

Abstract
IκB kinase/NF-κB (IKK/NF-κB) signaling pathways play critical roles in a variety of physiological and pathological processes. One function of NF-κB is promotion of cell survival through induction of target genes, whose products inhibit components of the apoptotic machinery in normal and cancerous cells. NF-κB can also prevent programmed necrosis by inducing genes encoding antioxidant proteins. Regardless of mechanism, many cancer cells, of either epithelial or hematopoietic origin, use NF-κB to achieve resistance to anticancer drugs, radiation, and death cytokines. Hence, inhibition of IKK-driven NF-κB activation offers a strategy for treatment of different malignancies and can convert inflammation-induced tumor growth to inflammation-induced tumor regression.
NF-κB proteins and IκB kinase signaling pathways
The mammalian NF-κB family contains 5 members: NF-κB1 (p105 and p50), NF-κB2 (p100 and p52), c-Rel, RelB, and RelA (p65). These proteins share a Rel homology domain (RHD), which mediates DNA binding, dimerization, and interactions with specific inhibitory factors, the IκBs, which retain NF-κB dimers in the cytoplasm. Many stimuli activate NF-κB, mostly through IκB kinase–dependent (IKK-dependent) phosphorylation and subsequent degradation of IκB proteins. The liberated NF-κB dimers enter the nucleus, where they regulate transcription of diverse genes encoding cytokines, growth factors, cell adhesion molecules, and pro- and antiapoptotic proteins (1, 2). The IKK complex consists of 2 highly homologous kinase subunits, IKKα and IKKβ, and a nonenzymatic regulatory component, IKKγ/NEMO (3).
Two NF-κB activation pathways exist (Figure 1). The first, the classical pathway, is normally triggered in response to microbial and viral infections or exposure to proinflammatory cytokines that activate the tripartite IKK complex, leading to phosphorylation-induced IκB degradation. This pathway, which mostly targets p50:RelA and p50:c-Rel dimers, depends mainly on IKKβ activity (4). The other pathway, the alternative pathway, leads to selective activation of p52:RelB dimers by inducing processing of the NF-κB2/p100 precursor protein, which mostly occurs as a heterodimer with RelB in the cytoplasm. This pathway is triggered by certain members of the TNF cytokine family, through selective activation of IKKα homodimers by the upstream kinase NIK (5). Both pathways regulate cell survival and death (6); the classical pathway is responsible for inhibition of programmed cell death (PCD) under most conditions (2, 3). The alternative pathway is important for survival of premature B cells and development of secondary lymphoid organs (7). The antiapoptotic activity of the IKKβ-driven classical pathway is important for various immunoreceptors, including T and B cell receptors, TLR4, and type 1 TNF-α receptor (TNFR1), all of which generate pro-survival and pro-death signals upon ligation (8, 9). Under most circumstances, the survival signals dominate, but under conditions where IKKβ or NF-κB activities have been compromised, receptor activation results in cell death (10–12).
Figure 1.

IKK/NF-κB signaling pathways. The classical pathway is activated by a variety of inflammatory signals, resulting in coordinate expression of multiple inflammatory and innate immune genes. The alternative pathway is strictly dependent on IKKα homodimers and is activated by lymphotoxin β receptor (LTβR), B cell–activating factor belonging to the TNF family (BAFF), and CD40 ligand (CD40L). The alternative pathway plays a central role in the expression of genes involved in development and maintenance of secondary lymphoid organs. BLC, B lymphocyte chemoattractant; ELC, Epstein-Barr virus–induced molecule 1 ligand CC chemokine; MCP-1, monocyte chemoattractant protein-1; MIP-1α, macrophage inflammatory protein-1α; PLA2, phospholipase A2; SDF-1, stromal cell–derived factor-1α; SLC, secondary lymphoid tissue chemokine.
The survival function of NF-κB: mechanisms and mediators
Pathways of cell death.
PCD can be either apoptotic or necrotic. Apoptosis is characterized by membrane blebbing, shrinking, and condensation of the cell and its organelles (13, 14). Two well-established pathways lead to apoptosis: the death receptor (DR) (extrinsic) pathway and the mitochondrial (intrinsic) pathway (15). Both pathways depend on cysteine proteases called caspases (15, 16). However, apoptosis-like PCD can sometimes proceed without caspase activation (17, 18). Furthermore, caspase activation does not always lead to cell death (19), and caspase-8 also has pro-survival functions (20, 21). Necrosis is characterized by swelling of the cell and its organelles, culminating in membrane disruption and cell lysis, often accompanied by inflammation. Failure of energy metabolism and massive generation of ROS are each thought to cause necrosis (22).
NF-κB suppresses both PCD types, although initially it was thought to antagonize only apoptosis. The first clear evidence for NF-κB as a PCD inhibitor was provided by RelA knockout mice that die mid-gestation by massive liver apoptosis (23). The role of NF-κB in embryonic liver survival, brought about by inhibition of TNFR1-mediated apoptosis (24), is underscored by the very similar phenotypes of mice lacking IKKβ (4, 25) or IKKγ (26). A protective role for NF-κB in adult liver was confirmed in mouse models of liver damage (10, 27, 28) and involves inhibition of both apoptosis and necrosis (9). We will discuss the various mechanisms by which NF-κB suppresses PCD (Figure 2).
Figure 2.
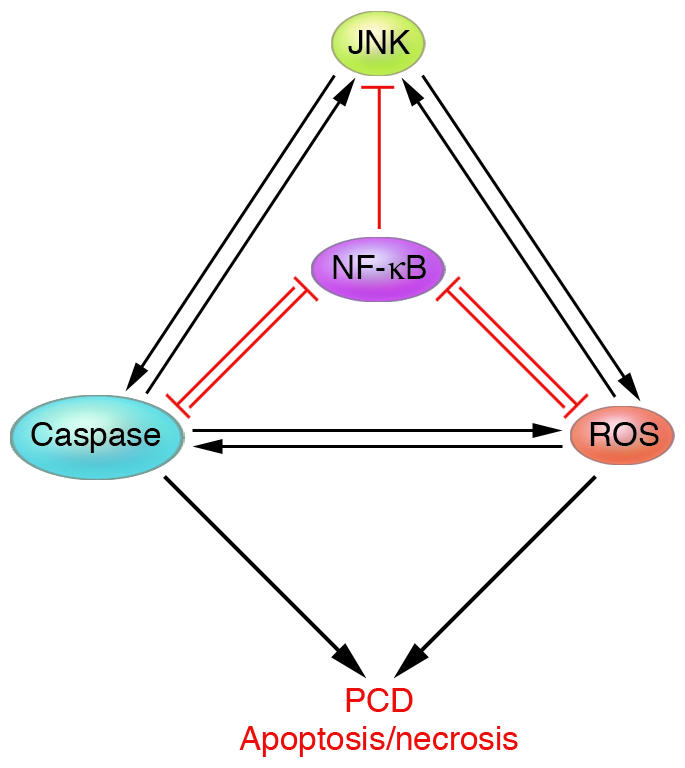
Control of cell survival and death through NF-κB–JNK cross-talk. Positive feedback loops exist between ROS and caspases, caspases and JNK, and JNK and ROS. Negative feedback loops exist between NF-κB and caspases, and NF-κB and ROS. NF-κB functions as a pro-survival transcription factor by inducing the expression of antiapoptotic genes, such as the Bcl-2 family members and caspase inhibitors, and antioxidant genes, such as MnSOD and FHC. Activation of NF-κB also results in inhibition of prolonged JNK activation, mostly through inhibition of ROS accumulation. Inhibition of NF-κB enhances PCD, which can be either apoptotic or necrotic, by removing the negative feedback loops.
NF-κB and caspases.
There are 2 groups of DRs, based on their signaling complexes. The first group comprises Fas, DR4, and DR5, which directly recruit the death domain–containing (DD-containing) adaptor Fas-associated death domain (FADD), procaspase-8, procaspase-10, and the cellular FLICE-inhibitory protein (FLIP) to form death-inducing signaling complexes (DISCs) (29). The second group comprises TNFR1, DR3, DR6, and ectodysplasin A receptor (EDAR). TNFR1 forms a signaling complex (complex I) at the plasma membrane by recruiting the adaptor TNFR1-associated DD protein (TRADD) and the signaling proteins TNFR-associated factor 2 (TRAF2), TRAF5, and receptor-interacting protein 1 (RIP1). After assembly, complex I dissociates from TNFR1, which can then recruit FADD and caspase-8 and trigger an apoptotic response (30).
NF-κB as a transcription factor induces genes whose products prevent PCD. An elicitor of NF-κB activation is TNF-α, which is a rather poor inducer of PCD. TNF-α triggers PCD only when new protein or RNA synthesis is inhibited or in NF-κB–deficient cells. NF-κB exerts its pro-survival activity through several antiapoptotic proteins, including FLIP, Bcl-XL, A1/Bfl-1, cellular inhibitor of apoptosis (c-IAP), X chromosome–linked inhibitor of apoptosis (XIAP), TRAF1, and TRAF2 (2, 31). FLIP inhibits apoptosis by interfering with caspase-8 activation (30). c-IAP and XIAP directly bind and inhibit effector caspases, acting downstream of initiator caspases.
NF-κB and Bcl-2 family members.
NF-κB induces expression of several Bcl-2 family members, most notably Bcl-XL and A1/Bfl-1, which prevent apoptosis by inhibiting permeability transition and depolarization of mitochondria, and cytochrome c release (2, 31). DRs can trigger apoptosis through different pathways (32). In certain cells, activated caspase-8 directly activates effector caspases, while in cells with poor DISC formation, death signaling requires an additional amplification loop, based on caspase-8–mediated Bid cleavage and generation of truncated tBid that triggers cytochrome c release (33, 34) and activation of caspase-9 and caspase-3 (15). This type of DR signaling can be blocked by antiapoptotic Bcl-2 family members, such as Bcl-2 and Bcl-XL (15).
ROS and the NF-κB–JNK cross-talk.
The role of JNK in PCD has been controversial, because it has both survival and death-enhancing effects. The clearest evidence for JNK as regulator of PCD comes from analysis of knockout mice: JNK1- or JNK2-deficient mice are relatively resistant to induction of fulminant hepatitis in response to concanavalin A, a pathology that depends on activation of TNFR1 and other DRs (10).
The ratio between JNK and NF-κB activities controls cell survival or death, not only in response to TNFR1 but also in response to other death stimuli (35–37). Whereas TNF-α leads to transient JNK activation in WT cells, it leads to prolonged JNK activation in cells that cannot activate NF-κB (9, 38–40). The pro-survival activity of NF-κB depends on this ability to prevent prolonged JNK activation (9, 38–40). Prolonged JNK activation following concanavalin A administration was also seen in mice lacking IKKβ in liver cells, resulting in massive TNFR1-dependent hepatocyte death (10). In the liver, however, TNFR1 and JNK signaling is also required for regeneration or compensatory hepatocyte proliferation following partial hepatectomy or chemically induced injury (41, 42). Thus, NF-κB may be a critical regulator of cell survival and death through its ability to control the duration of JNK activation (Figure 2).
Prolonged JNK activation in NF-κB–deficient cells implies that NF-κB induces expression of JNK inhibitors. Such a function was proposed for GADD45β (43) and XIAP (39). However, analysis of GADD45β- or XIAP-deficient fibroblasts failed to reveal changes in the kinetics of JNK activation (31, 44), suggesting that NF-κB regulates JNK activation through a different mediator.
ROS are likely the mediators (40). ROS, including H2O2, O2–, and HO• radicals, are generated through many enzymatic pathways, but their major source is leakage of electrons from the mitochondrial respiratory chain (45). ROS activate kinases through oxidation of kinase-interacting molecules. For instance, ROS activate tyrosine kinases by inactivating protein tyrosine phosphatases through oxidation of a highly reactive cysteine residue at their catalytic site (46–49). Similarly, ROS mediate the NF-κB–JNK cross-talk through their ability to inactivate various MAPK phosphatases (MKPs) involved in JNK inactivation (45).
TNF-α induces ROS accumulation in many cell types, and these ROS are important mediators of PCD (22). TNF-α–induced ROS accumulation is seen in NF-κB–deficient cells, but not in NF-κB–competent cells (9, 40). Treatment of cells with the antioxidant butylated hydroxyanisole (BHA) has no effect on transient JNK activation triggered by TNF-α, but it suppresses prolonged JNK activation and PCD in TNF-α–treated NF-κB–deficient cells (9, 40). This protective effect is due to BHA’s ability to prevent oxidation of MKPs, ensuring transient JNK activation (9). Expression of dominant-negative mutants of MKPs leads to prolonged JNK activation and allows killing by TNF-α of NF-κB–competent cells, which otherwise are TNF-α–resistant (9).
The loss of NF-κB activity results in ROS accumulation because NF-κB induces expression of several antioxidant genes such as manganese superoxide dismutase (MnSOD), ferritin heavy chain (FHC), glutathione-S-transferase, and metallothionein (50). Overexpression of mitochondrial MnSOD protects cells from TNF-α–induced cytotoxicity (9, 51). Overexpression of FHC also suppresses TNF-α–induced PCD along with attenuation of prolonged JNK activation (52). Another interesting observation is that TNF-α induces expression of a number of cytochrome p450 family members, such as CYP1B1, that enhance ROS production in NF-κB–deficient fibroblasts (37). Taken together, these findings show that NF-κB protects cells from oxidative stress by activating expression of various antioxidant systems, whose failure enhances TNF-α–induced PCD.
The mechanism of TNF-α–induced ROS production is unclear. One possible source of ROS is the cytosolic phospholipase A2 (53). However, several lines of evidence suggest that mitochondria are the main source of ROS during TNF-α–induced PCD (22). Compared with our understanding of DR-induced caspase activation, the mechanism of DR-induced ROS production is obscure. TNF-α does not induce ROS accumulation and programmed necrosis in FADD- or RIP1-deficient cells, indicating essential roles for FADD and RIP1 (54). In contrast to the established function of RIP1 as an adaptor molecule in NF-κB activation, its kinase activity is necessary for Fas-induced necrosis, which mostly occurs in caspase-8–deficient cells (55). Interestingly, inhibition of caspases potentiates ROS accumulation and cell death (53, 56). Although DRs can induce ROS accumulation without caspase activation in certain cell types, caspase activation can also lead to mitochondrial damage and ROS accumulation (33, 34, 57–59). Thus, caspase-dependent and -independent mechanisms might be involved in ROS accumulation.
JNK activation may also enhance ROS accumulation, potentiating TNF-α–stimulated necrosis (60). Although the mechanism by which JNK potentiates ROS accumulation is unclear, a positive feedback loop between ROS accumulation and JNK activation may exist (Figure 2). Such a loop may also involve caspase activation. Although caspases are not involved in TNF-α–induced prolonged JNK activation in NF-κB–deficient cells (40), caspase-mediated cleavage of upstream MAP3Ks may cause constitutive JNK activation (61). JNK activation also contributes to caspase activation, an effect mediated through enhanced cytochrome c release, during UV-induced apoptosis (62). Alternatively, JNK causes caspase activation through jBid formation during TNFR1 signaling (63). Importantly, NF-κB suppresses all of these amplification loops by inducing expression of caspase inhibitors, Bcl-2 family members, and antioxidants (Figure 2). Interestingly, negative feedback loops exist between NF-κB and various death-promoting proteins. Caspase-mediated cleavage of RelA and IKKβ can prevent NF-κB activation (64, 65). Caspases can also cleave IκB to generate a degradation-resistant NF-κB inhibitor (66). Oxidation of a cysteine residue in the RHD of RelA prevents its binding to DNA (67), whereas oxidation of another cysteine within the activation loop of IKKβ interferes with its activation (68). It is unlikely, however, that all of these regulatory loops and modifications take place simultaneously, and a major challenge for the future is to sort out the events that do take place during different physiological and pathophysiological conditions. It is possible to use some of these regulatory loops in designing drugs and therapeutic strategies to kill cancer cells. Fas and TNF-α can induce both apoptosis and necrosis, and so do anticancer drugs. In L929 cells, for instance, TNF-α triggers mostly necrosis, whereas Fas can induce necrosis only when the apoptotic pathway is suppressed (69). FADD and RIP play central roles in controlling the choice between the 2 death pathways (70). NF-κB activation also inhibits programmed necrosis, in addition to its role in prevention of apoptosis.
Proapoptotic functions of NF-κB?
NF-κ B may induce apoptosis in a cell type– and stimulus-dependent manner.
Most commonly, NF-κB activation inhibits PCD, as evidenced by several knockout mouse models (4, 23, 26, 71). However, under certain circumstances activation of NF-κB may promote cell death. For instance, NF-κB may mediate doxorubicin-induced cell death in N-type neuroblastoma cells (72). NF-κB is also required for anti-CD3–induced apoptosis of double-positive thymocytes (73). Apoptosis in HL-60 cells induced by etoposide or 1-β-D-arabinofuranosylcytosine correlates with NF-κB activation (74). Human melanoma cells were protected from UV-induced apoptosis by NF-κB downregulation (75). More recently, it was reported that NF-κB induced by UV light or daunorubicin/doxorubicin is functionally distinct from the response elicited by TNF-α, and under such conditions NF-κB may become a repressor of antiapoptotic genes (76). Furthermore, UV light and daunorubicin inhibit TNF-α–induced NF-κB transcriptional activity, which is antiapoptotic, by enhancing association of RelA with histone deacetylases (76). These results suggest that NF-κB may mediate apoptosis under certain conditions. However, the pathophysiological relevance of these observations is not clear, and it remains to be demonstrated that NF-κB has proapoptotic functions in vivo. It appears that those agents or stimuli that were reported to induce apoptosis by activating NF-κB are neither strong nor typical NF-κB activators, as opposed to TNF-α, IL-1, or LPS, and that they also activate another signaling pathway(s), which may be more relevant to cell killing than NF-κB.
Tumor suppressors interact with NF-κB pathway.
Suppression of cell proliferation, induction of premature senescence, and/or induction of apoptosis are some mechanisms through which tumor suppressors inhibit cancer development. In general, NF-κB acts antagonistically to tumor suppressors, based on its ability to promote cell survival, inhibit PCD, and enhance cell proliferation (77). However, in some cases NF-κB may collaborate with, rather than antagonize, certain tumor suppressors.
Although p53 stabilization decreases upon NF-κB activation (78), under special circumstances apoptosis induced by p53 may involve activation of NF-κB (79). Similar to situations in which NF-κB activation promotes apoptosis, NF-κB induction by p53 does not involve classical IKK activation and IκB degradation. Instead, p53 may stimulate the serine/threonine kinase ribosomal S6 kinase 1 (RSK1), which in turn phosphorylates RelA (80). The lower affinity of RSK1-phosphorylated RelA for IκBα decreases IκBα-mediated nuclear export, prolonging RelA nuclear residence (80). NF-κB also plays an essential role in activation of p53 to initiate proapoptotic signaling in response to ROS accumulation. Consequently, NF-κB–dependent p53 activity induces p53-regulated genes, such as Puma and p21waf1 (81). However, a more common observation, seen in vivo, is that NF-κB activation counteracts p53-induced apoptosis by destabilizing p53, perhaps through enhanced Mdm2 expression (78, 82).
Another tumor suppressor, BRCA1, can bind RelA to serve as a coactivator (83). Treatment of 293T cells with TNF-α induces an interaction between endogenous RelA and BRCA1, mediated by the RHD of RelA and the N-terminal region of BRCA1. Forced BRCA1 expression significantly enhances the ability of TNF-α or IL-1β to induce NF-κB target genes, and inhibition of NF-κB by the chemical inhibitor SN-50 blocks this effect (83). Nonetheless, it remains to be seen whether any of these responses documented in vitro occurs in vivo.
NF-κB and proapoptotic genes.
NF-κB has been implicated as a transcriptional activator of some proapoptotic genes, such as Fas/CD95 (84), FasL (85), DR4, and DR5 (86). FasL is expressed in activated T cells and represents a major cytotoxic effector through which T cells kill their targets. FasL expression is under the stringent control of various transcription factors, including NF-κB (85). Recently, it was reported that certain types of cancer cells also express FasL, which may contribute to their ability to escape immune surveillance and resist immunotherapy. Overexpression of the Myc family member Max in non–small cell lung cancer cell lines markedly increases basal FasL promoter activity and enhances NF-κB–mediated FasL induction. Thus, high levels of Max and stress-induced NF-κB activation may elevate FasL expression in human lung cancer cells (85). TNF-α combined with IFN-α accelerates NF-κB–mediated apoptosis by enhancing Fas expression in human colon adenocarcinoma RPMI4788 cells (84). However, there may be another explanation for these results, as type I IFNs and related cytokines, such as IL-10, may actually function as NF-κB inhibitors (87). Another TNF family member, TRAIL, triggers apoptosis through engagement of DR4 and DR5. The c-Rel subunit of NF-κB induces expression of both receptors, while a degradation-resistant mutant of IκBα (IκB super-repressor) or a transactivation-deficient mutant of c-Rel reduces DR expression (86). However, NF-κB was shown to be a major impediment to TRAIL-mediated tumor killing (88).
IKK/NF-κB and cancer
IKK/NF-κB links inflammation to cancer.
Based on many functions of NF-κB target genes, a close relationship between NF-κB and cancer was proposed (89) and recently reviewed (89–94). The association of NF-κB activation with inflammation-associated tumor promotion, progression, and metastasis is well documented and was demonstrated in several mouse models (88, 95, 96). The IKKβ-dependent NF-κB activation pathway is a critical molecular link between inflammation and colon cancer in a mouse model (95). Activation of IKKβ in enterocytes, which give rise to the malignant component of this tumor, suppresses apoptosis of preneoplastic cells, whereas its activation in myeloid cells promotes production of various cytokines that serve as growth factors for the transformed enterocytes. Inhibition of 1 of these factors, IL-6, interferes with tumor growth but has no effect on tumor cell survival (97). Conversely, inactivation of IKKβ in enterocytes results in a dramatic decrease in tumor number due to increased apoptosis but has no effect on proliferation of transformed enterocytes or tumor growth (95).
The role of NF-κB in inflammation-associated cancer was also demonstrated in Mdr2-deficient mice, which develop cholestatic hepatitis followed by hepatocellular carcinoma (96). In this model, the inflammatory process triggered chronic activation of NF-κB in hepatocytes, most likely through enhanced production of TNF-α by adjacent endothelial and inflammatory cells. Switching NF-κB off in Mdr2–/– mice from birth to 7 months of age had no effect on the course of hepatitis or early phases of tumorigenesis (96). By contrast, suppressing chronic NF-κB activation at later stages resulted in the apoptotic death of transformed hepatocytes and failure to progress to hepatocellular carcinoma (96).
NF-κB activation also plays a critical role in inflammation-driven tumor progression as demonstrated in a syngeneic colon and mammary cancer xenograft mouse model (88). Cancer cells in this model were introduced into syngeneic immunocompetent mice to form metastatic growths in the lungs. Once the metastases were established, the mice were given a sublethal dose of LPS to elicit systemic inflammation, which stimulated tumor growth. Remarkably, inhibition of NF-κB in cancer cells converted LPS-induced tumor growth to LPS-induced tumor regression without affecting the ability of the cancer cells to migrate to the lung and establish metastatic growths (88). Further investigation revealed that inflammation-induced tumor growth in this model was mediated by TNF-α produced by host immune cells, whereas LPS-induced regression of NF-κB–deficient tumors was mediated by TRAIL, whose synthesis is induced by IFNs in host inflammatory cells in response to LPS-mediated activation of TLR4 (Figure 3). These results indicate that NF-κB is a major mediator of inflammation-induced tumor progression through overcoming the potential tumor-killing by TRAIL induction (88). Given that NF-κB activation in cancer cells may be a major hindrance to TRAIL-induced apoptosis, NF-κB or IKK inhibitors may potentiate the activity of either recombinant TRAIL or TRAIL inducers, such as type I and type II IFNs, to achieve enhanced tumor killing (Figure 3).
Figure 3.

Inhibition of NF-κB in cancer cells converts inflammation-induced tumor growth to tumor regression. Activation of the innate and adaptive immune system can have profound influence on tumor growth and development. In addition to its role in activation of immune cells, NF-κB within the malignant cell is a major modulator of the tumor response to inflammation. Activation of NF-κB promotes tumor growth and confers resistance to death cytokines, such as TRAIL. Conversely, inhibition of NF-κB prevents inflammation-stimulated tumor growth and enhances inflammation-induced tumor regression mediated by TRAIL.
Whereas the role of NF-κB in inflammation-induced tumor promotion, growth, and progression is becoming clear (88, 95, 96), its role in tumor initiation is still ambiguous (98–101). As NF-κB regulates a large group of genes that have different functions, some of which display cell-type specificity, NF-κB may have distinct roles in different cell types. For instance, in normal epidermal keratinocytes, NF-κB proteins are present in the cytoplasm of basal cells but are nuclear in more differentiated suprabasal cells, suggesting that NF-κB activation is linked to growth arrest (98, 102). Indeed, inhibition of NF-κB signaling in the murine epidermis results in an increased apoptosis, hyperproliferation of surviving cells, and spontaneous development of squamous cell carcinomas (99, 103). Correspondingly, application of a pharmacological NF-κB inhibitor to mouse skin induced epidermal hyperplasia (98). In contrast, overexpression of active NF-κB subunits in transgenic epithelium produced hypoplasia and growth inhibition (98). Contrary to the requirement of an intact IKK/NF-κB pathway for H-ras–mediated fibroblast transformation (104), NF-κB inhibition synergized with oncogenic H-ras to induce transformation of primary human keratinocytes (100). Congruously, activation of NF-κB in normal human epidermal keratinocytes triggered cell-cycle arrest (100). These results suggest that the IKKβ-dependent NF-κB pathway in epidermal keratinocytes promotes keratinocyte growth arrest and differentiation to maintain the barrier function of the epidermis, whose perturbation may result in severe inflammation (105). Interestingly, formation of mouse squamous cell carcinomas in response to a chemical carcinogen (106) and following inhibition of NF-κB (107) is dependent on TNF-α. Similar observations were recently made in a mouse model of chemically induced hepatocellular carcinoma, where deletion of IKKβ in hepatocytes promoted tumor development by enhancing compensatory proliferation, whereas an additional deletion of IKKβ in liver myeloid cells prevented tumor development by depriving the transformed hepatocytes of essential growth factors (108).
NF-κB inhibitors in cancer therapy.
The pivotal role of the IKKβ/NF-κB signaling pathway in inhibition of PCD, tumor promotion, and tumor progression, together with the occurrence of constitutively activated NF-κB in various solid and hematopoietic malignancies, strongly suggests that IKKβ and/or NF-κB inhibitors would be useful in cancer therapy. In fact, much effort is currently invested in developing various IKKβ and/or NF-κB inhibitors and testing their efficacy in both animal models and human cancer (109, 110). Many inhibitors currently available are not specific for either IKKβ or NF-κB. These include antiinflammatory agents such as sulfasalazine and trans-resveratrol, NSAIDs such as aspirin and sulindac sulfide, cyclopentenone prostaglandins, proteasome inhibitors, and glucocorticoids (90, 109–112). However, specific IKKβ inhibitors are being developed, and a few publications have documented their efficacy in triggering apoptosis in cancer cell lines in combination with either death-inducing cytokines or chemotherapeutic drugs (113–115).
Even nonspecific IKKβ/NF-κB inhibitors may be effective when used as adjuvants with conventional anticancer treatments. As many signaling pathways may be simultaneously activated and/or inactivated in a given malignant cell, collectively contributing to its neoplastic phenotype, nonspecific IKKβ/NF-κB inhibitors may affect several signaling pathways at once and lead to much more effective killing of such cells. The anticancer drug arsenic trioxide (ATO), which is useful for treating promyelocytic leukemia (116) and possibly multiple myeloma (117), is a noteworthy example. ATO is not a specific inhibitor for IKKβ or NF-κB and may have several molecular targets, since it was found that trivalent arsenicals, a chemical class to which ATO belongs, are potent JNK activators (118) as well as IKKβ inhibitors (68). JNK activation in this case is mostly due to the ability of trivalent arsenicals to directly interact with the catalytic cysteine of JNK phosphatases, whereas in the case of IKKβ the target is the aforementioned reactive cysteine within the activation loop. An additional effect of ATO on JNK activity may be due to NF-κB inhibition and accumulation of ROS (35). Thus, by inhibiting IKK and activating JNK, ATO may trigger apoptosis in many different types of cancers. Since NF-κB inhibition usually does not result in spontaneous apoptosis, it is unlikely that even specific IKKβ/NF-κB inhibitors would be functional as monotherapeutic agents in most cancers. Indeed, using Jurkat cells as a model, the IKKβ inhibitor AS602868 was not cytocidal on its own but strongly potentiated killing by TNF-α (113). Based on our analysis of knockout mice and tumor models, we predict that IKKβ/NF-κB inhibitors will be useful adjuvants for conventional chemotherapeutic drugs, ionizing radiation, or tumoricidal cytokines, such as IFNs or TRAIL (Figure 3).
NF-κB regulates PCD through a cross-talk with JNK, ROS, and caspases, and an important pro-survival factor regulated by NF-κB is the antioxidant enzyme MnSOD (9, 51). Thus, MnSOD2 inhibitors may target a particular NF-κB function, the suppression of ROS production and PCD, while leaving other functions, such as innate immunity, intact. In fact, inhibition of superoxide dismutase (SOD) in human leukemia cells caused accumulation of O2–, which was followed by ROS-mediated mitochondrial damage, cytochrome c release, and apoptosis (119). Given its regulation by NF-κB, whose activity is elevated in most types of cancer, it is likely that MnSOD expression is higher in malignant cells than in normal cells, and therefore the former may be more sensitive to SOD inhibitors. In fact, certain estrogen derivatives, acting as SOD inhibitors, selectively kill human leukemia cells but not normal lymphocytes (119). In case such compounds are not sufficiently potent on their own, they need to be tested as adjuvants for more conventional chemotherapeutic and radiotherapeutic approaches.
Concluding remarks
The inhibition of IKKβ/NF-κB appears to be a promising strategy for cancer therapy when combined with established cytocidal drugs, death cytokines, or therapeutic radiation. Certain anticancer drugs may work much better with IKKβ/NF-κB inhibitors than others. For instance, the combined application of TRAIL or TRAIL inducers, such as IFNs (Figure 3), with antiinflammatory or anti–TNF-α therapy alongside IKKβ/NF-κB inhibitors may result in selective killing of malignant cells not achieved by either agent alone (88). An important advantage of IKKβ/NF-κB inhibitors over conventional therapeutics is their ability to block NF-κB activation also in infiltrating inflammatory cells, which are an important source of tumor growth and survival factors. It should be noted, however, that, given the critical role of NF-κB in innate and adaptive immune responses, there may be a certain amount of risk due to induced immunodeficiency caused by long-term use of IKKβ/NF-κB inhibitors. Hence, alternative approaches should be considered. For instance, an approach based on selective inhibition of antiapoptotic targets of NF-κB, without affecting target genes required for immune responses, would be particularly attractive.
Acknowledgments
Work in the authors’ laboratory was supported by grants from the NIH (ES04151, AI43477, CA76188), the US Army Medical Research and Materiel Command (W8IXWH-04-1-0120), and the Prostate Cancer Foundation. M. Karin is an American Cancer Society Research Professor.
Footnotes
Nonstandard abbreviations used: ATO, arsenic trioxide; DD, death domain; DISC, death-inducing signaling complex; DR, death receptor; FADD, Fas-associated death domain; FHC, ferritin heavy chain; FLIP, FLICE-inhibitory protein; IKK, IκB kinase; MKP, MAPK phosphatase; MnSOD, manganese superoxide dismutase; PCD, programmed cell death; RHD, Rel homology domain; RIP, receptor-interacting protein; SOD, superoxide dismutase; TNFR1, type 1 TNF-α receptor; TRAF, TNFR-associated factor; XIAP, X chromosome–linked inhibitor of apoptosis.
Conflict of interest: The authors have declared that no conflict of interest exists.
References
- 1.Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle [review] Cell. 2002;109(Suppl.):S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 2.Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat. Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 3.Karin M, Delhase M. The I kappa B kinase (IKK) and NF-kappa B: key elements of proinflammatory signalling. Semin. Immunol. 2000;12:85–98. doi: 10.1006/smim.2000.0210. [DOI] [PubMed] [Google Scholar]
- 4.Li ZW, et al. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J. Exp. Med. 1999;189:1839–1845. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Senftleben U, et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 6.Senftleben U, Karin M. The IKK/NF-kappa B pathway. Crit. Care Med. 2002;30(1 Suppl.):S18–S26. [Google Scholar]
- 7.Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 8.Varfolomeev EE, Ashkenazi A. Tumor necrosis factor: an apoptosis JuNKie [review]? Cell. 2004;116:491–497. doi: 10.1016/s0092-8674(04)00166-7. [DOI] [PubMed] [Google Scholar]
- 9.Kamata H, et al. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 10.Maeda S, et al. IKKbeta is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFalpha. Immunity. 2003;19:725–737. doi: 10.1016/s1074-7613(03)00301-7. [DOI] [PubMed] [Google Scholar]
- 11.Weil R, Israel A. T-cell-receptor- and B-cell-receptor-mediated activation of NF-kappaB in lymphocytes. Curr. Opin. Immunol. 2004;16:374–381. doi: 10.1016/j.coi.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 12.Hsu LC, et al. The protein kinase PKR is required for macrophage apoptosis after activation of Toll-like receptor 4. Nature. 2004;428:341–345. doi: 10.1038/nature02405. [DOI] [PubMed] [Google Scholar]
- 13.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics [review] Br. J. Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wyllie AH, Kerr JF, Currie AR. Cell death: the significance of apoptosis [review] Int. Rev. Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 15.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 16.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 17.Leist M, Jaattela M. Four deaths and a funeral: from caspases to alternative mechanisms [review] Nat. Rev. Mol. Cell Biol. 2001;2:589–598. doi: 10.1038/35085008. [DOI] [PubMed] [Google Scholar]
- 18.Jaattela M, Tschopp J. Caspase-independent cell death in T lymphocytes. Nat. Immunol. 2003;4:416–423. doi: 10.1038/ni0503-416. [DOI] [PubMed] [Google Scholar]
- 19.Abraham MC, Shaham S. Death without caspases, caspases without death. Trends Cell Biol. 2004;14:184–193. doi: 10.1016/j.tcb.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 20.Yu L, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 21.Kang TB, et al. Caspase-8 serves both apoptotic and nonapoptotic roles. J. Immunol. 2004;173:2976–2984. doi: 10.4049/jimmunol.173.5.2976. [DOI] [PubMed] [Google Scholar]
- 22.Fiers W, Beyaert R, Declercq W, Vandenabeele P. More than one way to die: apoptosis, necrosis and reactive oxygen damage [review] Oncogene. 1999;18:7719–7730. doi: 10.1038/sj.onc.1203249. [DOI] [PubMed] [Google Scholar]
- 23.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 24.Doi TS, et al. Absence of tumor necrosis factor rescues RelA-deficient mice from embryonic lethality. Proc. Natl. Acad. Sci. U. S. A. 1999;96:2994–2999. doi: 10.1073/pnas.96.6.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science. 1999;284:321–325. doi: 10.1126/science.284.5412.321. [DOI] [PubMed] [Google Scholar]
- 26.Makris C, et al. Female mice heterozygous for IKK gamma/NEMO deficiencies develop a dermatopathy similar to the human X-linked disorder incontinentia pigmenti. Mol. Cell. 2000;5:969–979. doi: 10.1016/s1097-2765(00)80262-2. [DOI] [PubMed] [Google Scholar]
- 27.Chaisson ML, Brooling JT, Ladiges W, Tsai S, Fausto N. Hepatocyte-specific inhibition of NF-kappaB leads to apoptosis after TNF treatment, but not after partial hepatectomy. J. Clin. Invest. 2002;110:193–202. doi:10.1172/JCI200215295. doi: 10.1172/JCI15295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lavon I, et al. High susceptibility to bacterial infection, but no liver dysfunction, in mice compromised for hepatocyte NF-kappaB activation. Nat. Med. 2000;6:573–577. doi: 10.1038/75057. [DOI] [PubMed] [Google Scholar]
- 29.Peter ME, Krammer PH. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003;10:26–35. doi: 10.1038/sj.cdd.4401186. [DOI] [PubMed] [Google Scholar]
- 30.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 31.Kucharczak J, Simmons MJ, Fan Y, Gelinas C. To be, or not to be: NF-kappaB is the answer. Role of Rel/NF-kappaB in the regulation of apoptosis [review] Oncogene. 2003;22:8961–8982. doi: 10.1038/sj.onc.1207230. [DOI] [PubMed] [Google Scholar]
- 32.Scaffidi C, et al. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998;17:1675–1687. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 34.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y, Chen F. Reactive oxygen species (ROS), troublemakers between nuclear factor-kappaB (NF-kappaB) and c-Jun NH(2)-terminal kinase (JNK) Cancer Res. 2004;64:1902–1905. doi: 10.1158/0008-5472.can-03-3361. [DOI] [PubMed] [Google Scholar]
- 36.Papa S, Zazzeroni F, Pham CG, Bubici C, Franzoso G. Linking JNK signaling to NF-kappaB: a key to survival. J. Cell Sci. 2004;117:5197–5208. doi: 10.1242/jcs.01483. [DOI] [PubMed] [Google Scholar]
- 37.Chen F, Castranova V, Li Z, Karin M, Shi X. Inhibitor of nuclear factor kappaB kinase deficiency enhances oxidative stress and prolongs c-Jun NH2-terminal kinase activation induced by arsenic. Cancer Res. 2003;63:7689–7693. [PubMed] [Google Scholar]
- 38.De Smaele E, et al. Induction of gadd45beta by NF-kappaB downregulates pro-apoptotic JNK signalling. Nature. 2001;414:308–313. doi: 10.1038/35104560. [DOI] [PubMed] [Google Scholar]
- 39.Tang G, et al. Inhibition of JNK activation through NF-kappaB target genes. Nature. 2001;414:313–317. doi: 10.1038/35104568. [DOI] [PubMed] [Google Scholar]
- 40.Sakon S, et al. NF-kappaB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J. 2003;22:3898–3909. doi: 10.1093/emboj/cdg379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwabe RF, et al. c-Jun-N-terminal kinase drives cyclin D1 expression and proliferation during liver regeneration. Hepatology. 2003;37:824–832. doi: 10.1053/jhep.2003.50135. [DOI] [PubMed] [Google Scholar]
- 42.Yamada Y, Kirillova I, Peschon JJ, Fausto N. Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc. Natl. Acad. Sci. U. S. A. 1997;94:1441–1446. doi: 10.1073/pnas.94.4.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Papa S, et al. Gadd45 beta mediates the NF-kappa B suppression of JNK signalling by targeting MKK7/JNKK2. Nat. Cell Biol. 2004;6:146–153. doi: 10.1038/ncb1093. [DOI] [PubMed] [Google Scholar]
- 44.Amanullah A, et al. Cell signalling: cell survival and a Gadd45-factor deficiency [comment] Nature. 2003;424:741; discussion, 742. doi: 10.1038/424741b. [DOI] [PubMed] [Google Scholar]
- 45.Kamata H, Hirata H. Redox regulation of cellular signalling. Cell. Signal. 1999;11:1–14. doi: 10.1016/s0898-6568(98)00037-0. [DOI] [PubMed] [Google Scholar]
- 46.Salmeen A, et al. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 47.van Montfort RL, Congreve M, Tisi D, Carr R, Jhoti H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature. 2003;423:773–777. doi: 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- 48.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 49.Meng TC, Buckley DA, Galic S, Tiganis T, Tonks NK. Regulation of insulin signaling through reversible oxidation of the protein-tyrosine phosphatases TC45 and PTP1B. J. Biol. Chem. 2004;279:37716–37725. doi: 10.1074/jbc.M404606200. [DOI] [PubMed] [Google Scholar]
- 50.Sasazuki T, et al. Genome wide analysis of TNF-inducible genes reveals that antioxidant enzymes are induced by TNF and responsible for elimination of ROS. Mol. Immunol. 2004;41:547–551. doi: 10.1016/j.molimm.2004.03.030. [DOI] [PubMed] [Google Scholar]
- 51.Wong GH, Elwell JH, Oberley LW, Goeddel DV. Manganous superoxide dismutase is essential for cellular resistance to cytotoxicity of tumor necrosis factor. Cell. 1989;58:923–931. doi: 10.1016/0092-8674(89)90944-6. [DOI] [PubMed] [Google Scholar]
- 52.Pham CG, et al. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell. 2004;119:529–542. doi: 10.1016/j.cell.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 53.Cauwels A, Janssen B, Waeytens A, Cuvelier C, Brouckaert P. Caspase inhibition causes hyperacute tumor necrosis factor-induced shock via oxidative stress and phospholipase A2. Nat. Immunol. 2003;4:387–393. doi: 10.1038/ni914. [DOI] [PubMed] [Google Scholar]
- 54.Lin Y, et al. Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J. Biol. Chem. 2004;279:10822–10828. doi: 10.1074/jbc.M313141200. [DOI] [PubMed] [Google Scholar]
- 55.Holler N, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 56.Liu CY, et al. Broad-spectrum caspase inhibition paradoxically augments cell death in TNF-alpha-stimulated neutrophils. Blood. 2003;101:295–304. doi: 10.1182/blood-2001-12-0266. [DOI] [PubMed] [Google Scholar]
- 57.Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat. Cell Biol. 2000;2:156–162. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- 58.Waterhouse NJ, et al. Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J. Cell Biol. 2001;153:319–328. doi: 10.1083/jcb.153.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ricci JE, et al. Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell. 2004;117:773–786. doi: 10.1016/j.cell.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 60.Ventura JJ, Cogswell P, Flavell RA, Baldwin AS, Jr, Davis RJ. JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev. 2004;18:2905–2915. doi: 10.1101/gad.1223004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cardone MH, Salvesen GS, Widmann C, Johnson G, Frisch SM. The regulation of anoikis: MEKK-1 activation requires cleavage by caspases. Cell. 1997;90:315–323. doi: 10.1016/s0092-8674(00)80339-6. [DOI] [PubMed] [Google Scholar]
- 62.Tournier C, et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288:870–874. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 63.Deng Y, Ren X, Yang L, Lin Y, Wu X. A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell. 2003;115:61–70. doi: 10.1016/s0092-8674(03)00757-8. [DOI] [PubMed] [Google Scholar]
- 64.Levkau B, Scatena M, Giachelli CM, Ross R, Raines EW. Apoptosis overrides survival signals through a caspase-mediated dominant-negative NF-kappa B loop. Nat. Cell Biol. 1999;1:227–233. doi: 10.1038/12050. [DOI] [PubMed] [Google Scholar]
- 65.Tang G, Yang J, Minemoto Y, Lin A. Blocking caspase-3-mediated proteolysis of IKKbeta suppresses TNF-alpha-induced apoptosis. Mol. Cell. 2001;8:1005–1016. doi: 10.1016/s1097-2765(01)00380-x. [DOI] [PubMed] [Google Scholar]
- 66.Reuther JY, Baldwin AS., Jr Apoptosis promotes a caspase-induced amino-terminal truncation of IkappaBalpha that functions as a stable inhibitor of NF-kappaB. J. Biol. Chem. 1999;274:20664–20670. doi: 10.1074/jbc.274.29.20664. [DOI] [PubMed] [Google Scholar]
- 67.Toledano MB, Leonard WJ. Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc. Natl. Acad. Sci. U. S. A. 1991;88:4328–4332. doi: 10.1073/pnas.88.10.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kapahi P, et al. Inhibition of NF-kappa B activation by arsenite through reaction with a critical cysteine in the activation loop of Ikappa B kinase. J. Biol. Chem. 2000;275:36062–36066. doi: 10.1074/jbc.M007204200. [DOI] [PubMed] [Google Scholar]
- 69.Denecker G, et al. Death receptor-induced apoptotic and necrotic cell death: differential role of caspases and mitochondria. Cell Death Differ. 2001;8:829–840. doi: 10.1038/sj.cdd.4400883. [DOI] [PubMed] [Google Scholar]
- 70.Vanden Berghe T, et al. Differential signaling to apoptotic and necrotic cell death by Fas-associated death domain protein FADD. J. Biol. Chem. 2004;279:7925–7933. doi: 10.1074/jbc.M307807200. [DOI] [PubMed] [Google Scholar]
- 71.Schmidt-Supprian M, et al. NEMO/IKK gamma-deficient mice model incontinentia pigmenti. Mol. Cell. 2000;5:981–992. doi: 10.1016/s1097-2765(00)80263-4. [DOI] [PubMed] [Google Scholar]
- 72.Bian X, et al. NF-kappa B activation mediates doxorubicin-induced cell death in N-type neuroblastoma cells. J. Biol. Chem. 2001;276:48921–48929. doi: 10.1074/jbc.M108674200. [DOI] [PubMed] [Google Scholar]
- 73.Hettmann T, DiDonato J, Karin M, Leiden JM. An essential role for nuclear factor kappaB in promoting double positive thymocyte apoptosis. J. Exp. Med. 1999;189:145–158. doi: 10.1084/jem.189.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bessho R, et al. Pyrrolidine dithiocarbamate, a potent inhibitor of nuclear factor kappa B (NF-kappa B) activation, prevents apoptosis in human promyelocytic leukemia HL-60 cells and thymocytes. Biochem. Pharmacol. 1994;48:1883–1889. doi: 10.1016/0006-2952(94)90586-x. [DOI] [PubMed] [Google Scholar]
- 75.Ivanov VN, Ronai Z. p38 protects human melanoma cells from UV-induced apoptosis through down-regulation of NF-kappaB activity and Fas expression. Oncogene. 2000;19:3003–3012. doi: 10.1038/sj.onc.1203602. [DOI] [PubMed] [Google Scholar]
- 76.Campbell KJ, Rocha S, Perkins ND. Active repression of antiapoptotic gene expression by RelA(p65) NF-kappa B. Mol. Cell. 2004;13:853–865. doi: 10.1016/s1097-2765(04)00131-5. [DOI] [PubMed] [Google Scholar]
- 77.Webster GA, Perkins ND. Transcriptional cross talk between NF-kappaB and p53. Mol. Cell. Biol. 1999;19:3485–3495. doi: 10.1128/mcb.19.5.3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tergaonkar V, Pando M, Vafa O, Wahl G, Verma I. p53 stabilization is decreased upon NFkappaB activation: a role for NFkappaB in acquisition of resistance to chemotherapy. Cancer Cell. 2002;1:493–503. doi: 10.1016/s1535-6108(02)00068-5. [DOI] [PubMed] [Google Scholar]
- 79.Ryan KM, Ernst MK, Rice NR, Vousden KH. Role of NF-kappaB in p53-mediated programmed cell death. Nature. 2000;404:892–897. doi: 10.1038/35009130. [DOI] [PubMed] [Google Scholar]
- 80.Bohuslav J, Chen LF, Kwon H, Mu Y, Greene WC. p53 induces NF-kappaB activation by an IkappaB kinase-independent mechanism involving phosphorylation of p65 by ribosomal S6 kinase 1. J. Biol. Chem. 2004;279:26115–26125. doi: 10.1074/jbc.M313509200. [DOI] [PubMed] [Google Scholar]
- 81.Fujioka S, et al. Stabilization of p53 is a novel mechanism for proapoptotic function of NF-kappaB. J. Biol. Chem. 2004;279:27549–27559. doi: 10.1074/jbc.M313435200. [DOI] [PubMed] [Google Scholar]
- 82.Egan LJ, et al. IkappaB-kinasebeta-dependent NF-kappaB activation provides radioprotection to the intestinal epithelium. Proc. Natl. Acad. Sci. U. S. A. 2004;101:2452–2457. doi: 10.1073/pnas.0306734101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Benezra M, et al. BRCA1 augments transcription by the NF-kappaB transcription factor by binding to the Rel domain of the p65/RelA subunit. J. Biol. Chem. 2003;278:26333–26341. doi: 10.1074/jbc.M303076200. [DOI] [PubMed] [Google Scholar]
- 84.Kimura M, et al. TNF combined with IFN-alpha accelerates NF-kappaB-mediated apoptosis through enhancement of Fas expression in colon cancer cells. Cell Death Differ. 2003;10:718–728. doi: 10.1038/sj.cdd.4401219. [DOI] [PubMed] [Google Scholar]
- 85.Wiener Z, et al. Synergistic induction of the Fas (CD95) ligand promoter by Max and NFkappaB in human non-small lung cancer cells. Exp. Cell Res. 2004;299:227–235. doi: 10.1016/j.yexcr.2004.05.031. [DOI] [PubMed] [Google Scholar]
- 86.Ravi R, et al. Regulation of death receptor expression and TRAIL/Apo2L-induced apoptosis by NF-kappaB. Nat. Cell Biol. 2001;3:409–416. doi: 10.1038/35070096. [DOI] [PubMed] [Google Scholar]
- 87.Driessler F, Venstrom K, Sabat R, Asadullah K, Schottelius AJ. Molecular mechanisms of interleukin-10-mediated inhibition of NF-kappaB activity: a role for p50. Clin. Exp. Immunol. 2004;135:64–73. doi: 10.1111/j.1365-2249.2004.02342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Luo JL, Maeda S, Hsu LC, Yagita H, Karin M. Inhibition of NF-kappaB in cancer cells converts inflammation-induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Cancer Cell. 2004;6:297–305. doi: 10.1016/j.ccr.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 89.Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit [review] Nat. Rev. Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 90.Lin A, Karin M. NF-kappaB in cancer: a marked target. Semin. Cancer Biol. 2003;13:107–114. doi: 10.1016/s1044-579x(02)00128-1. [DOI] [PubMed] [Google Scholar]
- 91.Greten FR, Karin M. The IKK/NF-kappaB activation pathway: a target for prevention and treatment of cancer. Cancer Lett. 2004;206:193–199. doi: 10.1016/j.canlet.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 92.Gilmore TD. The Re1/NF-kappa B/I kappa B signal transduction pathway and cancer. Cancer Treat. Res. 2003;115:241–265. [PubMed] [Google Scholar]
- 93.Shishodia S, Aggarwal BB. Nuclear factor-kappaB: a friend or a foe in cancer [review]? Biochem. Pharmacol. 2004;68:1071–1080. doi: 10.1016/j.bcp.2004.04.026. [DOI] [PubMed] [Google Scholar]
- 94.Perkins ND. NF-kappaB: tumor promoter or suppressor? Trends Cell Biol. 2004;14:64–69. doi: 10.1016/j.tcb.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 95.Greten FR, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 96.Pikarsky E, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 97.Becker C, et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004;21:491–501. doi: 10.1016/j.immuni.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 98.Seitz CS, Lin Q, Deng H, Khavari PA. Alterations in NF-kappaB function in transgenic epithelial tissue demonstrate a growth inhibitory role for NF-kappaB. Proc. Natl. Acad. Sci. U. S. A. 1998;95:2307–2312. doi: 10.1073/pnas.95.5.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.van Hogerlinden M, Rozell BL, Ahrlund-Richter L, Toftgard R. Squamous cell carcinomas and increased apoptosis in skin with inhibited Rel/nuclear factor-kappaB signaling. Cancer Res. 1999;59:3299–3303. [PubMed] [Google Scholar]
- 100.Dajee M, et al. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature. 2003;421:639–643. doi: 10.1038/nature01283. [DOI] [PubMed] [Google Scholar]
- 101.Zhang JY, Green CL, Tao S, Khavari PA. NF-kappaB RelA opposes epidermal proliferation driven by TNFR1 and JNK. Genes Dev. 2004;18:17–22. doi: 10.1101/gad.1160904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Seitz CS, Freiberg RA, Hinata K, Khavari PA. NF-κB determines localization and features of cell death in epidermis. J. Clin. Invest. 2000;105:253–260. doi: 10.1172/JCI7630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.van Hogerlinden M, Auer G, Toftgard R. Inhibition of Rel/nuclear factor-kappaB signaling in skin results in defective DNA damage-induced cell cycle arrest and Ha-ras- and p53-independent tumor development. Oncogene. 2002;21:4969–4977. doi: 10.1038/sj.onc.1205620. [DOI] [PubMed] [Google Scholar]
- 104.Finco TS, et al. Oncogenic Ha-Ras-induced signaling activates NF-kappaB transcriptional activity, which is required for cellular transformation. J. Biol. Chem. 1997;272:24113–24116. doi: 10.1074/jbc.272.39.24113. [DOI] [PubMed] [Google Scholar]
- 105.Pasparakis M, et al. TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature. 2002;417:861–866. doi: 10.1038/nature00820. [DOI] [PubMed] [Google Scholar]
- 106.Moore RJ, et al. Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat. Med. 1999;5:828–831. doi: 10.1038/10552. [DOI] [PubMed] [Google Scholar]
- 107.Lind MH, et al. Tumor necrosis factor receptor 1-mediated signaling is required for skin cancer development induced by NF-kappaB inhibition. Proc. Natl. Acad. Sci. U. S. A. 2004;101:4972–4977. doi: 10.1073/pnas.0307106101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 109.Monks NR, Biswas DK, Pardee AB. Blocking anti-apoptosis as a strategy for cancer chemotherapy: NF-kappaB as a target [review] J. Cell. Biochem. 2004;92:646–650. doi: 10.1002/jcb.20080. [DOI] [PubMed] [Google Scholar]
- 110.Ravi R, Bedi A. NF-kappaB in cancer: a friend turned foe. Drug Resist. Updat. 2004;7:53–67. doi: 10.1016/j.drup.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 111.Karin M, Yamamoto Y, Wang QM. The IKK NF-kappa B system: a treasure trove for drug development [review] Nat. Rev. Drug Discov. 2004;3:17–26. doi: 10.1038/nrd1279. [DOI] [PubMed] [Google Scholar]
- 112.Orlowski RZ, Baldwin AS., Jr NF-kappaB as a therapeutic target in cancer. Trends Mol. Med. 2002;8:385–389. doi: 10.1016/s1471-4914(02)02375-4. [DOI] [PubMed] [Google Scholar]
- 113.Frelin C, et al. AS602868, a pharmacological inhibitor of IKK2, reveals the apoptotic potential of TNF-alpha in Jurkat leukemic cells. Oncogene. 2003;22:8187–8194. doi: 10.1038/sj.onc.1206963. [DOI] [PubMed] [Google Scholar]
- 114.Frelin C, et al. Targeting NF-kappaB activation via pharmacologic inhibition of IKK2-induced apoptosis of human acute myeloid leukemia cells. Blood. 2005;105:804–811. doi: 10.1182/blood-2004-04-1463. [DOI] [PubMed] [Google Scholar]
- 115.Ziegelbauer K, et al. A selective novel low-molecular-weight inhibitor of IkappaB kinase-beta (IKK-beta) prevents pulmonary inflammation and shows broad anti-inflammatory activity. Br. J. Pharmacol. 2005;145:178–192. doi: 10.1038/sj.bjp.0706176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Niu C, et al. Studies on treatment of acute promyelocytic leukemia with arsenic trioxide: remission induction, follow-up, and molecular monitoring in 11 newly diagnosed and 47 relapsed acute promyelocytic leukemia patients. Blood. 1999;94:3315–3324. [PubMed] [Google Scholar]
- 117.Rousselot P, et al. A clinical and pharmacological study of arsenic trioxide in advanced multiple myeloma patients. Leukemia. 2004;18:1518–1521. doi: 10.1038/sj.leu.2403424. [DOI] [PubMed] [Google Scholar]
- 118.Cavigelli M, et al. The tumor promoter arsenite stimulates AP-1 activity by inhibiting a JNK phosphatase. EMBO J. 1996;15:6269–6279. [PMC free article] [PubMed] [Google Scholar]
- 119.Huang P, Feng L, Oldham EA, Keating MJ, Plunkett W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature. 2000;407:390–395. doi: 10.1038/35030140. [DOI] [PubMed] [Google Scholar]