

Abstract
The visualization of autophagosomes in dying cells has led to the belief that autophagy is a nonapoptotic form of programmed cell death. This concept has now been evaluated using cells and organisms deficient in autophagy genes. Most evidence indicates that, at least in cells with intact apoptotic machinery, autophagy is primarily a pro-survival rather than a pro-death mechanism. This review summarizes the evidence linking autophagy to cell survival and cell death, the complex interplay between autophagy and apoptosis pathways, and the role of autophagy-dependent survival and death pathways in clinical diseases.
Cell biologists have long recognized the possibility that eukaryotic cells may undergo nonapoptotic forms of programmed cell death. Autophagy, a lysosomal pathway involving the bulk degradation of cytoplasmic contents, has been identified as a prime suspect in such death, and recent studies have implicated the autophagy pathway as a cause of nonapoptotic cellular demise. However, most evidence linking autophagy to cell death is circumstantial. Now, with new tools to assess causality, it is an opportune time to revisit the case of autophagy in cell death. Is autophagy an innocent bystander, a direct death execution pathway, a defense mechanism that ultimately fails in its mission to preserve cell viability, and/or a garbage disposal mechanism that cleans up remnants of a cell already committed to die?
Autophagy is a regulated lysosomal degradation pathway
The term autophagy (Greek, “to eat oneself”) does not refer to a death process; it denotes the process of self-cannibalization through a lysosomal degradation pathway. Autophagy is the cell’s major regulated mechanism for degrading long-lived proteins and the only known pathway for degrading organelles (reviewed in refs. 1, 2). During autophagy, an isolation membrane forms, presumably arising from a vesicular compartment known as the preautophagosomal structure, invaginates, and sequesters cytoplasmic constituents including mitochondria, endoplasmic reticulum, and ribosomes (Figure 1). The edges of the membrane fuse to form a double or multimembranous structure, known as the autophagosome or autophagic vacuole. The outer membrane of the autophagosome fuses with the lysosome (in mammalian cells) or vacuole (in yeast and plants) to deliver the inner membranous vesicle to the lumen of the degradative compartment. Degradation of the sequestered material generates nucleotides, amino acids, and free fatty acids that are recycled for macromolecular synthesis and ATP generation.
Figure 1.

The autophagy pathway and its role in cellular adaptation to nutrient deprivation. Starvation or growth factor deprivation results in a decrease in intracellular nutrients and activation of nutrient-sensing signaling pathways (reviewed in ref. 97) that stimulate autophagy. Autophagy involves the sequestration of cytoplasmic material by an isolation membrane (derived from the preautophagosomal structure) to form a double-membrane vacuole, the autophagosome. The autophagosome undergoes fusion with a late endosome or lysosome, to form an autolysosome, in which the sequestered material is degraded. Degradation of membrane lipids and proteins by the autolysosome generates free fatty acids and amino acids that can be reused by the cell to maintain mitochondrial ATP energy production and protein synthesis and thereby promote cell survival. Disruption of this pathway by autophagy gene inactivation prevents cell survival in diverse organisms (Table 2). The same molecular machinery and overlapping dynamic membrane rearrangement events that occur during starvation may also be used in other settings to degrade unwanted cytoplasmic contents, including damaged mitochondria, protein aggregates, and intracellular pathogens. See text for discussion. TCA cycle, tricarboxylic acid cycle.
Autophagy occurs at low basal levels in all cells to perform homeostatic functions (e.g., cytoplasmic and organelle turnover) but is rapidly upregulated when cells need to generate intracellular nutrients and energy (e.g., during starvation or trophic factor withdrawal), undergo architectural remodeling (e.g., during developmental transitions), or rid themselves of damaging cytoplasmic components (e.g., during oxidative stress, infection, and accumulation of protein aggregates). Nutritional status, hormonal factors, and other cues like temperature, oxygen concentrations, and cell density are important in the control of autophagy. Two evolutionarily conserved nutrient sensors play roles in autophagy regulation: (a) the target of rapamycin (TOR) kinase is the major inhibitory signal that shuts off autophagy during nutrient abundance (reviewed in ref. 3), and (b) the eukaryotic initiation factor 2α (eIF2α) kinase Gcn2 and its downstream target Gcn4, a transcriptional transactivator of autophagy genes, turn on autophagy during nutrient depletion (4). The class I PI3K/Akt signaling molecules link receptor tyrosine kinases to TOR activation and thereby repress autophagy in response to insulin-like and other growth factor signals (reviewed in ref. 3).
Downstream of TOR kinase, there are approximately 17 gene products essential for autophagy and related pathways in yeast, referred to as the ATG genes (5), and most yeast ATG genes have orthologs in higher eukaryotes (reviewed in ref. 2). The ATG genes encode proteins needed for the induction of autophagy, and the generation, maturation, and recycling of autophagosomes. These proteins are composed of 4 functional groups, including a protein serine/threonine kinase complex that responds to upstream signals such as TOR kinase (Atg1, Atg13, Atg17), a lipid kinase signaling complex that mediates vesicle nucleation (Atg6, Atg14, Vps34, and Vps15), 2 novel ubiquitin-like conjugation pathways that mediate vesicle expansion (the Atg8 and Atg12 systems), and a recycling pathway that mediates the disassembly of Atg proteins from matured autophagosomes (Atg2, Atg9, Atg18) (Figure 2).
Figure 2.
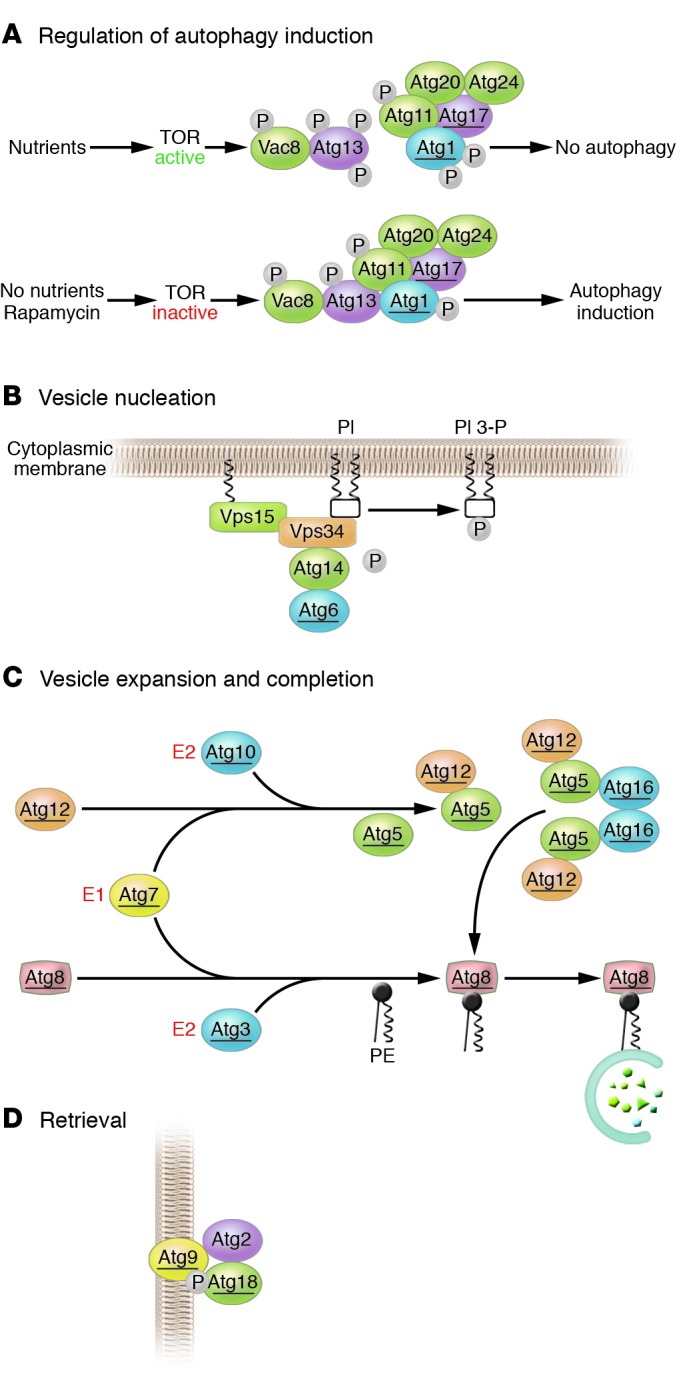
The molecular mechanisms of autophagy. The autophagy (Atg) proteins can be divided into 4 functional groups, including (A) a protein kinase autophagy regulatory complex that responds to upstream signals, including nutrient limitation; (B) a lipid kinase signaling complex that mediates vesicle nucleation; (C) ubiquitin-like protein conjugation pathways that are required for vesicle expansion and completion; and (D) a retrieval pathway required for the disassembly of Atg protein complexes from matured autophagosomes. Shown are the yeast Atg proteins that participate in each functional group. Yeast Atg proteins with known orthologs in higher eukaryotes are underlined. PI, phosphatidylinositol; PI3-P, phosphatidylinositol 3-phosphate; PE, phosphatidylethanolamine.
Autophagy as a cell death mechanism
The term “autophagic cell death” describes a form of programmed cell death morphologically distinct from apoptosis and presumed to result from excessive levels of cellular autophagy (6). In classical apoptosis, or type I programmed cell death, there is early collapse of cytoskeletal elements but preservation of organelles until late in the process. In contrast, in autophagic, or type II, programmed cell death, there is early degradation of organelles but preservation of cytoskeletal elements until late stages. Whereas apoptotic cell death is caspase-dependent and characterized by internucleosomal DNA cleavage, caspase activation and DNA fragmentation occur very late (if at all) in autophagic cell death (Figure 3). In contrast with necrosis, both apoptotic and autophagic cell death are characterized by the lack of a tissue inflammatory response.
Figure 3.
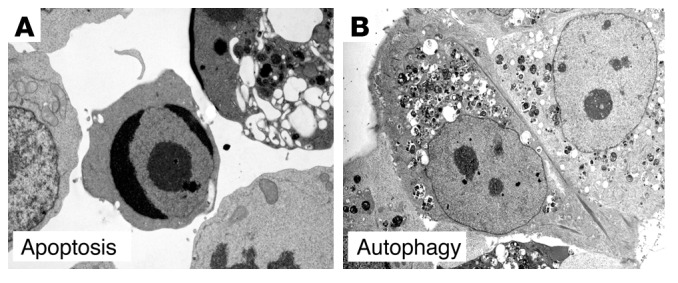
Ultrastructural examples of apoptotic and autophagic cell death. Electron micrographs of a FasL-treated Jurkat cell undergoing cell death with apoptotic features (A) and of a tamoxifen-treated MCF7 human breast carcinoma cell undergoing cell death with autophagic features (B). In A, note chromatin condensation (cell in center) and cytoplasmic vacuolization (cell in upper right). In B, note absence of chromatin condensation and presence of numerous autophagosomes. Images in A and B reproduced with permission from Nature Cell Biology (98) and Landes Bioscience (90), respectively.
Large numbers of autophagic vacuoles have been observed in dying cells of animals of diverse taxa (reviewed in refs. 6–9) (Table 1). The consensus view has been that autophagic cell death occurs primarily when the developmental program (e.g., insect metamorphosis) or homeostatic processes in adulthood (e.g., mammary gland postlactational involution, prostate involution following castration) require massive cell elimination. Recently, studies have also described autophagic cell death in diseased mammalian tissues and in tumor cell lines treated with chemotherapeutic agents (Table 1). In many of these cases, morphologic features of autophagic and apoptotic cell death or of autophagic and necrotic cell death are observed in the same cell.
Table 1.
Examples of cell death with morphologic features of autophagyA

What is the evidence that autophagy is a death execution mechanism in autophagic cell death? If cell death is truly due to autophagy, then pharmacologic or genetic inhibition of autophagy should prevent the death. Yet, for most of the developmental, disease-associated, and toxic stimulus-induced deaths that are presumed to be autophagic (Table 1), the evidence for its role is only correlative. Moreover, in certain cases of autophagic cell death, the available evidence calls into question a causative role of autophagy. For example, in Drosophila, autophagic cell death but not autophagy observed during salivary gland regression is prevented by mutations in the ecdysone-regulated transcription factors BR-C and E74A (10). In the slime mold Dictyostelium, a null mutation in the autophagy gene atg1 blocks vacuolization but not cell death in an in vitro model of autophagic cell death (11). Thus, in these model systems, autophagy per se is neither sufficient nor required for autophagic cell death. Furthermore, the caspase inhibitor p35 blocks metamorphic cell death in Drosophila without complete inhibition of autophagy, suggesting that it is caspase-mediated apoptosis, rather than autophagy, that plays a key role in this death process (10).
There is, however, some evidence in certain in vitro settings that pharmacologic or genetic inhibition of autophagy can prevent cell death. The pharmacologic inhibitor of autophagy 3-methyladenine (3–MA), a nucleotide derivative that blocks class III PI3K activity (12–14), delays or partially inhibits death in starved hepatocytes from carcinogen-treated rats (15), in anti-estrogen–treated human mammary carcinoma cells (16), in chloroquine-treated cortical neurons (17), in nerve growth factor–deprived sympathetic neurons (18), in serum- and potassium-deprived cerebellar granule cells (19), in serum-deprived PC12 cells (20), and in TNF-treated human T lymphoblastic leukemia cells (21). However, in several of these studies, autophagy occurred in cells thought to die by apoptosis, and it was presumed that autophagy triggered apoptosis, rather than playing a direct role in the death process. Moreover, 3-MA can inhibit kinases other than class III PI3K (18), some of which may independently affect death signaling, as well as inhibit the permeability transition in mitochondria (22). Thus, it is not possible to directly implicate autophagy in death execution from these 3-MA inhibitor studies.
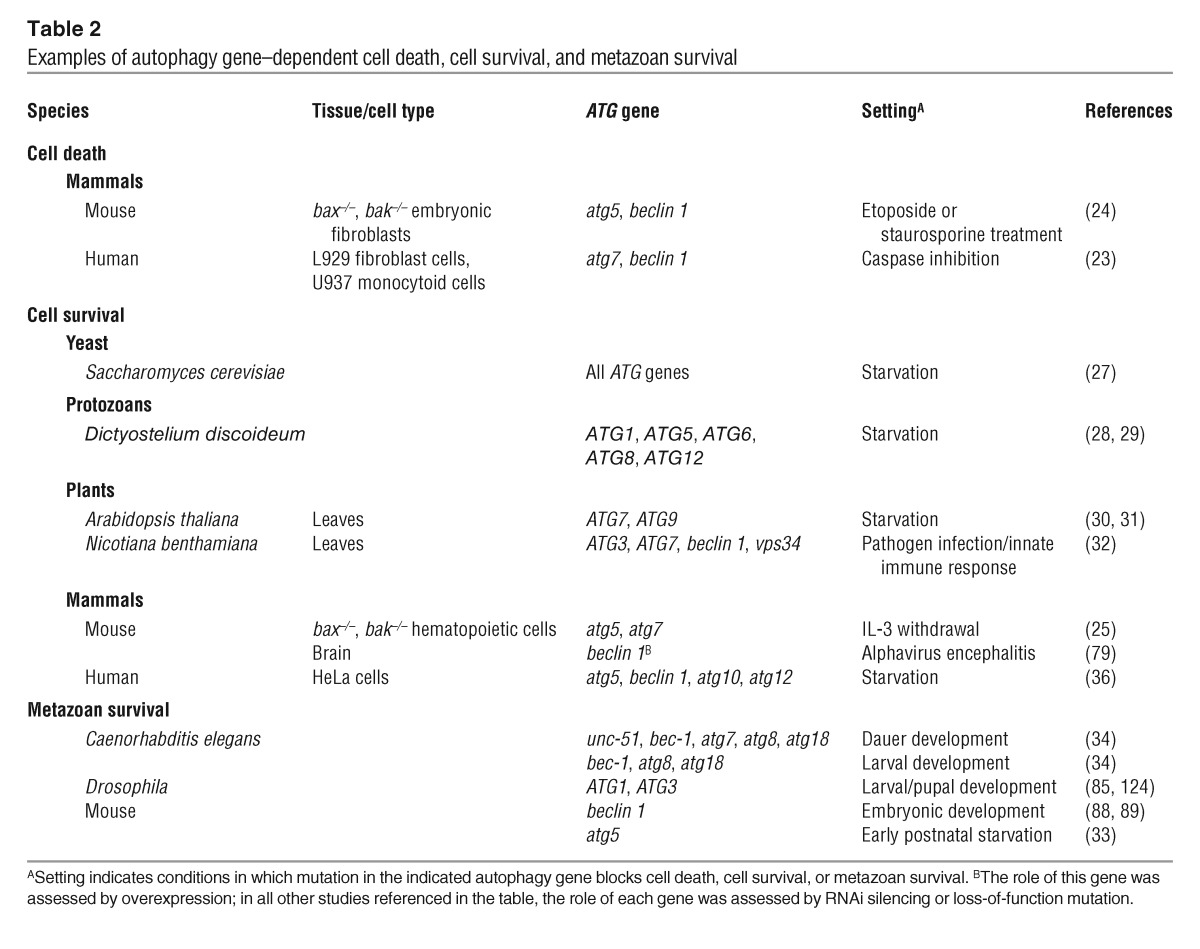
Two recent studies provide the first genetic evidence that the autophagy pathway is capable of killing cells (Table 2). RNA interference (RNAi) directed against 2 autophagy genes, atg7 and beclin 1, blocked cell death in mouse L929 cells treated with the caspase inhibitor zVAD (23). Further, RNAi against autophagy genes atg5 and beclin 1 blocked death of bax–/–, bak–/– murine embryonic fibroblasts (MEFs) treated with staurosporine or etoposide (24). Notably, in both of these studies, atg gene RNAi blocked the death of cells whose apoptotic pathway had been crippled. Although these findings exclude the possibility that autophagy is triggering death through apoptosis induction, they raise the question of whether autophagy is a death mechanism in cells whose apoptotic machinery is intact.
Table 2.
Examples of autophagy gene–dependent cell death, cell survival, and metazoan survival
Interestingly, in etoposide-treated wild-type MEFs (which die by apoptosis), only minimal autophagic activity and no inhibition of death by 3-MA is seen, indicating that autophagy is not involved in the death process unless apoptosis is blocked (24). These data are consistent with the theory previously proposed by Lockshin and Zakeri that cells preferentially die by apoptosis but will die by any alternative available route, including autophagy, if exposed to harsh enough stimuli (9). A related possibility is that apoptotic death is faster than autophagic death and, therefore, autophagy is only witnessed playing a role in cell death in apoptotic-deficient cells. This hypothesis is consistent with recent data indicating that growth factor–deprived wild-type cells undergo a rapid apoptotic death, whereas growth factor–deprived bax–/–, bak–/– cells undergo a slow demise characterized by progressive self-cannibalization (25).
Given the uncertain physiologic relevance of autophagy gene–dependent cell death in zVAD-treated cells or in bax–/–, bak–/– cells, it seems premature to conclude that autophagy is a physiologically important cause of cell death. To prove that autophagy is an important cell death pathway in normal cells, it will be necessary to demonstrate cell death resistance phenotypes in apoptotic-competent cells lacking autophagy genes.
Autophagy as a cell survival mechanism
A contradictory, but equally plausible, explanation for the presence of autophagy in dying cells is that activation of autophagy is a cellular survival strategy. This concept was first proposed in 1977 (26) and considered radical (8) but now is supported by studies demonstrating increased death in cells or organisms lacking gene products essential for autophagy (Table 2). The pro-survival function of autophagy is an evolutionarily ancient process, conserved from yeast to mammals, and best characterized in nutrient deficiency. During nutrient deficiency, degradation of membrane lipids and proteins by the autolysosome generates free fatty acids and amino acids that can be reused to fuel mitochondrial ATP energy production and maintain protein synthesis (Figure 1). Presumably, this recycling function of autophagy is linked mechanistically to its ability to sustain life during starvation.
Unicellular organisms with null mutations in autophagy genes are viable in normal growth conditions; however, unlike their wild-type counterparts, they die rapidly during starvation (Table 2) (27–29). In plants, deletion of autophagy genes (e.g., ATG6, ATG7, and ATG9) results in loss of chlorophyll and accelerated senescence following nutrient deprivation (30–32). Mice lacking Atg5, an acceptor molecule for the ubiquitin-like molecule Atg12, die during the neonatal period, when the placental blood supply is interrupted and they undergo a form of starvation (33). Atg5–/– mice have decreased amino acid levels, decreased cardiac ATP production, and myocardial damage. Although the death of individual cells has not yet been assessed in atg5–/– mice, it is predicted that the recycling function of autophagy is critical to maintain cellular energy homeostasis and cellular survival during the neonatal period. This is particularly likely in tissues such as the heart and diaphragm that have sudden increases in energy needs and exhibit increased autophagy immediately following birth. Similarly, the inability of Caenorhabditis elegans with RNAi-silenced autophagy genes (e.g., unc-51, bec-1, atg8, and atg18) to survive during dauer diapause (34) likely reflects an inability to recycle nutrients at the organismal level.
Autophagy genes may also be critical for maintaining cellular bioenergetics and survival when cells are unable to take up external nutrients (i.e., during growth factor deprivation). In the absence of growth factors such as IL-3, there is decreased surface expression of nutrient transporters, decreased nutrient uptake, and an intracellular deficiency of nutrients (35). Growth factor withdrawal usually results in rapid apoptotic cell death, but recent studies in apoptotic-deficient bax–/–, bak–/– cells have unraveled an essential role for autophagy genes (e.g., atg5, atg7) in maintaining cellular survival following IL-3 deprivation (25). As in nutrient starvation in yeast, autophagy is a self-limited survival strategy during growth factor deprivation. IL-3–deprived bax–/–, bak–/– cells eventually die, presumably because of excessive self-consumption and bioenergetic failure. However, at any point before death, the addition of growth factor reverses the catabolic process and maintains cell viability. These observations are consistent with the concept that autophagy is a self-limited survival strategy, rather than a primary or irreversible death execution program.
Like the pro-death function of autophagy genes in etoposide-treated bax–/–, bak–/– knockout cells, it will be important to determine whether this pro-survival function of autophagy genes during growth factor deprivation in bax–/–, bak–/– cells is conserved in cells with intact apoptotic machinery. It will also be interesting to examine whether autophagy genes play a similar cytoprotective role during withdrawal of hormonal support or growth factors besides IL-3. Perhaps, rather than contributing to death execution, autophagy delays initiation of the apoptotic death pathway in cells deprived of trophic support. Although studies have not been performed in apoptosis-competent cells deprived of trophic factors, autophagy genes prevent the onset of apoptosis during nutrient deprivation. RNAi against beclin 1, atg5, atg10, and atg12 enhances starvation-induced, but not staurosporine-induced, apoptotic cell death (36). Thus, the mechanism by which autophagy genes promote survival during nutrient deprivation may involve suppression of the canonical apoptotic death pathway.
The mechanisms by which autophagy promotes cell survival are not restricted to its role in maintaining cellular energy homeostasis during starvation. Autophagy is also involved in removing damaged mitochondria and other organelles, in degrading intracellular pathogens, and in degrading protein aggregates too large to be removed by the ubiquitin-proteasomal system. These functions of autophagy could promote cellular survival during aging, infectious diseases, and neurodegenerative processes. In addition to a cell-autonomous role for autophagy in promoting survival, autophagy may regulate programmed cell death during physiologic processes in vivo. For example, during the plant innate immune response, silencing of autophagy genes beclin 1, vps34, atg3, and atg7 does not alter the death of infected cells or pathogen spread but results in uncontrolled spread of programmed cell death beyond sites of pathogen infection (32). This suggests that autophagy limits cell death to the site of infection, allowing plant innate immunity to contain pathogen spread without death of innocent bystander cells. It is not known whether autophagy alters the production of death-promoting signals, prevents the movement of death-promoting signals into uninfected tissues, or protects uninfected tissues against death induced by these signals, or whether a similar function of autophagy plays a role in the spatial restriction of development and stress-induced programmed cell death in other eukaryotic organisms.
Autophagy as a self-clearance mechanism
A third explanation for high levels of autophagy in dying cells is that it is a clean-up or self-clearance mechanism in cells committed to die by apoptosis or necrosis. This theory might explain why only selected populations of dying apoptotic cells have morphologic features of autophagy. The dogma is that most apoptotic cells are engulfed by phagocytes, with the lysosomes of the phagocyte responsible for the final degradation of dead cell bodies. However, in some forms of developmental programmed cell death (e.g., embryogenesis, insect metamorphosis), the availability of engulfment cells may be insufficient for clearance of dead cells. In such cases, dying cells may activate autophagy to target the cell’s contents for degradation by its own lysosomes.
This need might contribute to the overlap between signaling pathways that activate apoptosis and autophagy. It has been shown that the proapoptotic signaling molecule TNF-related apoptosis-inducing ligand (TRAIL) regulates autophagy in an in vitro model of mammary gland formation (37). Here, TRAIL-dependent induction of autophagy occurs in parallel with apoptosis. Suppression of either apoptosis alone or TRAIL signaling does not prevent lumen formation, but simultaneous inhibition of apoptosis and TRAIL signaling prevents cell clearance.
These findings suggest that both apoptosis and autophagy may be involved in cavitation during mammary gland morphogenesis. However, to confirm a role for autophagy, it will be important to observe whether luminal filling occurs if autophagy genes are inactivated in cells with intact TRAIL signaling. It is not clear whether autophagy is required for luminal cell death or for removal of cells committed to death by an apoptotic pathway. Similarly, it is not known whether autophagy is required for caspase-dependent death and/or for the clearance of dying cells during insect metamorphosis.
Cross-talk between apoptotic signaling, autophagy, and mitochondria
Several proapoptotic signals induce autophagy — e.g., components of the extrinsic apoptosis pathway, TRAIL, TNF, and FADD (21, 37–40); the calcium/calmodulin–regulated serine/threonine kinases DRP-1 and DAPk (41); and ceramide (42, 43). Conversely, antiapoptotic signaling pathways suppress autophagy — e.g., the class I PI3K/Akt/TOR signaling pathway (reviewed in refs. 3, 44). Coordinated regulation of apoptosis and autophagy is also reflected in the results of genome-wide analyses of transcriptional changes during developmental programmed cell death of the Drosophila salivary gland (45, 46).
The mitochondrion may integrate cell death signals and autophagy activation. Mitochondria generate apoptotic signals but are removed when damaged by autophagy; therefore, mitochondria represent a nexus at which autophagy and apoptosis pathways may interact. Accordingly, genes involved in mitochondrial physiology and/or mitochondrial regulation of apoptosis interact with the autophagy pathway. One example is the yeast gene, UTH1, that encodes an outer mitochondrial membrane protein involved in mitochondrial biogenesis and stress responses. Uth1 mutants are defective in degrading mitochondria during autophagy (47) and survive and proliferate when expressing the mammalian proapoptotic cell death gene bax or when treated with the autophagy inducer rapamycin (48). These findings led Camougrand and colleagues to suggest that Uth1p mediates mitochondrial autophagy and autophagic death. However, it is not yet clear whether rapamycin induces cell death versus cell cycle arrest in wild-type yeast, and whether the phenotype of rapamycin-treated uth1 mutant yeasts is due to direct effects of UTH1 and the autophagy pathway in death regulation.
In mammalian cells, Bcl-2 family members in the outer mitochondrial membrane modulate autophagy. Bcl-2 downregulation increases autophagy in a caspase-independent manner in human leukemic cells (49), and Bcl-2 overexpression inhibits both autophagy and caspase-independent death in growth factor–deprived neural progenitor cells and in serum- and potassium-deprived cultured cerebellar granule cells (19, 50). Recent evidence suggests that Bcl-2 inhibits autophagy through a direct interaction with the Beclin 1 autophagy protein and that the interaction between Bcl-2 and Beclin 1 may function as a rheostat that maintains autophagy at levels that are compatible with cell survival rather than cell death (51). In contrast, Bcl-2 or Bcl-xL overexpression potentiates autophagy and autophagy gene–dependent death in MEFs treated with the proapoptotic stimulus etoposide (24). The basis for the opposite effects of Bcl-2 family members on autophagy in different settings is unclear. Furthermore, it is not yet clear that Bcl-2 proteins function at the mitochondrion to regulate autophagy, since autophagy is inhibited by Bcl-2 targeted to endoplasmic reticulum but not by Bcl-2 targeted to mitochondria (51).
The role of proapoptotic Bcl-2 family members in autophagy gene–dependent life-and-death decisions is also controversial. As discussed earlier, bax–/–, bak–/– cells undergo autophagy gene–dependent death when treated with etoposide (24) but undergo autophagy gene–dependent survival when deprived of trophic factor support (25). It is possible that in the setting of bax/bak deficiency, the stimulus plays a critical role in determining cell fate, and that etoposide, but not growth factor deprivation, can target an intracellular pathway that turns autophagy into a deadly process. Some atypical Bcl-2 family members, including BNIP3 and Hspin, also activate autophagy and nonapoptotic cell death (52–54), but it is not yet known whether this caspase-independent cell death requires autophagy genes.
Another question is how the autophagy pathway recognizes damaged mitochondria. The mitochondrial permeability transition (MPT) may trigger the engulfment of depolarized mitochondria by autophagy (55). However, it is not known whether inhibition of autophagy increases the numbers of depolarized mitochondria in mammalian cells or how a depolarized mitochondrion might be targeted to autophagosomes. The MPT may represent a point of convergence of apoptotic and autophagy pathways, since Bcl-2 family members regulate the MPT. The proapoptotic family member Bax interacts with the voltage-dependent anion channel (56) and/or the adenine nucleotide translocator (57) to induce the MPT in cells and isolated mitochondria upon induction of apoptosis. It is currently not clear, however, whether the MPT regulated by Bax triggers mitochondrial turnover by autophagy.
In many canonical apoptosis pathways, the MPT is caspase-dependent (58). Thus, if the MPT is a critical signal for mitochondrial degradation through autophagy, inhibition of caspases should prevent the loss of mitochondria. However, Tolkovsky and coworkers reported that, although caspase inhibitors effectively inhibit neuronal cell death, they fail to prevent the formation of autophagosomes or the degradation of mitochondria. In fact, the long-term culturing of neurons in the presence of proapoptotic stimuli and caspase inhibitors leads to the loss of mitochondria (59, 60). Thus, the relationship among MPT, caspase-dependent cell death, and mitochondrial autophagy remains unclear.
Autophagy in neurodegenerative diseases
The accumulation of mutant or toxic proteins plays a major role in chronic neurodegenerative diseases (61). Morphologic evidence of autophagy has been reported in neurodegenerative diseases including Parkinson, Huntington, and Alzheimer diseases, and transmissible spongiform encephalopathies (62–65). It is possible that autophagy activation contributes to neurodegeneration (66, 67), but the evidence is correlative. A contrasting view is that autophagy may be a protective mechanism to degrade mutant or toxic proteins. According to this model, defects in autophagy-related pathways contribute to the accumulation of neurotoxic proteins and the ensuing neuronal cell death. Although the exact roles of autophagy in neurodegenerative diseases are not fully defined, recent studies have provided some insights.
Τhe protein α-synuclein is a major component of neuronal cytoplasmic inclusions that characterize Parkinson and other neurodegenerative diseases (68). Although earlier studies suggested that α-synuclein is degraded through both the proteasome and classical autophagy pathways (69), a recent study demonstrated that the turnover of α-synuclein is regulated by chaperone-mediated autophagy, which involves the direct lysosomal targeting of proteins containing specific pentapeptide recognition motifs (70). Interestingly, pathogenic α-synuclein mutants associated with familial, autosomal-dominant forms of Parkinson disease (71, 72) are inefficiently degraded by chaperone-mediated autophagy. Since the accumulation of wild-type α-synuclein in neuronal inclusions is common in adult-onset neurodegenerative diseases, these experiments suggest that defects in autophagy-related pathways may contribute to multiple neurodegenerative diseases.
Consistent with this hypothesis, autophagy has also been implicated in regulating the turnover of Huntingtin (Htt), the protein involved in Huntington disease, an autosomal-dominant neurodegenerative disorder caused by the expansion of a polyglutamine (polyQ) tract in Htt. Although the mechanism of neurotoxicity mediated by expanded polyQ is still controversial, expanded polyQ provokes a dominant gain-of-function neurotoxicity, regardless of the specific protein context within which it resides. The accumulation of expanded polyQ-containing proteins in insoluble aggregates in affected neurons is a hallmark feature of Huntington and other polyQ expansion diseases (73).
Although neuronal Htt proteins in inclusions are highly ubiquitinated, polyQ is a poor substrate for proteasomes (74). Thus, the highly ubiquitinated state of Htt inclusions may indicate the inability of proteasomes in affected neurons to clear abnormal Htt proteins. In contrast, there is pharmacologic evidence to suggest a role for autophagy in the degradation of the N-terminus of Htt. For example, 3-MA increases the aggregation of Htt with expanded polyQ in clonal striatal cells (62). Rapamycin, an inducer of autophagy, reduces the aggregation of expanded polyQ in transfected cells (75), protects against neurodegeneration in a fly model of Huntington disease, and improves performance on behavioral tests and decreases aggregate formation in a mouse model of Huntington disease (76). These results suggest a possible role of autophagy in the turnover of expanded polyQ proteins and in protection of neurons against their toxicity.
Autophagy and infectious diseases
The autophagic machinery is used to degrade intracellular pathogens (reviewed in refs. 77, 78) including intracellular bacteria (e.g., Shigella flexneri and Mycobacterium tuberculosis), mammalian viruses that produce encephalitis (e.g., alphaviruses and herpes simplex virus), and plant viruses (32, 78–81). It also may be used to degrade invading extracellular pathogens such as group A Streptococcus (82). It is reasonable to propose that autophagy might promote cellular survival during pathogen invasion because of either enhanced degradation of intracellular pathogens and consequent decreases in microbial replication; enhanced degradation of specific cytotoxic microbial virulence products; or preservation of cellular nutrient status during a period of microbial parasitism (which mimics nutrient starvation). However, with the exception of the finding that forced expression of the beclin 1 autophagy gene protects against Sindbis virus–induced apoptosis in mouse brains (in parallel with decreasing viral replication) (79), direct proof of a cell-autonomous, pro-survival role of autophagy in pathogen infection is lacking. Moreover, it has been proposed that a virulence protein, SipB, of the intracellular pathogen Salmonella enterica causes macrophage death by inducing autophagy, perhaps by triggering mitochondrial fusion with autophagosomes (83). Yet, in this study, there was no direct evidence that macrophage cell death was caused by, rather than simply associated with, autophagy. Further studies in autophagy-deficient host organisms are required to determine the role of autophagy in life-and-death decisions during pathogen infection.
Autophagy and cancer
Cancer results from the dysregulation of pathways that regulate cell differentiation, cell proliferation, and cell survival. Autophagy may protect against cancer by sequestering damaged organelles, permitting cellular differentiation, increasing protein catabolism, and/or promoting autophagic death. Alternatively, autophagy may contribute to cancer by promoting the survival of nutrient-starved cells. Recent data are most consistent with a model in which autophagy contributes to tumor suppression and defects in autophagy contribute to oncogenesis. Biochemical evidence in mammalian cells and genetic evidence in C. elegans and Drosophila indicate that autophagy is positively regulated by the PTEN tumor suppressor gene and negatively regulated by the oncogenic class I PI3K signaling pathway (14, 34, 84, 85). Furthermore, the mammalian autophagy gene beclin 1 has tumor suppressor activity in breast carcinoma cells (86), is commonly deleted in human breast ovarian and prostate cancer (87), and is a haploinsufficient tumor suppressor gene in mice (88, 89).
Several theories regarding the role of autophagy-dependent death and autophagy-dependent survival in cancer biology have been proposed (3, 66, 90–94). One is that autophagy-dependent death is a mechanism of tumor suppression. However, there are no direct data to support this hypothesis. In contrast, studies in cells and animals with a deficiency in beclin 1 suggest that death induction may not be involved in the tumor suppressor function of this autophagy gene. Beclin 1–/– ES cells are not resistant to death triggered by UV irradiation or serum withdrawal, and beclin 1–/– null animals die early during embryogenesis with massive cell death (89). Moreover, in beclin 1 heterozygous-deficient mice (with reduced tissue levels of autophagy), there is hyperproliferation of mammary epithelial cells during glandular morphogenesis and increased antigen-driven proliferation of B cells without decreased cell death (88). Together, these observations suggest that the role of the beclin 1 autophagy gene in tumor suppression is related not to cell death induction, but rather to inhibition of cellular proliferation.
It is possible that autophagy is involved in the spontaneous or chemotherapy-induced death of existing tumor cells. Although the role of autophagy in cell death in apoptosis-competent cells is unclear, autophagy gene–dependent death in cells crippled in apoptosis (e.g., zVAD-treated cells; bax–/–, bak–/– cells) may have relevance for cancer biology and therapy, since human tumor cells frequently contain mutations that render them resistant to apoptosis. One prediction is that such cells have an increased dependency on autophagy pathways for self-destruction, and that the impact of decreased autophagy-dependent cell death on tumor progression may be greater in tumor cells that are resistant to apoptosis. Another prediction is that the enhanced autophagy-dependent death potential of apoptosis-resistant tumor cells might be exploited therapeutically by the administration of autophagy-inducing agents. Indeed, there are examples of putative autophagic cell death in cancer cell lines treated with chemotherapeutic agents (Table 1). During tamoxifen-induced death of MCF7 cells (a cell type that contains a mutation in caspase-3), there is a marked upregulation of Beclin 1 autophagy protein expression (42, 90), and, in some examples, chemotherapy-induced autophagic cell death is inhibited by 3-MA (16, 42, 95). However, evidence proving that autophagy is a bona fide death pathway in chemotherapy-treated cancer cells is lacking. In addition, rapamycin, an inhibitor of TOR kinase that has promising antitumor effects in human clinical trials (96), is one of the most potent known inducers of autophagy but is not known to induce autophagic cell death.
In contrast to potential pro-death effects, more clearly established pro-survival effects of autophagy during nutrient starvation might foster tumor initiation and/or progression (3, 91, 93). As tumor cells grow beyond their blood supply, they are exposed to nutrient-limiting conditions, and it is possible that transformed cells use autophagy as a survival strategy in this setting. It has been proposed that such a need for autophagy in tumor initiation might explain the retention of the wild-type allele in all tumors arising in beclin 1+/– mice (92). However, the role of autophagy in tumor cell survival in vivo has not been tested experimentally. Moreover, in considering the net effect of autophagy on tumorigenesis, it is important to recognize its other functions that could contribute to restricting tumorigenesis (e.g., the degradation of certain proteins or organelles required for cell growth and/or the degradation of damaged mitochondria and other organelles that generate genotoxic stress and increase the likelihood of oncogenic mutations).
Conclusion
Autophagy functions across a diverse range of species as a pro-survival pathway during nutrient deprivation and other forms of cellular stress. Paradoxically, in cells that cannot die by apoptosis and, more speculatively, in cells that cannot be removed by engulfment cells, the autophagic machinery may also be used for self-destruction. The challenge for scientists will be to understand the molecular basis of this paradox. The challenge for clinicians will be to selectively turn on or turn off autophagy gene–dependent survival and death pathways in the treatment of different clinical diseases.
Acknowledgments
The work from the authors’ laboratories was supported by NIH grants RO1 CA084254O1, RO1 AI151367, and RO1 CA109618 (to B. Levine) and R37 NIA12859 (to J. Yuan); American Cancer Society grant RSG 98-339 (to B. Levine); and an Ellison Medical Foundation Senior Scholar Award in Infectious Diseases (to B. Levine). We thank Alexi Degetrev for providing electron micrographs and Sophie Pattingre and Renee Talley for help with manuscript preparation.
Footnotes
Nonstandard abbreviations used: Htt, Huntingtin; 3-MA, 3-methyladenine; MEF, murine embryonic fibroblast; MPT, mitochondrial permeability transition; polyQ, polyglutamine; RNAi, RNA interference; TOR, target of rapamycin; TRAIL, TNF-related apoptosis-inducing ligand.
Conflict of interest: The authors have declared that no conflict of interest exists.
References
- 1.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 3.Lum JJ, DeBarardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty [review] Nat. Rev. Mol. Cell Biol. 2005;6:439–448. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- 4.Talloczy Z, et al. Regulation of starvation- and virus-induced autophagy by the eIF2α kinase signaling pathway. Proc. Natl. Acad. Sci. U. S. A. 2002;99:190–195. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klionsky DJ, et al. A unified nomenclature for yeast autophagy-related genes. Dev. Cell. 2003;5:539–545. doi: 10.1016/s1534-5807(03)00296-x. [DOI] [PubMed] [Google Scholar]
- 6.Schweichel J-U, Merker H-J. The morphology of various types of cell death in prenatal tissues. Teratology. 1973;7:253–266. doi: 10.1002/tera.1420070306. [DOI] [PubMed] [Google Scholar]
- 7.Bursch W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 2001;8:569–581. doi: 10.1038/sj.cdd.4400852. [DOI] [PubMed] [Google Scholar]
- 8.Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms [review] Anat. Embryol. (Berl.) 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 9.Lockshin RA, Zakeri Z. Apoptosis, autophagy, and more. Int. J. Biochem. Cell Biol. 2004;36:2405–2419. doi: 10.1016/j.biocel.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 10.Lee CY, Baehrecke EH. Steroid regulation of autophagic programmed cell death during development. Development. 2001;128:1443–1455. doi: 10.1242/dev.128.8.1443. [DOI] [PubMed] [Google Scholar]
- 11.Kosta A, et al. Autophagy gene disruption reveals a non-vacuolar cell death pathway in dictyostelium. J. Biol. Chem. 2004;279:48404–48409. doi: 10.1074/jbc.M408924200. [DOI] [PubMed] [Google Scholar]
- 12.Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. U. S. A. 1982;79:1889–1892. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. 1997;243:240–246. doi: 10.1111/j.1432-1033.1997.0240a.x. [DOI] [PubMed] [Google Scholar]
- 14.Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 2000;275:992–998. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- 15.Schwarze PE, Seglen PO. Reduced autophagic activity, improved protein balance and enhanced in vitro survival of hepatocytes isolated from carcinogen-treated rats. Exp. Cell Res. 1985;157:15–28. doi: 10.1016/0014-4827(85)90148-x. [DOI] [PubMed] [Google Scholar]
- 16.Bursch W, et al. Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: the role of autophagy. Carcinogenesis. 1996;17:1595–1607. doi: 10.1093/carcin/17.8.1595. [DOI] [PubMed] [Google Scholar]
- 17.Kaidi AU, et al. Chloroquine-induced neuronal cell death is p53 and Bcl-2 family-dependent but caspase-independent. J. Neuropathol. Exp. Neurol. 2001;60:937–945. doi: 10.1093/jnen/60.10.937. [DOI] [PubMed] [Google Scholar]
- 18.Xue L, Fletcher GC, Tolkovsky AM. Autophagy is activated by apoptotic signalling in sympathetic neurons: an alternative mechanism of death execution. Mol. Cell. Neurosci. 1999;14:180–198. doi: 10.1006/mcne.1999.0780. [DOI] [PubMed] [Google Scholar]
- 19.Canu N, et al. Role of the autophagic-lysosomal system on low potassium-induced apoptosis in cultured cerebellar granule cells. J. Neurochem. 2005;92:1228–1242. doi: 10.1111/j.1471-4159.2004.02956.x. [DOI] [PubMed] [Google Scholar]
- 20.Uchiyama Y. Autophagic cell death and its execution by lysosomal cathepsins. Arch. Histol. Cytol. 2001;64:233–246. doi: 10.1679/aohc.64.233. [DOI] [PubMed] [Google Scholar]
- 21.Jia L, et al. Inhibition of autophagy abrogates tumour necrosis factor alpha induced apoptosis in human T-lymphoblastic leukaemic cells. Br. J. Haematol. 1997;98:673–685. doi: 10.1046/j.1365-2141.1997.2623081.x. [DOI] [PubMed] [Google Scholar]
- 22.Xue L, Borutaite V, Tolkovsky AM. Inhibition of mitochondrial permeability transition and release of cytochrome c by anti-apoptotic nucleoside analogues. Biochem. Pharmacol. 2002;64:441–449. doi: 10.1016/s0006-2952(02)01181-4. [DOI] [PubMed] [Google Scholar]
- 23.Yu L, et al. Regulation of an ATG7-beclin 1program of autophagic cell death by caspase 8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 24.Shimizu S, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 2004;6:1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 25.Lum JJ, et al. Growth factor regulation of autophagy and cell survival in the absence of autophagy. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 26.Hourdry J. Cytological and cytochemical changes in the intestinal epithelium during anuran metamorphosis. Int. Rev. Cytol. Suppl. 1977;5:337–385. [Google Scholar]
- 27.Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333:169–174. doi: 10.1016/0014-5793(93)80398-e. [DOI] [PubMed] [Google Scholar]
- 28.Otto GP, Wu MY, Kazgan N, Anderson OR, Kessin RH. Macroautophagy is required for multicellular development of the social amoeba Dictyostelium discoideum. J. Biol. Chem. 2003;278:17636–17645. doi: 10.1074/jbc.M212467200. [DOI] [PubMed] [Google Scholar]
- 29.Otto GP, Wu MY, Kazgan N, Anderson OR, Kessin RH. Dictyosteliummacroautophagy mutants vary in the severity of their developmental defects. J. Biol. Chem. 2004;279:15621–15629. doi: 10.1074/jbc.M311139200. [DOI] [PubMed] [Google Scholar]
- 30.Doelling JH, Walker JM, Friedman EM, Thompson AR, Vierstra RD. The APG8/12-activating enzyme APG7 is required for proper nutrient recycling and senescence in Arabidopsis thaliana. J. Biol. Chem. 2002;277:33105–33114. doi: 10.1074/jbc.M204630200. [DOI] [PubMed] [Google Scholar]
- 31.Hanaoka H, et al. Leaf senescence and starvation-induced chlorosis are accelerated by the disruption of an Arabidopsisautophagy gene. Plant Physiol. 2002;129:1181–1193. doi: 10.1104/pp.011024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Y, et al. Autophagy genes are essential for limiting the spread of programmed cell death associated with plant innate immunity. Cell. 2005;121:567–577. [Google Scholar]
- 33.Kuma A, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 34.Melendez A, et al. Autophagy genes are essential for dauer development and lifespan extension in C. elegans. Science. 2003;301:1387–1391. doi: 10.1126/science.1087782. [DOI] [PubMed] [Google Scholar]
- 35.Edinger AL, Cinnalli RM, Thompson CB. Rab7 prevents growth factor-independent survival by inhibiting cell-autonomous nutrient transport expression. Dev. Cell. 2003;5:571–582. doi: 10.1016/s1534-5807(03)00291-0. [DOI] [PubMed] [Google Scholar]
- 36.Boya P, et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 2005;25:1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mills KR, Reginato M, Debnath J, Queenan B, Brugge JS. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is required for induction of autophagy during lumen formation in vitro. Proc. Natl. Acad. Sci. U. S. A. 2004;101:3438–3443. doi: 10.1073/pnas.0400443101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thorburn J, et al. Selective inactivation of FADD-dependent apoptosis and autophagy pathway in immortal epithelial cells. Mol. Biol. Cell. 2005;16:1189–1199. doi: 10.1091/mbc.E04-10-0906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prins J, et al. Tumour necrosis factor induced autophagy and mitochondrial morphological abnormalities are mediated by TNFR-1 and/or TNFR-II and do not invariably lead to cell death. Biochem. Soc. Trans. 1998;26:S314. doi: 10.1042/bst026s314. [DOI] [PubMed] [Google Scholar]
- 40.Pyo J-O, et al. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J. Biol. Chem. 2005;280:20722–20729. doi: 10.1074/jbc.M413934200. [DOI] [PubMed] [Google Scholar]
- 41.Inbal B, Bialik S, Sabanay I, Shani G, Kimchi A. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J. Cell Biol. 2002;157:455–468. doi: 10.1083/jcb.200109094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scarlatti F, et al. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and upregulation of Beclin 1. J. Biol. Chem. 2004;279:18384–18391. doi: 10.1074/jbc.M313561200. [DOI] [PubMed] [Google Scholar]
- 43.Daido S, et al. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res. 2004;64:4286–4293. doi: 10.1158/0008-5472.CAN-03-3084. [DOI] [PubMed] [Google Scholar]
- 44.Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. Int. J. Biochem. Cell Biol. 2004;36:2445–2462. doi: 10.1016/j.biocel.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 45.Lee C-Y, et al. Genome-wide analyses of steroid- and radiation-triggered programmed cell death in Drosophila. Curr. Biol. 2003;13:350–357. doi: 10.1016/s0960-9822(03)00085-x. [DOI] [PubMed] [Google Scholar]
- 46.Gorski SM, et al. A SAGE approach to discovery of genes involved in autophagic cell death. Curr. Biol. 2003;13:358–363. doi: 10.1016/s0960-9822(03)00082-4. [DOI] [PubMed] [Google Scholar]
- 47.Kissova I, Deffieu M, Manon S, Camougrand N. Uth1p is involved in the autophagic degradation of mitochondria. J. Biol. Chem. 2004;279:39068–39074. doi: 10.1074/jbc.M406960200. [DOI] [PubMed] [Google Scholar]
- 48.Camougrand N, et al. The product of the UTH1gene, required for Bax-induced cell death in yeast, is involved in the response to rapamycin. Mol. Microbiol. 2003;47:495–506. doi: 10.1046/j.1365-2958.2003.03311.x. [DOI] [PubMed] [Google Scholar]
- 49.Saeki K, et al. Bcl-2 down-regulation causes autophagy in caspase-independent manner in human leukemic HL60 cells. Cell Death Differ. 2000;7:1263–1269. doi: 10.1038/sj.cdd.4400759. [DOI] [PubMed] [Google Scholar]
- 50.Cardenas-Aguayo MDC, Santa-Olalla J, Baizabal JM, Salgado LM, Covarrubias L. Growth factor deprivation induces an alternative non-apoptotic death mechanism that is inhibited by Bcl2 in cells derived from neural precursor cells. J. Hematother. Stem Cell Res. 2003;12:735–748. doi: 10.1089/15258160360732759. [DOI] [PubMed] [Google Scholar]
- 51.Pattingre, S., et al. 2005. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. In press. [DOI] [PubMed]
- 52.Velde CV, et al. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol. Cell. 2000;20:5454–5468. doi: 10.1128/mcb.20.15.5454-5468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yanagisawa H, Miyashita T, Nakano Y, Yamamoto D. HSpin1, a transmembrane protein interacting with Bc-2/Bcl-xL, induces a caspase-independent autophagic cell death. Cell Death Differ. 2003;10:798–807. doi: 10.1038/sj.cdd.4401246. [DOI] [PubMed] [Google Scholar]
- 54.Nakano Y, et al. Mutations in the novel membrane protein spinster interfere with programmed cell death and cause neural degeneration in Drosophila melanogaster. Mol. Cell. Biol. 2001;21:3775–3788. doi: 10.1128/MCB.21.11.3775-3788.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001;15:2286–2287. doi: 10.1096/fj.01-0206fje. [DOI] [PubMed] [Google Scholar]
- 56.Jacotot E, et al. Mitochondrial membrane permeabilization during the apoptotic process. Ann. N. Y. Acad. Sci. 1999;887:18–30. doi: 10.1111/j.1749-6632.1999.tb07919.x. [DOI] [PubMed] [Google Scholar]
- 57.Marzo I, et al. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- 58.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 59.Tolkovsky AM, Xue L, Fletcher GC, Borutaite V. Mitochondrial disappearance from cells: a clue to the role of autophagy in programmed cell death and disease [review]? Biochimie. 2002;84:233–240. doi: 10.1016/s0300-9084(02)01371-8. [DOI] [PubMed] [Google Scholar]
- 60.Xue L, Fletcher GC, Tolkovsky AM. Mitochondria are selectively eliminated from eukaryotic cells after blockade of caspases during apoptosis. Curr. Biol. 2001;11:361–365. doi: 10.1016/s0960-9822(01)00100-2. [DOI] [PubMed] [Google Scholar]
- 61.Grune T, Jung T, Merker K, Davies KJ. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and “aggresomes” during oxidative stress, aging, and disease. Int. J. Biochem. Cell Biol. 2004;36:2519–2530. doi: 10.1016/j.biocel.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 62.Qin ZH, et al. Autophagy regulates the processing of amino terminal huntingtin fragments. Hum. Mol. Genet. 2003;12:3231–3244. doi: 10.1093/hmg/ddg346. [DOI] [PubMed] [Google Scholar]
- 63.Anglade P, et al. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- 64.Yu WH, et al. Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for beta-amyloid peptide over-production and localization in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2004;36:2531–2540. doi: 10.1016/j.biocel.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 65.Liberski PP, Sikorska B, Bratosiewicz-Wasik J, Gajdusek DC, Brown P. Neuronal cell death in transmissible spongiform encephalopathies (prion diseases) revisited: from apoptosis to autophagy. Int. J. Biochem. Cell Biol. 2004;36:2473–2490. doi: 10.1016/j.biocel.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 66.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yue Z, et al. A novel protein complex linking the δ2 glutamate receptor and autophagy: implications for neurodegeneration in lurcher mice. Neuron. 2002;35:921–933. doi: 10.1016/s0896-6273(02)00861-9. [DOI] [PubMed] [Google Scholar]
- 68.Maries E, Dass B, Collier TJ, Kordower JH, Steece-Collier K. The role of alpha-synuclein in Parkinson’s diseases: insights from animal models. Nat. Rev. Neurosci. 2003;4:727–738. doi: 10.1038/nrn1199. [DOI] [PubMed] [Google Scholar]
- 69.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 70.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 71.Polymeropoulos MH, et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 72.Kruger R, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 73.DiFiglia M, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 74.Venkatraman P, Wetzel R, Tanaka M, Nukina N, Goldberg AL. Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol. Cell. 2004;14:95–104. doi: 10.1016/s1097-2765(04)00151-0. [DOI] [PubMed] [Google Scholar]
- 75.Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002;11:1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 76.Ravikumar B, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 77.Kirkegaard K, Taylor MP, Jackson WT. Cellular autophagy: surrender, avoidance and subversion by microorganisms [review] Nat. Rev. Microbiol. 2004;2:301–314. doi: 10.1038/nrmicro865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Seay, M., Dinesh-Kumar, S., and Levine, B. 2005. Digesting oneself and digesting microbes: autophagy as a host response to viral infection. In Modulation of host gene expression and innate immunity by viruses. P. Palese, editor. Springer. Dordrecht, The Netherlands. 245–279.
- 79.Liang XH, et al. Protection against fatal Sindbis virus encephalitis by Beclin, a novel Bcl-2-interacting protein. J. Virol. 1998;72:8586–8596. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ogawa M, et al. Escape of intracellular Shigella from autophagy. Science. 2005;307:727–731. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- 81.Gutierrez MG, et al. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosissurvival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 82.Nakagawa I, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 83.Hernandez LD, Pypaert M, Flavell RA, Galan JE. A Salmonellaprotein causes macrophage cell death by inducing autophagy. J. Cell Biol. 2003;163:1123–1131. doi: 10.1083/jcb.200309161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rusten TE, et al. Programmed autophagy in the Drosophilafat body is induced by ecdysone through regulation of the PI3K pathway. Dev. Cell. 2004;7:179–192. doi: 10.1016/j.devcel.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 85.Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophilafat body. Dev. Cell. 2004;7:167–178. doi: 10.1016/j.devcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 86.Liang XH, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 87.Aita VM, et al. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59:59–65. doi: 10.1006/geno.1999.5851. [DOI] [PubMed] [Google Scholar]
- 88.Qu X, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1gene. J. Clin. Invest. 2003;112:1809–1820. doi:10.1172/JCI200320039. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. U. S. A. 2003;100:15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Furuya, N., Liang, X.H., and Levine, B. 2004. Autophagy and cancer. In Autophagy. D.J. Klionsky, editor. Landes Bioscience. Georgetown, Texas, USA. 244–253.
- 91.Ogier-Denis E, Codogno P. Autophagy: a barrier or an adaptive response to cancer? . Biochim. Biophys. Acta. 2003; 1603:113–128. doi: 10.1016/s0304-419x(03)00004-0. [DOI] [PubMed] [Google Scholar]
- 92.Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr. Opin. Cell Biol. 2004;16:663–669. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 93.Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–2906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 94.Nelson DA, White E. Exploiting different ways to die. Genes Dev. 2004;18:1223–1226. doi: 10.1101/gad.1212404. [DOI] [PubMed] [Google Scholar]
- 95.Kanzawa T, Kondo Y, Ito H, Kondo S, Germano I. Induction of autophagic cell death in malignant glioma cells by arsenic trioxide. Cancer Res. 2003;63:2103–2108. [PubMed] [Google Scholar]
- 96.Huang S, Houghton PJ. Targeting mTOR signaling for cancer therapy. Curr. Opin. Pharmacol. 2003;3:371–377. doi: 10.1016/s1471-4892(03)00071-7. [DOI] [PubMed] [Google Scholar]
- 97.Codogno, P., and Meijer, A.J. 2004. Signaling pathways in mammalian autophagy. In Autophagy. D.J. Klionsky, editor. Landes Bioscience. Georgetown, Texas, USA. 26–47.
- 98.Degterev A, et al. Identification of small-molecule inhibitors of interaction between the BH3 domain and BclxL. Nat. Cell Biol. 2001;3:173–182. doi: 10.1038/35055085. [DOI] [PubMed] [Google Scholar]
- 99.Kinch G, Hoffman KL, Rodrigues EM, Zee MC, Weeks JC. Steroid-triggered programmed cell death of a motoneuron is autophagic and involves structural changes in mitochondria. J. Comp. Neurol. 2003;457:384–403. doi: 10.1002/cne.10563. [DOI] [PubMed] [Google Scholar]
- 100.Muller F, Adori C, Sass M. Autophagic and apoptotic features during programmed cell death in the fat body of the tobacco hornworm (Manduca sexta). Eur. J. Cell Biol. 2004;83:67–78. doi: 10.1078/0171-9335-00359. [DOI] [PubMed] [Google Scholar]
- 101.Romer F, Martau T. Degeneration of moulting glands in male crickets. J. Insect Physiol. 1998;44:981–989. doi: 10.1016/s0022-1910(98)00021-3. [DOI] [PubMed] [Google Scholar]
- 102.Bowen ID, Mullarkey K, Morgan SM. Programmed cell death during metamorphosis in the blow-fly Calliphora vomitoria. Microsc. Res. Tech. 1996;34:202–217. doi: 10.1002/(SICI)1097-0029(19960615)34:3<202::AID-JEMT3>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 103.Jones HE, Bown ID. Acid phosphatase activity in the larval salivary glands of developing Drosophila melanogaster. Cell Biol. Int. 1993;17:305–315. doi: 10.1006/cbir.1993.1066. [DOI] [PubMed] [Google Scholar]
- 104.Lee C-Y, Cooksey BAK, Baehrecke EH. Steroid regulation of midgut cell death during Drosophiladevelopment. Dev. Biol. 2002;250:101–111. doi: 10.1006/dbio.2002.0784. [DOI] [PubMed] [Google Scholar]
- 105.Butterworth FM, LaTendresse BL. Quantitative studies of cytochemical and cytological changes during cell death in the larval fat body of Drosophila melanogaster. J. Insect. Physiol. 1973;19:1487–1500. [Google Scholar]
- 106.Debnath J, et al. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell. 2002;111:29–40. doi: 10.1016/s0092-8674(02)01001-2. [DOI] [PubMed] [Google Scholar]
- 107.Kegel KB, et al. Huntingtin expression stimulates endolysosomal-activity, endosome tubulation, and autophagy. J. Neurosci. 2000;20:7268–7278. doi: 10.1523/JNEUROSCI.20-19-07268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. Expression of A53T mutant but not wild-type α-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation, loss of dopamine release, and autophagic cell death. J. Neurosci. 2001;21:9549–9560. doi: 10.1523/JNEUROSCI.21-24-09549.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nitatori T, et al. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J. Neurosci. 1995;2:1001–1011. doi: 10.1523/JNEUROSCI.15-02-01001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kostin S. Types and mechanisms of myocyte cell death in diseased human heart. Cardiovasc. J. S. Afr. 2004;15(4 Suppl. 1):S1. [Google Scholar]
- 111.Hein S, et al. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation. 2003;107:984–991. doi: 10.1161/01.cir.0000051865.66123.b7. [DOI] [PubMed] [Google Scholar]
- 112.Bera A, Singh S, Nagaraj R, Vaidya T. Induction of autophagic cell death in Leishmania donovaniby antimicrobial peptides. Mol. Biochem. Parasitol. 2003;127:23–35. doi: 10.1016/s0166-6851(02)00300-6. [DOI] [PubMed] [Google Scholar]
- 113.Christensen ST, et al. Staurosporine-induced cell death in Tetrahymena thermophilahas mixed characteristics of both apoptotic and autophagic degeneration. Cell Biol. Int. 1998;22:591–598. doi: 10.1006/cbir.1998.0320. [DOI] [PubMed] [Google Scholar]
- 114.Cornillon S, et al. Programmed cell death in Dictyostelium. J. Cell Sci. 1994;107:2691–2704. doi: 10.1242/jcs.107.10.2691. [DOI] [PubMed] [Google Scholar]
- 115.Akazawa H, et al. Diphtheria toxin-induced autophagic cardiomyocyte death plays a pathogenic role in mouse model of heart failure. J. Biol. Chem. 2004;279:41095–41103. doi: 10.1074/jbc.M313084200. [DOI] [PubMed] [Google Scholar]
- 116.Guimaraes CA, Benchimo M, Amarante-Mendes GP, Linden R. Alternative programs of cell death in developing retinal tissue. J. Biol. Chem. 2003;278:41938–41946. doi: 10.1074/jbc.M306547200. [DOI] [PubMed] [Google Scholar]
- 117.Borsello T, Croquelois K, Hornung JP, Clarke PGH. N-methyl-D-aspartate triggered neuronal death in organotypic hippocampal cultures is endocytic, autophagic and mediated by the c-Jun N-terminal kinase pathway. Eur. J. Neurosci. 2003;18:473–485. doi: 10.1046/j.1460-9568.2003.02757.x. [DOI] [PubMed] [Google Scholar]
- 118.Ohsawa Y, et al. An ultrastructural and immunohistochemical study of PC12 cells during apoptosis induced by serum deprivation with special reference to autophagy and lysosomal cathepsins. Arch. Histol. Cytol. 1998;61:395–403. doi: 10.1679/aohc.61.395. [DOI] [PubMed] [Google Scholar]
- 119.von Bultzingslowen I, Jontell M, Hurst P, Nannmark U, Kardos T. 5-Fluorouracil induces autophagic degeneration in rat oral keratinocytes. Oral Oncol. 2001;37:537–544. doi: 10.1016/s1368-8375(01)00009-4. [DOI] [PubMed] [Google Scholar]
- 120.Opipari AW, Jr, et al. Resveratrol-induced autophagocytosis in ovarian cancer cells. Cancer Res. 2004;64:696–703. doi: 10.1158/0008-5472.can-03-2404. [DOI] [PubMed] [Google Scholar]
- 121.Bursch W, et al. Autophagic and apoptotic types of programmed cell death exhibit different fates of cytoskeletal filaments. J. Cell Sci. 2000;113:1189–1198. doi: 10.1242/jcs.113.7.1189. [DOI] [PubMed] [Google Scholar]
- 122.Kanzawa T, et al. Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene. 2005;24:980–991. doi: 10.1038/sj.onc.1208095. [DOI] [PubMed] [Google Scholar]
- 123.Chau Y-P, Lin S-Y, Chen JH-C, Tai M-H. Endostatin induced autophagic cell death in EAhy926 human endothelial cells. Histol. Histopathol. 2003;18:715–726. doi: 10.14670/HH-18.715. [DOI] [PubMed] [Google Scholar]
- 124.Juhasz G, Csikos G, Sinka R, Erdelyi M, Sass M. The Drosophilahomolog of Aut1 is essential for autophagy and development. FEBS Lett. 2003;543:154–158. doi: 10.1016/s0014-5793(03)00431-9. [DOI] [PubMed] [Google Scholar]