

Abstract
Transposons have been used in invertebrates for transgenesis and insertional mutagens in genetic screens. We tested a functional transposon called Sleeping Beauty in the one-cell mouse embryo. In this report, we describe experiments in which transposon vectors were injected into one-cell mouse embryos with mRNA expressing the SB10 transposase enzyme. Molecular evidence of transposition was obtained by cloning of insertion sites from multiple transgenic mice produced by SB10 mRNA/transposon coinjection. We also demonstrate germ-line transmission and expression from transposed elements. This technique has promise as a germ-line transgenesis method in other vertebrate species and for insertional mutagenesis in the mouse.
Keywords: transposon‖insertional mutagenesis‖Sleeping Beauty
Germ-line transgenesis can be used to gain insight into the biological function of genes in vivo, examine the potential of promoter sequences to function in specific tissues, and endow animals with new traits. New technologies for germ-line transgenesis could allow similar work to be done routinely in other species. Although transgenesis is relatively facile in the laboratory mouse, similar levels of success are not easily achieved in large animals important to the agribusiness industry (1, 2).
The availability of genomic sequence data from complex mammalian organisms such as the mouse and human has far outpaced biologist's ability to assign function to these sequences. This dilemma has spawned a field of inquiry often called “functional genomics” (3). Germ-line DNA transgenesis can potentially be used for functional genomics by introducing insertional mutagens into chromosomal DNA. The laboratory mouse has many distinct advantages as a mammalian genetic model for functional genomics. These advantages include the ability to introduce foreign DNA into the germ line (4) and to selectively delete or alter endogenous DNA sequences in the germ line by using homologous recombination (5). However, a high throughput system for insertional mutagenesis of the mouse germ line is still lacking. Such a system would allow facile functional analysis of mammalian genes with phenotype-driven genetic screens. A particularly efficient method of insertional mutagenesis in lower organisms including yeast, bacteria, Drosophila melanogaster, and Caenorhabditis elegans has been the transposable elements (6–9). Recently a transposon system, called Sleeping Beauty, was resurrected by site-directed mutagenesis of inactive elements in salmonid genomes (10). The Sleeping Beauty transposase (SB10) is active in a wide range of vertebrate cells, including human cultured cells (10), mouse somatic tissues (11), and the mouse germ line (12–14). In this report, we investigate the potential of Sleeping Beauty to function in the one-cell mouse embryo as a potential tool for insertional mutagenesis and as an adjuvant for germ-line transgenesis in general.
Methods
Preparation and Testing of SB10 mRNA.
SB10 mRNA was synthesized from NotI linearized SB10 template DNA by using T7 RNA polymerase (Ambion mMessage mMachine high-yield capped RNA transcritption kit). Homozygous E-line fish were set up in single-pair matings. One-cell embryos were injected with 300 pg of SB10 mRNA in a 3-nl volume as described (20). Embryos were raised at 31°C and collected 48 h after fertilization for DNA isolation. Embryos were lysed in 500 μl of lysis buffer (100 mM Tris⋅HCl, pH 8.5/5 mM EDTA/0.2% SDS/200 mM NaCl/100 μg/ml Proteinase K) and incubated for 4 h at 55°C with occasional vortexing. Lysates were then treated with RNase A and phenol/chloroform-extracted. Genomic DNA was precipitated with 1 vol of isopropanol. DNA was resuspended in 10 mM Tris⋅Cl/0.1 mM EDTA (pH 7.5). Information from the genomic sequences flanking the E-line insertion locus were used to design primers for the PCR excision assay (F1Ep1: 5′-GTGATGTCCCATCTGATACAGAACG-3′ and F1Ep2: 5′-CGAGGAGATGCGTCAAGTGGC TTA-3′). PCR conditions used were 1.75 mM MgCl2/16 pmol F1Ep1/16 pmol F1Ep2/0.35 mM of each dNTP/0.5 units of expand HiFi Taq DNA polymerase (Roche). The PCR profile used was: (i) 4 min at 94°C, (ii) 30 s at 94°C, (iii) 30 s at 60°C, (iv) 4 min at 68°C, GOTO step ii and loop 30 times, (v) 7 min at 68°C, then hold at 4°C.
Preparation of DNA for Pronuclear Injection.
A 2.9-kb PvuII fragment of pT/cytomegalovirus–green fluorescent protein (CMV–GFP) was isolated on a crystal violet gel. The fragment was electroeluted onto a DEAE membrane (NA45, Schleicher & Schuell). The membrane was then eluted overnight in 700 μl of a solution containing 1 M NaCl and 0.05 M arginine-free base. The fragment was then phenol/chloroform extracted and precipitated twice with 2 vol of ethanol. The DNA was then resuspended in injection buffer (5 mM Tris/0.1 mM EDTA, pH 7.4). A 6.0-kb fragment of the pT/K14A was isolated in the same manner. Both linear transposon fragments were brought to a final concentration of 4 ng/μl in injection buffer. Just before injection, SB10 mRNA was added to the DNA fragment at a final concentration of 10 ng/μl. Pronuclear injection was performed by using standard techniques.
Southern Blotting.
Southern blotting was performed essentially as described (21). Briefly, embryonic DNA was digested with NcoI, run out on a 1% agarose gel, and transferred to a membrane. The GFP probe used was an 816-bp EcoRV fragment from the pT/CMV-GFP plasmid. The plasmid probes used were an 104-bp PvuII/KpnI fragment and a 207-bp XbaI/PvuII fragment from the pT/CMV-GFP plasmid. For the K14A experiments, genomic DNA from the founders was digested with SspI. The K14 probe used was a 1,253-bp EcoRV fragment from the pT/K14A plasmid. The plasmid probe was a 218-bp SspI/KpnI fragment from the pT/K14A plasmid.
Inverse PCR of Embryonic DNA.
Genomic DNAs from transgenic embryos were digested with BglII restriction enzyme. digested DNAs were then phenol/chloroform extracted, ethanol precipitated, and ligated in a large volume. Ligated DNAs were precipitated and used in an PCR with the Expand Long Template PCR kit (Roche Molecular Biochemicals). The PCR protocol followed manufacturer's instructions and used primer Rp1 (5′-CTCGGATTAAATGTCAGGAATTGTG-3′) for inverted repeat/directed repeat (IR/DR)-(R) and Lp1 (5′-GTGTCATGCACAAAGTAGATGTCC-3′) for IR/DR(L).
Splinkerette PCR of Transgenic Founders.
Genomic DNAs from tail-clips of the offspring of the founder transgenic for the agouti transposon were each digested with both Sau3AI and NlaIII at a concentration of 50 ng/μg. The Sau3AI digestions are useful for cloning from the IR/DR(L) by using the primers listed below, whereas NlaIII is used to clone from the IR/DR(R). Splinkerettes were made by heating equimolar amounts of the primerette-long (5′-CCTCCACTACGACTCACTGAAGGGCAAGCAGTCCTAACAACCATG-3′) with the appropriate splink to 80°C for 5 min and allowing to cool to room temperature. Splink BglII (5′-GATCCATGGTTGTTAGGACTGGAGGGGAAATCAATCCCCT-3′, 5′-phosphate) was used for the IR/DR(L) and splink SphI (5′-GTTGTTAGGACTGCTTGGAGGGGAAATCAATCCCCT-3′, 5′-phosphate) for the IR/DR(R). Appropriate splinkerettes were then ligated to the ends of the restriction endonuclease treated genomic DNA. Ligation of the appropriate “splinkerette” to the digested genomic DNA was performed at a splinkerette concentration of 7.5 μM and a DNA concentration of 25 ng/μl by using T4 DNA Ligase. Primary PCR entailed primerette-short (5′-CCTCCACTACGACTCACTGAAGGGC-3′) in conjunction with either long IR/DR(L2) (5′-CTGGAATTTTCCAAGCTGTTTAAAGGCACAGTCAAC-3′) for IR/DR(L) or long IR/DR(R) (5′-GCTTGTGGAAGGCTACTCGAAATGTTTGACCC-3′) for IR/DR(R). Primary PCR involved 10 cycles of 95°C for 5 s and 70°C (−0.5 degrees per cycle) for 2 min followed by 20 cycles of 95°C for 5 s and 65°C for 2 min. A secondary “nested” PCR was performed by using the primary PCR products diluted 1/250 within the nested PCR reaction. The second PCR entailed primerette-nested (5′-GGGCAAGCAGTCCTAACAACCA TG-3′) in conjunction with either new L1 (5′-GACTTGTGTCATGCACAAAGTAGATGTCC-3′) for IR/DR(L) or IR/DR(R)KJC1 (5′-CCACTGGGAATGTGATGAAAGAAATAAAAGC-3′) for the right IR/DR. Nested PCR involved 30 cycles of 95°C for 5 s, 61°C for 30 s, and 70°C for 90 s. Both primary and nested PCRs incorporated a hot-start at 95°C for 1 min and a final extension at 70°C for 10 min.
PCR Genotyping of Agouti Offspring.
Approximately 15- to 30-ng genomic DNA from a founder mouse transgenic for the Agouti transposon and each of his offspring was used for PCR. Primers specific for cloned junction sequences on either side of transposon-mediated insertion E (as shown by Southern blot) were used to amplify the endogenous genomic locus. PCR was also performed with one of the junction-sequence-specific primers in combination with a transposon-sequence-specific primer. For B insertion genotyping, PCR entailed 40 cycles of denaturation at 94°C for 1 min, annealing at 55°C for 1 min, and extension at 72°C for 2 min with a hot-start at 95°C for 10 min, and a final extension at 72°C for 7 min. Primers used were 36L2 (5′CTCTCTTGCCTGCCTGTG-3′) and IR/DR(L) (5′GTAAACTTCTGACCCACTGGAAT-3′) The endogenous B locus was amplified by using primer 36L2 and primer 36R (5′-AGTATTGGTTGCCTGGATTG-3′) with a 58°C annealing temperature. A transposon-specific primer Rp2 (5′-GTGGTGATCCTAACTGACCTTAAGAC-3′; specific for the right IR/DR) was used with primer 38R and an annealing temperature of 51° to amplify the transposon-genomic DNA junction (if present). For E insertion genotyping, PCR entailed 40 cycles of denaturation at 94°C for 1 min, annealing at 52.4°C for 1 min, and extension at 72°C for 2 min with a hot-start at 95°C for 10 min, and a final extension at 72°C for 7 min. The endogenous locus was amplified by using primer 38L (5′-GGGAAGAGGGCAGGTTTG-3′) and primer 38R (5′-CCACATCCTCCTTTAGATCTCAGA-3′). A transposon-specific primer Rp2 (5′-GTGGTGATCCTAACTGACCTTAAGAC-3′; specific for the right IR/DR) was used with primer 38R and an annealing temperature of 51° to amplify the transposon-genomic DNA junction (if present).
Results
Pronuclear Coinjection of Transposon DNA and SB10 mRNA.
To determine whether the Sleeping Beauty transposon system could be adapted for mouse transgenesis, we prepared in vitro-transcribed and capped mRNA for both SB10 transposase and control GFP mRNA. The activity of the SB10 mRNA preparation was verified by using a transposon-mobilization assay developed for zebrafish (Fig. 1). In this assay, zebrafish embryos homozygous for an SB10-mediated transposon vector insertion, called line E, are injected with in vitro prepared SB10 mRNA. Injected embryos were pooled and harvested after 24 h. Primers flanking the transposon insertion site are used in a PCR to detect mobilization out of the line E insertion site. PCR on homozygous line E DNA generates a 4.0-kb fragment. However, if the line E transposon has been mobilized and excision repair occurs, the PCR will detect a 0.6-kb fragment. Sequences of these 0.6-kb fragments show a three nucleotide footprint separating two TA dinucleotides, which is characteristic of true transposon transposition mobilization events (Fig. 1).
Figure 1.

Verifying activity of SB10 mRNA. In vitro-transcribed SB10 mRNA was injected into zebrafish embryos homozygous for a transposon insertion (line E). Embryos are then collected after 24 h, and genomic DNA is extracted from them (a). PCR is then performed by using primers that flank the site of transposon insertion. Transposition can then be detected by appearance of the PCR product from the transposon insertion site after excision (b). Excision can be confirmed by sequencing the PCR product to detect the three nucleotide footprint (c). The sequence of the insertion site following transposon excision is indicated (*). Two different base-pair footprints are found in roughly equal proportion, CAG or CTG.
Activity-verified SB10 mRNA or GFP mRNA was injected at 10 ng/μl into the pronuclei FVB/N strain one-cell mouse embryos with the pT/CMV-GFP plasmid fragment. The pT/CMV-GFP plasmid contains a transposon, a DNA sequence flanked by IR/DRs with the CMV promoter driving a GFP cDNA and a polyadenylation site (Fig. 2a). These embryos were implanted into pseudopregnant ICR strain stock females. A subset of the embryos was examined under fluorescence microscopy or cultured overnight. Embryos injected with GFP mRNA showed GFP fluorescence (data not shown). Injection of these mRNA/DNA combinations did not result in significant toxicity as assessed by the ability of one-cell embryos to develop in vitro for 2 days (data not shown). In initial experiments, injected and implanted embryos were killed at 13.5 days of gestation and total, high molecular weight genomic DNA was prepared from whole embryos. Genomic DNAs were digested with NcoI and analyzed by Southern blotting with either a GFP probe to detect transposon sequences or a pUC19-derived probe to detect the plasmid sequences adjacent to the transposon vectors. Digestion with NcoI and hybridization with the GFP probe should detect a 0.6-kb internal fragment and a variable junction fragment in transgenic animals. Transgenes generally integrate as head-to-tail concatomers, which would generate a 2.4-kb NcoI fragment (Fig. 2b). Intense multicopy 2.4-kb bands were detected in some embryos from both the GFP mRNA/transposon- and SB10 mRNA/transposon-injected groups. In contrast, only in SB10 mRNA/transposon-injected group were animals observed that contained multiple, variably sized, GFP-hybridizing bands in addition to the 0.6 kb band (Fig. 2b). The intensities of these bands were similar and suggested a single copy of each per cell (Fig. 2c). These variably sized NcoI bands did not hybridize to the plasmid vector probe; in contrast to the intense 2.4 kb bands present in both groups (Fig. 2c), and therefore represent potential SB10-mediated transposon insertions. A summary of the data from several large experiments is shown in Table 1. There was a trend toward a higher overall transgenesis frequency when transposon vector DNA was coinjected with transposase mRNA compared with injection with control GFP mRNA. The increase is accounted for by the presence of embryos harboring multiple single-copy transposon insertions. On average, each of these embryos had three single-copy transposon insertions.
Figure 2.

Structure and analysis of transposon integration sites. (a) The structure of the linear transposon that was used for pronuclear injection is shown. The transposon contains a GFP reporter (gray arrow) driven by the immediate early CMV promoter (black arrow) followed by the bovine GH polyadenylation site (black box) and flanked by the IR/DRs (white arrows). (b Upper) The structure of a Sleeping Beauty-mediated integration site including the expected fragments from a NcoI (N) digest. (b Lower) The structure of a random integration site and NcoI restriction map. The probes used for Southern blot analysis are also defined as black bars. (c) Examples of three embryonic DNAs analyzed by Southern blotting are shown. The results shown were achieved with the GFP probe (Left) and the plasmid probe (Right).
Table 1.
Summary of Southern blot analysis
| mRNA injected | No. of embryos
|
Transgenic frequency | Type of insertion
|
Avg. SB-mediated copy number | ||
|---|---|---|---|---|---|---|
| Total | Transgenic | SB-mediated | Random | |||
| SB | 42 | 19 | 45% | 6 | 13 | 3 |
| GFP | 21 | 6 | 29% | 0 | 6 | NA |
Data from 4 days of pronuclear coinjection are shown. The data in the left portion of the table shows the total number of embryos analyzed for each type of mRNA injected along with the number of transgenic embryos determined by Southern blotting. The right portion of the table summarizes the Southern blot analysis. Transgenic embryos were classified as having SB-mediated insertions if they had a band on Southern blot that hybridized to the GFP probe but not the plasmid probe. Also shown is the average transposon copy number for each of the 6 transgenic embryos containing SB-mediated insertions.
Cloning and Analysis on Transposon Insertion Sites.
We molecularly cloned several transposon insertion sites to verify that SB10-mediated transposition of the transposon from introduced plasmids to chromosomal DNA had occurred. We used two different techniques. We digested genomic DNA from transgenic animals with BglII, which does not cut within the GFP transposon, and performed inverse PCR using transposon-specific primers to simultaneously clone both sides of any transposon insertion. Alternately, we used splinkerette-mediated PCR on genomic DNA from transgenic mice digested with NlaIII or Sau3AI to clone individual left- or right-hand transposon junction fragments. Three two-sided transposon insertions and 21 left- or right-hand transposon insertions were sequenced (Fig. 3). These clones show that the transposons were flanked by TA dinucleotides and lacked pUC19 sequences adjacent to the transposon. Some of the insertion sites were mapped electronically by comparing the junction sequence to the whole mouse genome database (Celera Corporation). Transposon insertion occurred on a variety of chromosomes and chromosomal regions. In subsequent experiments, SB10 mRNA/transposon-injected embryos were implanted and allowed to develop to term. In these experiments, increasing doses of SB10 mRNA were injected with the transposon. A significant drop in litter size was noted between doses of 200 ng/μl and 300 ng/μl of SB10 messenger RNA (data not shown).
Figure 3.

Molecular analysis of flanking genomic DNA sequences. The flanking genomic DNA sequences from transposon insertions were cloned by using either inverse PCR or splinkerette PCR. The last 10 nucleotides of the IR/DR (bold and underlined) next to the flanking genomic DNA is shown. Some clones were mapped by using the whole mouse genome database from Celera Coproration. The map position for some clones is listed. Chr., chromosome; cM, centimorgan.
Germ-line Transmission and Expression of Transposed Elements.
A transposon vector harboring the K14A expression cassette was constructed to determine whether founder animals and their offspring could express a transgene contained within a transposon that was introduced into the germ line by transposition (Fig. 4a). The K14A construct expresses the agouti gene from a keratin-14 promoter and in non-agouti mice, such as C57BL/6J, will cause varying degrees of a yellow coat color (15). Other effects, such as obesity and diabetes, seen when agouti is expressed ubiquitously (16), are not observed in K14A transgenic mice. The pT/K14A transposon was coinjected with SB10 mRNA into C57BL/6J one-cell mouse embryos to create transgenic founder animals. Six of twenty offspring (30%) had some yellow pigmentation in their coats. The range of pigmentation seen in these animals is similar to that reported previously with the K14A cassette not contained within a transposon. Therefore, the presence of the IR/DRs of the transposon flanking the transgene did not have a detectable deleterious effect on expression in transgenic mice. Southern blot analysis of tail DNA from these animals revealed that 9 of the 20 mice were positive for the transposon—a 45% transgenesis frequency (data not shown). Hybridization of SspI-digested DNA from these transgenic animals with the keratin-14 or plasmid vector probe was used to identify potential single copy, Sleeping Beauty-mediated transposon insertions. As shown in Fig. 4a, random insertion of the transposon should create a 4.2-kb fragment. Transposons integrated into chromosomes by transposition should be single copy and the keratin-14 probe will detect a variably sized SspI fragment whose size depends on the location of the next SspI site in the flanking genomic DNA. Southern blot analysis of the nine transposition-positive mice indicated that three had Sleeping Beauty-mediated insertions, three had random insertions, and three had both types of insertions. Interestingly, the three animals with only Sleeping Beauty-mediated insertions had only one or two copies of the transposon and did not have yellow coats.
Figure 4.

Structure of pT/K14A transposon reporter and analysis of a T/K14A+ mouse line. (a) The structure of the linear pT/K14A transposon that was used for pronuclear injection is shown. The transposon contains an agouti reporter (Ag,gray arrow) driven by the keratin-14 gene promoter (K14, black arrow) followed by the human GH polyadenylation site (pA, black box) and flanked by the IR/DRs (white arrows). Also shown are the SspI restriction enzyme sites (S) and probes used for Southern blotting. The three different coat color phenotypes observed in the offspring and founder are also shown: B = normal C57BL/6, I = intermediate expression in the skin but not the coat, Y = yellow coat color in the skin and coat. (b) Southern blot analysis of one T/K14A+ founder (red box) and 14 offspring generated by backcrossing to C57BL/6 is shown. Four independent transposon insertion sites are indicated by arrows B–E at the left. Band A represents a junction fragment that so-segregates with bands C and D. The coat color phenotype is indicated above each lane corresponding to a mouse in the top panel.
One of the transgene-positive founders with a yellow coat showed 5 SspI fragments that hybridized with the keratin-14 probe after Southern blotting (Fig. 4b). This founder was bred to nontransgenic C57BL/6 animals, and offspring displayed one of three different coat color phenotypes: non-agouti, yellow pigmented skin with black hair, or yellow pigmented skin and hair (Fig. 4b). Three of the SspI bands cosegregated in offspring and were responsible for the yellow skin with black hair phenotype (bands A, C, and D in Fig. 4b). One of these three bands also hybridized with the plasmid vector probe indicating that these bands resulted from at least on random insertion event (Fig. 4b). Two other SspI bands (bands B and E in Fig. 4b) segregated independently of each other and the first three SspI bands. Neither of these bands hybridized to the plasmid vector probe, and one of them conferred the yellow skin and hair phenotype, whereas the other was associated with no phenotype.
The genotypes of a select group of agouti transposon-positive backcross mice with several different transposon-mediated insertion patterns were determined by PCR analysis. After determining the junction sequences of each unique transposon-mediated insertion, we designed primers that would amplify either the endogenous locus or the rearranged locus containing transposon insertion B or E by using an IR/DR specific primer. This PCR approach was used to genotype the founder mouse and its offspring for insertion E (Fig. 5a) and insertion B (Fig. 5b). All PCR products were of the expected size demonstrating that the cloned junction sequences are actually those that flank the transposon after its insertion into the genome. The E insertion was also bred to homozygousity, and these mice appear to have a normal phenotype (Fig. 5c).
Figure 5.
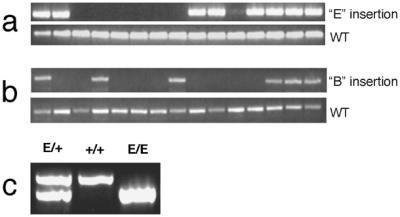
PCR genotyping of founder and offspring genomic DNA. Offspring of an agouti founder were genotyped by using primers based on flanking sequences cloned from insertion site E (a) or insertion site B (b). In both cases, a second reaction was performed by using a transposon-specific primer and a primer in the flanking genomic DNA to detect the transposon insertion. (c) Mice heterozygous for the E insertion were intercrossed. Offspring were genotyped by PCR, and an example of a mouse with each genotype is shown.
The B band agouti transposon insertion is associated with yellow pigmentation, whereas the E band insertion causes no yellow pigmentation (Fig. 4). Both transposon vectors seem to be intact by Southern analysis. These data suggest that transgenes present within single-copy transposons can be expressed but are also subject to the same position effects as standard transgenes in mice. A comparison of the sequence context of the E and B band insertions suggests nothing about why these insertions differ in expression. The E band insertion, although silent, is within just a few kilobases of a gene represented in the expressed sequence tag database (data not shown).
Discussion
In summary, the SB10 transposase enzyme can function, in the context of the one-cell mouse embryo, to mediate transposition of transposons into chromosomes. This method allows the introduction of multiple single-copy transposons into mice genomes. Recent work reported by Yant et al. (11) demonstrated that SB10 also mediates transposition into somatic cells of mice. Thus, the SB10 system has potential for gene delivery in multiple settings. SB10-mediated transgenesis can increase the overall transgenesis rate. Moreover, because founders carry several independent transposon insertions, the number of potentially expressing transgenes is multiplied. It seems clear from our data and others (14) that transgenes embedded in transposon vectors can be expressed but are also subject to position effects. The inclusion of insulator elements within the transposon vector may allow more consistent expression of the transgene.
Sleeping Beauty transposons inserted into the genome, at TA dinucleotides, without changes in the adjacent sequence, except for the duplication of the TA dinucleotide (10, 11). The comparison of insertion site sequences in this report and in others that we have obtained shows no particular sequences adjacent to the TA dinucleotide are favored sites of transposon insertions. It appears likely that a very large number of TA dinucleotides are potential Sleeping Beauty transposon insertion sites.
Southern blotting results obtained in this work suggest coinjection of transposon vector DNA and transposase mRNA into the early mouse embryo results in the generation of offspring with single-copy transposon insertions and/or transposon-concatomer insertions. In this way, this technique differs from the generation of single-copy transgenes by retroviral infection of early mouse embryos that only yield single-copy insertions (17). Also, transposon integrants often express transgenes contained within the vector, unlike proviral integrants that are silenced when passed through the germ line (17). Retroviral infection of early mouse embryos also results in offspring showing a chimeric pattern of somatic proviral integration. We observed no evidence of chimerism in our experiments. Finally, some evidence suggests that hot and cold spots for proviral insertion exist and that integration favors the 5′ region of genes (18). It seems possible that transposon insertion may occur in some regions that are not favored for proviral insertion. For these reasons, the generation of single-copy, simple insertions of foreign DNA by transposition offers several advantages compared with retroviral infection.
Recent work demonstrates that the Sleeping Beauty transposase can function in transgenic mice (12–14). In these experiments, chromosomally resident transposon vectors were mobilized in transgenic mice that were ubiquitously expressing the transposase (13, 14) or were expressing the transposase in the male germ line (12). The approach described here results in a higher average number of transposon insertions per gamete then the other methods, 3 average transposition events per embryo versus 0.2–2 transposition events per offspring (12–14). Also, Sleeping Beauty transposition from chromosomally resident transposons tends to select for reinsertion at linked loci (12). Our data show that transposon insertion into one-cell mouse embryos, as described here, can result in inserts in many parts of the genome with equal likelihood. Indeed, transposon insertion into the one cell mouse embryo may be useful in obtaining many unlinked transposon insertions for subsequent remobilization in the germ line by using Sleeping Beauty transposase transgenes.
SB10-mediated transgenesis may be used to create transgenic animals in various strains of mice or in other species for which transgenesis is currently very inefficient or impossible. Sleeping Beauty transposons are very easily modified. Transposon vectors of varying size, up to 6 kb, have been constructed and shown to be functional, and the internal sequences can be extensively modified without affecting transgenesis (19). Thus, the SB system may have applications for generating large numbers of transgenic animals with simple, single copy insertions of foreign DNA.
Acknowledgments
We thank Deanna Mohn for her work in creating and characterizing the zebrafish E line. We would also like to thank Michael P. Rosenberg (Glaxo Research Institute) for providing the pK14A agouti expression plasmid.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Abbreviaitons: CMV, cytomegalovirus; GFP, green fluorescent protein; IR/DR, inverted repeat/directed repeat.
References
- 1.Ward K A, Nancarrow C D. Mol Biotechnol. 1995;4:167–178. doi: 10.1007/BF02921610. [DOI] [PubMed] [Google Scholar]
- 2.Pintado B, Gutierrez-Adan A. Reprod Nutr Dev. 1999;39:535–544. doi: 10.1051/rnd:19990502. [DOI] [PubMed] [Google Scholar]
- 3.Woychik R P, Klebig M L, Justice M J, Magnuson T R, Avner E D, Avrer E D. Mutat Res. 1998;400:3–14. doi: 10.1016/s0027-5107(98)00023-2. [DOI] [PubMed] [Google Scholar]
- 4.Gordon J W, Ruddle F H. Methods Enzymol. 1983;101:411–433. doi: 10.1016/0076-6879(83)01031-9. [DOI] [PubMed] [Google Scholar]
- 5.Capecchi M R. Science. 1989;244:1288–1292. doi: 10.1126/science.2660260. [DOI] [PubMed] [Google Scholar]
- 6.Garfinkel D J, Mastrangelo M F, Sanders N J, Shafer B K, Strathern J N. Genetics. 1988;120:95–108. doi: 10.1093/genetics/120.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tripathi A K. Trends Genet. 1995;11:340. doi: 10.1016/s0168-9525(00)89101-5. [DOI] [PubMed] [Google Scholar]
- 8.Sentry J W, Kaiser K. Trends Genet. 1992;8:329–331. doi: 10.1016/0168-9525(92)90267-8. [DOI] [PubMed] [Google Scholar]
- 9.Eide D, Anderson P. Proc Natl Acad Sci USA. 1985;82:1756–1760. doi: 10.1073/pnas.82.6.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ivics Z, Hackett P B, Plasterk R H, Izsvak Z. Cell. 1997;91:501–510. doi: 10.1016/s0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- 11.Yant S R, Meuse L, Chiu W, Ivics Z, Izsvak Z, Kay M A. Nat Genet. 2000;25:35–41. doi: 10.1038/75568. [DOI] [PubMed] [Google Scholar]
- 12.Fischer S E, Wienholds E, Plasterk R H. Proc Natl Acad Sci USA. 2001;98:6759–6764. doi: 10.1073/pnas.121569298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dupuy A J, Fritz S, Largaespada D A. Genesis. 2001;30:82–88. doi: 10.1002/gene.1037. [DOI] [PubMed] [Google Scholar]
- 14.Horie K, Kuroiwa A, Ikawa M, Okabe M, Kondoh G, Matsuda Y, Takeda J. Proc Natl Acad Sci USA. 2001;98:9191–9196. doi: 10.1073/pnas.161071798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kucera G T, Bortner D M, Rosenberg M P. Dev Biol. 1996;173:162–173. doi: 10.1006/dbio.1996.0014. [DOI] [PubMed] [Google Scholar]
- 16.Wolff G L, Roberts D W, Mountjoy K G. Physiol Genomics. 1999;1:151–163. doi: 10.1152/physiolgenomics.1999.1.3.151. [DOI] [PubMed] [Google Scholar]
- 17.Jaenisch R, Dausman J, Cox V, Fan H. Hamatol Bluttransfus. 1976;19:341–356. doi: 10.1007/978-3-642-87524-3_35. [DOI] [PubMed] [Google Scholar]
- 18.Vijaya S, Steffen D L, Robinson H L. J Virol. 1986;60:683–692. doi: 10.1128/jvi.60.2.683-692.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Izsvak Z, Ivics Z, Plasterk R H. J Mol Biol. 2000;302:93–102. doi: 10.1006/jmbi.2000.4047. [DOI] [PubMed] [Google Scholar]
- 20.Hyatt T M, Ekker S C. Methods Cell Biol. 1999;59:117–126. doi: 10.1016/s0091-679x(08)61823-3. [DOI] [PubMed] [Google Scholar]
- 21.Jenkins N A, Copeland N G, Taylor B A, Bedigian H G, Lee B K. J Virol. 1982;42:379–388. doi: 10.1128/jvi.42.2.379-388.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]