists (5, 7, 19, 23, 29, 31, 33). Two investigations of bacterial genomes revealed that differences in the SSU sequence occurred in 54 to 62% of organisms harboring at least two copies of the rRNA operon (1, 7). However, except in rare cases, the differences were less than 1% of the compared nucleotides, and both studies concluded that these minor differences did not have a significant effect on the determination of phylogenetic relationships.
A pioneering study by Martinez-Murcia et al. used the SSU sequence to reconstruct the phylogeny of Aeromonas species (20). This study revealed that some Aeromonas species, for example, A. trota and A. caviae, differ only by up to three nucleotides in the SSU sequence, even though DNA-DNA hybridization values for these species are low (30%), indicating that they are distantly related (20). In contrast, Aeromonas sobria and Aeromonas veronii differ by 12 nucleotides but have a DNA-DNA hybridization value of 60 to 65%, indicating that they are closely related (20). This contradiction and visual inspection of the SSU sequences led Sneath to suggest that lateral gene transfer and recombination could explain this lack of congruence (27). Previously, we undertook a restriction fragment length polymorphism-PCR analysis using multiple reference strains and discovered that several strains produced unexpected restriction patterns (10). In this report, we further analyzed Aeromonas veronii biovar sobria strains A132, A155, A916, AER28, CDC0437-84, and LMG13695, Aeromonas veronii biovar veronii strain AER397, and Aeromonas media strains A6 and CDC0862-83 by sequencing cloned SSU (10), determining the phylogenetic relationship of the different copies of the SSU and evaluating whether the SSU evolved as a single unit.
Sequence analysis of cloned 16S rRNA genes.
The strains were grown as previously described (10), and genomic DNA was isolated as described by Nelson and Selander (22), except that the resuspended sample was preheated for 3 min at 65°C. The 16S rRNA gene was amplified by using primers 27F and 1492R (15, 18). The PCR-amplified 16S rRNA genes were cloned into pGEM-T (Promega, Madison, WI) according to the manufacturer's instructions, and the cloned 16S rRNA genes were sequenced and analyzed (15).
In six Aeromonas strains, the copies of the 16S rRNA genes differed by up to 21 nucleotides out of 1,377 bp analyzed (Fig. 1). If differing sequences were detected in at least two plasmids containing SSUs from one strain, we considered them to be heterogeneous copies of SSU and for this discussion call them an allele without implying an associated functional difference. The analysis of the cloned SSUs suggested the presence of at least six alleles in the A. media strain CDC0862-83 and at least five different alleles in the A. veronii biovar sobria strain LMG13695 (Fig. 1). The alleles from A. media CDC0862-83 differed from each other by up to 19 bp and those from A. veronii LMG13695 by up to 21 bp out of 1,377 bp compared. In a recent publication, the presence of two heterogeneous copies of the SSU was suggested for Aeromonas trota but not substantiated (28). It is possible that any of the examined strains contains additional alleles.
FIG. 1.

Sequence differences among 16S rRNA gene alleles cloned from A. media and A. veronii strains. The diagram shows the variable sites detected in the strains analyzed in our study and in the 16S rRNA gene from Aeromonas species (modified from reference 27). The species names and the GenBank accession (Acc.) numbers are shown. The allele numbers are shown (Allele), as is how often each allele was cloned (Copies). The numbers in the top row correspond to the Escherichia coli numbering scheme. Yellow shading indicates the sequence of allele 1 from CDC0862-83 and red, green, or blue shading indicates a nucleotide substitution.
Verification of sequence differences by Southern analysis.
To confirm the presence of the different SSU sequences in the same genome in a PCR-independent manner, we used Southern analysis of genomic DNA from strain CDC0862-83. Oligonucleotide probes were designed to recognize allele 1 from CDC0862-83, (16S-230-232-A [5′-GGG TTC ATC CAA TCG CG-3′]), alleles 2 through 6 from CDC0836-83 (16S-230-232-B [5′-GGG CAT ATC CAA TCG CG-3′]), and, as a control, all alleles (27F [5′-AGA GTT TGA TCM TGG CTC AG-3′]). The hybridization was done by using a sodium chloride-sodium citrate protocol (3). Oligonucleotide probes (MWG, Münchenstein, Germany) were labeled with [α-32P]dCTP (3). The membranes were prehybridized for 1.5 h at 37°C, hybridized overnight at 37°C, washed in 6× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) with 0.05% pyrophosphate for 30 min at 48°C, and used to expose BioMax MS film (Kodak, Rochester, N.Y.).
The hybridization patterns of the allele-specific probes demonstrated that the polymorphism at nucleotides 230 through 232 of allele 1 was present in two copies and that the remaining copies hybridized with the probe for alleles 2 through 6. These results indicate that at least two different alleles were present in the genome of A. media strain CDC0862-83 (Fig. 2). The banding pattern suggests the presence of at least eight copies of SSU in CDC0862-83 (Fig. 2).
FIG. 2.
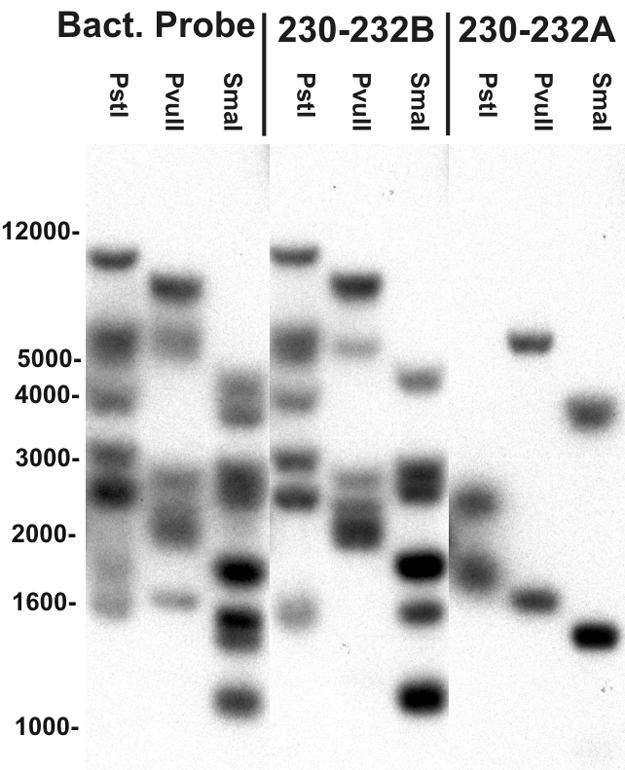
Southern analysis of CDC0826-83 demonstrates that the observed variation is not a PCR artifact but encoded on the chromosome. The bacterial (Bact.) probe recognizes most bacterial 16S rRNA genes and each of the 16S rRNA gene copies in Aeromonas. Probe 230-232A anneals only to allele 1 from CDC0862-83, and probe 230-232B anneals to alleles 2 through 6. The hybridization patterns of the membrane probed with 230-232A show two bands hybridizing with the genomic DNA digested with PstI, PvuII, or SmaI, indicating that two copies of this polymorphism characteristic of allele 1 are present in the genome of CDC0862-83.
Phylogenetic analysis.
We assessed the significance of the sequence differences by aligning sequences in ClustalW version 1.83 (30) and constructing phylogenetic trees using the PhyML program version 2.4.1 (13) under the HKY85 substitution model (14) with among-site rate variation described through a discrete approximation of the gamma distribution with four rate categories (32). One hundred bootstrap samples were generated and evaluated in PhyML under the same model, and the consensus tree was generated by using the CONSENSE program from the PHYLIP package (8).
The phylogenetic analysis shows that the phylogenetic placement of the SSU is affected by the intragenomic heterogeneity (Fig. 3). The SSU alleles from the A. media strain CDC0862-83 clustered either within the Aeromonas hydrophila-A. media clade that has a 77% bootstrap support value or within a large clade that contained Aeromonas encheleia, Aeromonas eucrenophila, and other species with a 75% bootstrap value. Alleles from the A. veronii strain LMG13695 grouped with sequences of A. veronii, Aeromonas jandaei, and Aeromonas allosaccharophila and with sequences of the A. hydrophila-A. media group. Another A. veronii biovar sobria strain, A155, clustered close to A. allosaccharophila, and A. veronii biovar sobria strain AER28 clustered near A. jandaei and A. veronii. All of these strains are well characterized and were previously identified to the species level by DNA-DNA hybridization (2, 6, 10). Restriction fragment length polymorphism analysis of 82 surveyed Aeromonas strains (10, 21) suggests that at least 21% had intragenomic heterogeneous SSU alleles as determined by the presence of faint bands that could not be removed by adding more restriction enzyme (data not shown) indicating that SSU heterogeneity is not a rare phenomenon in this genus. Our results suggest that the SSU sequences may not properly reflect the taxonomic relationship of these strains and are not the best choice for identifying Aeromonas species (10).
FIG. 3.

Maximum-likelihood tree from published 16S rRNA genes of Aeromonas species and cloned alleles from the CDC0862-83 and LMG13695 strains. Selected bootstrap support values and posterior probabilities are shown as the first and second numbers, respectively. The tree is calculated from the full-length alignment using PhyML. Individual alleles of the same strain sometimes form sister groups with other Aeromonas species. Moreover, the analyses of bipartitions show that alleles have different phylogenetic relationships among each other if different fragments of the alignment are analyzed (Table 2).
Mosaic evolution of SSU.
Visual inspection of the alignment (Fig. 1) suggested that different parts of the sequence from one allele were most similar to SSU sequences from distantly related Aeromonas species. These conflicting phylogenetic relationships suggest that some of the SSU sequences are mosaic. We tested the hypothesis by dividing the alignment into three fragments: nucleotides 1 through 264 (fragment W1), 457 through 476 (fragment W2), and 1009 through 1492 (fragment W3), and we compared the phylogenies derived from each fragment and from the entire sequence. The resulting trees were incongruent (topologies not shown). The approximately unbiased (AU) test (26) demonstrated that each of the trees calculated as optimal for one fragment had a significantly better fit to the data than the alternative trees calculated from the other fragments (Table 1). These results support the concept that the SSU evolved in a mosaic manner. This was further supported by a bipartition analysis that focuses on all significantly supported bipartitions (34) (Table 2). The different fragments strongly supported conflicting bipartitions, i.e., bipartitions that cannot coexist in a single phylogenetic tree.
TABLE 1.
Results of AU testa
| Fragment | Topology | P value |
|---|---|---|
| W1 | W1 | 0.997 |
| W2 | 2 × 10−13 | |
| W3 | 4 × 10−80 | |
| F | 0.003 | |
| W2 | W1 | 9 × 10−62 |
| W2 | 1.000 | |
| W3 | 1 × 10−40 | |
| F | 3 × 10−49 | |
| W3 | W1 | 5 × 10−07 |
| W2 | 6 × 10−52 | |
| W3 | 1.000 | |
| F | 5 × 10−06 | |
| F | W1 | 1 × 10−48 |
| W2 | 7 × 10−05 | |
| W3 | 1 × 10−118 | |
| F | 1.000 |
The fit of four tree topologies (calculated from full alignment [F] and three fragments) was evaluated for full-length alignment and three fragments. The significantly better topology per analyzed fragment is shown in bold. In all cases, the tree topology calculated for each data set is significantly better than the alternatives.
TABLE 2.
Supported bipartitions for relationships among allelesa
| Fragment | Supported bipartitions for indicated strain (bootstrap value [%])
|
|
|---|---|---|
| CDC0862-83 | LMG13695 | |
| Full alignment | 1, 2, 3, 4 | 5, 6 (70) | 1, 4 | 2, 3, 5 (97) |
| 1, 2, 3 | 4, 5, 6 (77) | 1, 4, 5 | 2, 3 (89) | |
| W1 | 1, 3 | 2, 4, 5, 6 (85) | 1, 4 | 2, 3, 5 (100) |
| W2 | 1, 4, 5, 6 | 2, 3 (100) | 1, 4, 5 | 2, 3 (100) |
| 1, 2, 3, 4 | 5, 6 (99) | ||
| 1, 4 | 2, 3, 5, 6 (69) | ||
| W3 | 1, 2, 4, 6 | 3, 5 (83) | 1, 2 | 3, 4, 5 (94) |
| 1, 2, 4 | 3, 5 (75) | ||
Alleles are numbered using Arabic numerals. Taxa on the opposite sides of bipartition are separated by the vertical bar: e.g., the bipartition “1, 4 | 2, 3, 5” stands for alleles 1 and 4 on one side of the bipartition and 2, 3, 5 on the other. The bootstrap support values for bipartitions are shown in parentheses. Only nontrivial bipartitions with bootstrap support values greater than 68% are shown. Note that each fragment supports only bipartitions that are compatible with the other bipartitions supported by this fragment; however, the sets of bipartitions supported by different fragments frequently are incompatible with one another.
One intriguing result is the clustering of A. media CDC0862-83 allele 1 in the branch containing A. encheleia and A. eucrenophila, because in the rpoD-derived phylogenetic tree, A. media clusters with these two species (28), while in the 16S rRNA-derived phylogenetic tree, A. media clusters with A. hydrophila (20). This discrepancy and the previously noted discrepancy between the phylogeny derived by SSU sequences and by DNA-DNA reassociation constants can be explained by the intragenomic heterogeneity. Two possible scenarios can explain the observed data. First, ancient polymorphisms could have preceded the speciation events in the genus Aeromonas, and these polymorphisms could have persisted throughout the history of the species. Our analysis shows that the different alleles do not cluster but intersperse in the reconstructed phylogeny. Although there was sufficient time for alleles from other lineages to be lost over evolution, for some reason, other alleles that are very similar to distantly related species were maintained. The alternative scenario suggested by Sneath (27), horizontal gene transfer, appears more likely. In either case, the alleles present today resulted from recombination between divergent SSUs, and thus, one would not expect that the SSU phylogeny always reflects the evolutionary history of the strain in this genus.
For the identification of Aeromonas spp., it is critical to use multiple molecular markers, such as rpoD and gyrB, in the phylogenetic analysis (28) or an even broader approach utilizing multilocus sequence typing. Whole-genome sequence comparisons of several strains from each species might provide the insight needed to resolve the complex evolutionary history that probably included frequent gene duplication, gene transfer, and recombination events.
Acknowledgments
We thank R. Troller for excellent technical assistance with the Southern hybridization, R. V. M. Rio and A. Laufer for helpful comments on the manuscript, and R. T. Papke for helpful discussion.
The National Science Foundation award MCB-0334627 to J.G., MCB-0237197 to J.P.G., a Sandoz Foundation grant to J.G., and a NASA Exobiology grant to J.P.G. supported this work.
REFERENCES
- 1.Acinas, S. G., L. A. Marcelino, V. Klepac-Ceraj, and M. F. Polz. 2004. Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J. Bacteriol. 186:2629-2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altwegg, M., M. W. Reeves, R. Altwegg-Bissig, and D. J. Brenner. 1991. Multilocus enzyme analysis of the genus Aeromonas and its use for species identification. Zentbl. Bakteriol. 275:28-45. [DOI] [PubMed] [Google Scholar]
- 3.Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.). 1998. Current protocols in molecular biology. Greene Publishing Associates and Wiley-Interscience, New York, N.Y.
- 4.Borrell, N., S. G. Acinas, M. J. Figueras, and A. J. Martinez-Murcia. 1997. Identification of Aeromonas clinical isolates by restriction fragment length polymorphism of PCR-amplified 16S rRNA genes. J. Clin. Microbiol. 35:1671-1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boucher, Y., C. J. Douady, A. K. Sharma, M. Kamekura, and W. F. Doolittle. 2004. Intragenomic heterogeneity and intergenomic recombination among haloarchaeal rRNA genes. J. Bacteriol. 186:3980-3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carnahan, A. M., and S. W. Joseph. 1993. Systematic assessment of geographically and clinically diverse Aeromonads. Syst. Appl. Microbiol. 16:72-84. [Google Scholar]
- 7.Coenye, T., and P. Vandamme. 2003. Intragenomic heterogeneity between multiple 16S ribosomal RNA operons in sequenced bacterial genomes. FEMS Microbiol. Lett. 228:45-49. [DOI] [PubMed] [Google Scholar]
- 8.Felsenstein, J. 1993. PHYLIP (Phylogeny Inference Package) version 3.6. Department of Genetics, University of Washington, Seattle.
- 9.Figueras, M. J., L. Soler, M. R. Chacon, J. Guarro, and A. J. Martinez-Murcia. 2000. Extended method for discrimination of Aeromonas spp. by 16S rDNA RFLP analysis. Int. J. Syst. Evol. Microbiol. 50:2069-2073. [DOI] [PubMed] [Google Scholar]
- 10.Graf, J. 1999. Diverse restriction fragment length polymorphism patterns of the PCR-amplified 16S rRNA genes in Aeromonas veronii strains and possible misidentification of Aeromonas species. J. Clin. Microbiol. 37:3194-3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graf, J. 2000. The symbiosis of Aeromonas and Hirudo medicinalis, the medicinal leech. ASM News 66:147-153. [Google Scholar]
- 12.Graf, J. 1999. Symbiosis of Aeromonas veronii biovar sobria and Hirudo medicinalis, the medicinal leech: a novel model for digestive tract associations. Infect. Immun. 67:1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guindon, S., and O. Gascuel. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52:696-704. [DOI] [PubMed] [Google Scholar]
- 14.Hasegawa, M., H. Kishino, and T. Yano. 1985. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 22:160-174. [DOI] [PubMed] [Google Scholar]
- 15.Iten, A., S. Graf, M. Egger, M. Täuber, and J. Graf. 2001. Helicobacter sp. flexispira bacteremia in an immunocompetent young adult. J. Clin. Microbiol. 39:1716-1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janda, J. M., and S. L. Abbott. 1998. Evolving concepts regarding the genus Aeromonas: an expanding panorama of species, disease presentations, and unanswered questions. Clin. Infect. Dis. 27:332-344. [DOI] [PubMed] [Google Scholar]
- 17.Klappenbach, J. A., J. M. Dunbar, and T. M. Schmidt. 2000. rRNA operon copy number reflects ecological strategies of bacteria. Appl. Environ. Microbiol. 66:1328-1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lane, D. J. 1991. 16S/23S rRNA sequencing. In E. Stackebrandt and M. Goodfellow (ed.), Nucleic acid techniques in bacterial systematics. John Wiley & Sons, New York, N.Y.
- 19.Marchandin, H., C. Teyssier, M. Simeon De Buochberg, H. Jean-Pierre, C. Carriere, and E. Jumas-Bilak. 2003. Intra-chromosomal heterogeneity between the four 16S rRNA gene copies in the genus Veillonella: implications for phylogeny and taxonomy. Microbiology 149:1493-1501. [DOI] [PubMed] [Google Scholar]
- 20.Martinez-Murcia, A. J., S. Benlloch, and M. D. Collins. 1992. Phylogenetic interrelationships of members of the genera Aeromonas and Plesiomonas as determined by 16S ribosomal DNA sequencing: lack of congruence with results of DNA-DNA hybridizations. Int. J. Syst. Bacteriol. 42:412-421. [DOI] [PubMed] [Google Scholar]
- 21.McLeod, E. S., Z. Dawood, R. MacDonald, M. C. Oosthuizen, J. Graf, P. L. Steyn, and V. S. Brözel. 1998. Isolation and identification of sulphite- and iron reducing, hydrogenase positive facultative anaerobes from cooling water systems. Syst. Appl. Microbiol. 21:297-305. [Google Scholar]
- 22.Nelson, K., and R. K. Selander. 1994. Analysis of genetic variation by polymerase chain reaction-based nucleotide sequencing. Methods Enzymol. 235:174-183. [DOI] [PubMed] [Google Scholar]
- 23.Pettersson, B., G. Bolske, F. Thiaucourt, M. Uhlen, and K. E. Johansson. 1998. Molecular evolution of Mycoplasma capricolum subsp. capripneumoniae strains, based on polymorphisms in the 16S rRNA genes. J. Bacteriol. 180:2350-2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pidiyar, V., A. Kaznowski, N. B. Narayan, M. Patole, and Y. S. Shouche. 2002. Aeromonas culicicola sp. nov., from the midgut of Culex quinquefasciatus. Int. J. Syst. Evol. Microbiol. 52:1723-1728. [DOI] [PubMed] [Google Scholar]
- 25.Pinus, M., and H. E. Muller. 1980. Enterobakterien bei Fledertieren (Chiroptera). Zentbl. Bakteriol. A 247:315-322. [PubMed] [Google Scholar]
- 26.Shimodaira, H. 2002. An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 51:492-508. [DOI] [PubMed] [Google Scholar]
- 27.Sneath, P. H. A. 1993. Evidence from Aeromonas for genetic crossing-over in ribosomal sequences. Int. J. Syst. Bacteriol. 43:626-629. [DOI] [PubMed] [Google Scholar]
- 28.Soler, L., M. A. Yanez, M. R. Chacon, M. G. Aguilera-Arreola, V. Catalan, M. J. Figueras, and A. J. Martinez-Murcia. 2004. Phylogenetic analysis of the genus Aeromonas based on two housekeeping genes. Int. J. Syst. Evol. Microbiol. 54:1511-1519. [DOI] [PubMed] [Google Scholar]
- 29.Teyssier, C., H. Marchandin, M. Simeon De Buochberg, M. Ramuz, and E. Jumas-Bilak. 2003. Atypical 16S rRNA gene copies in Ochrobactrum intermedium strains reveal a large genomic rearrangement by recombination between rrn copies. J. Bacteriol. 185:2901-2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ueda, K., T. Seki, T. Kudo, T. Yoshida, and M. Kataoka. 1999. Two distinct mechanisms cause heterogeneity of 16S rRNA. J. Bacteriol. 181:78-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang, Z. 1994. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J. Mol. Evol. 39:306-314. [DOI] [PubMed] [Google Scholar]
- 33.Yap, W. H., Z. Zhang, and Y. Wang. 1999. Distinct types of rRNA operons exist in the genome of the actinomycete Thermomonospora chromogena and evidence for horizontal transfer of an entire rRNA operon. J. Bacteriol. 181:5201-5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhaxybayeva, O., P. Lapierre, and J. P. Gogarten. 2004. Genome mosaicism and organismal lineages. Trends Genet. 20:254-260. [DOI] [PubMed] [Google Scholar]