

Abstract
V(D)J recombination is critical to the generation of a functional immune system. Intrinsic to the assembly of antigen receptor genes is the formation of endogenous DNA double-strand breaks, which normally are excluded from the cellular surveillance machinery because of their sequestration in a synaptic complex and/or rapid resolution. In cells deficient in double-strand break repair, such recombination-induced breaks fail to be joined promptly and therefore are at risk of being recognized as DNA damage. Poly(ADP-ribose) polymerase-1 is an important factor in the maintenance of genomic integrity and is believed to play a central role in DNA repair. Here we provide visual evidence that in a recombination inducible severe combined immunodeficient cell line poly(ADP-ribose) formation occurs during the resolution stage of V(D)J recombination where nascent opened coding ends are generated. Poly(ADP-ribose) formation appears to facilitate coding end resolution. Furthermore, formation of Mre11 foci coincide with these areas of poly(ADP-ribosyl)ation. In contrast, such a response is not observed in wild-type cells possessing a functional catalytic subunit of DNA-dependent protein kinase (DNA-PKcs). Thus, V(D)J recombination invokes a DNA damage response in cells lacking DNA-PKcs activity, which in turn promotes DNA-PKcs-independent resolution of recombination intermediates.
V(D)J recombination is the mechanism by which antigen receptor genes are assembled and diverse repertoires of immunoglobulins and T cell receptors are created. This reaction proceeds through two intimately linked steps: a site-specific cleavage to generate double-stranded DNA breaks and the religation of these breaks. Whereas the cleavage reaction is mediated by the lymphoid specific recombination-activating genes 1 and 2 (RAG1 and RAG2) (1–3) proteins, efficient joining of the recombination intermediates depends on several ubiquitous proteins involved in the nonhomologous end-joining pathway, including the DNA end binding Ku heterodimer, DNA-dependent protein kinase catalytic subunit (DNA-PKcs), XRCC4, and ligase IV (4–7).
Cells with a mutation in the DNA-PKcs gene, incurred in severe combined immunodeficient (scid) mice, carry a defect in the formation of coding joints. However, the presence of oligoclonal mature T and B lymphocytes (8, 9) suggests that an alternative cellular pathway may be used during V(D)J recombination, bypassing the requirement for a functional DNA-PKcs subunit. The existence of such a pathway was substantiated by the detection of DJ coding joints in DNA-PK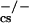 mutant mice (10–12).
mutant mice (10–12).
Poly(ADP-ribose) polymerase-1 (PARP-1) is a highly abundant nuclear protein implicated in the surveillance of genome integrity (13). Similar to DNA-PK, PARP-1 is catalytically activated by DNA strand breaks, which results in the rapid, covalent modification of nuclear proteins involved in DNA metabolism and chromatin architecture (including PARP-1 itself) with long branched polymers of negatively charged poly(ADP-ribose) (pADPr) from β-NAD+ (14). The poly(ADP-ribosyl)ated protein, in turn, dissociates from the DNA breaks and allows various enzymes to process and repair the DNA lesions. Recently, a number of additional pADPr polymerases have been identified, only some of which appear to be responsive to DNA damage, but detailed knowledge on their function is still lacking (15). Although DNA breakage and rejoining are intrinsic features of V(D)J recombination, PARP-1-deficient mice (Adprt1−/−) proved to be proficient in V(D)J recombination (16). Furthermore, thymocytes from scid/Adprt 1−/− mice are able to bypass the scid block from the double-negative stage to the double-positive stage through productive rearrangements of the T cell antigen receptor β locus (17). This finding indicates that PARP-1 may affect V(D)J recombination in scid, but not wild-type, lymphoid cells.
Mre11, Rad50, and NBS are components of a protein complex that is important in the DNA damage response of mammalian cells. This complex forms large nuclear foci in response to ionizing radiation and remains associated with the damaged sites until the majority of DNA lesions have been repaired (18). In vitro studies have suggested a possible role for this protein complex in V(D)J recombination through its ability to nick synthetic DNA hairpins and its exonuclease properties (19). This role is substantiated by a recent finding that NBS is colocalized with a rearranging T cell antigen receptor α locus (20). In this article, we provide evidence for the involvement of Mre11 and PARP in DNA-PK-independent V(D)J recombination.
Methods
Cell Culture.
The SP1 and S4 cell lines were derived from bcl-2 transgenic scid/+ and scid/scid mice, respectively, by transformation of fetal B cell precursors with temperature sensitive (ts)-Abelson (Ab)-murine leukemia virus (21). Cells were maintained at 33°C. To induce V(D)J recombination, cells were incubated at 39°C for 2–3 days, and to facilitate recombination resolution in the scid-ts (temperature sensitive) cells, cells were returned to 33°C for 1–2 days. 3-Aminobenzamide (3-AB) (Sigma), dissolved in H2O, was added to the culture medium immediately before shifting the cells to 39°C at the final concentrations as indicated in the text.
DNA Isolation and PCR.
Genomic DNA was prepared by using the EasyDNA kit (Invitrogen). The VJκ-coding joints were amplified by PCR with the primers previously described (21). The DNA was first denatured at 95°C for 5 min, followed by 20–27 cycles of amplification by using 0.5 unit of Taq polymerase per reaction. The level of α-actin products served as a control for the amount of input DNA. Serial dilutions of input DNA samples were tested to determine the linearity of the PCR. PCR products were separated by electrophoresis and analyzed by Southern blotting. A modified ligation-mediated PCR (LM-PCR) was performed as described (21).
Cell Fixation and Immunostaining.
Cell fixation was performed as described (22). Briefly, cells were pelleted at 1,500 rpm for 5 min, washed with cold PBS twice, and then fixed with 1% paraformaldehyde for 15 min on ice. Cells were then washed with PBS and resuspended in 70% ethanol at −20°C for 2 h. Fixed cells were then washed with PBS and permeabilized with 0.25% Triton X-100 for 5 min on ice followed by another wash with cold PBS. Cells were treated with 2% normal goat serum (Sigma) for 20 min at room temperature to block nonspecific binding sites. Cells were then incubated with primary Ab, anti-pADPr (10H, see refs. 23 and 25) and/or anti-Mre11 (Novus, Littleton, Co) Abs overnight at 4°C, diluted in staining buffer (1% BSA/0.05% Tween-20 in PBS) followed by three washes with staining buffer. Goat anti-rabbit-FITC (Sigma) was used as a secondary Ab against anti-Mre11 Ab, and either the Alexa-488 goat anti-mouse or Alexa-594 (Molecular Probes) goat anti-mouse was used as a secondary Ab against 10H. These incubations were carried out in the dark at room temperature for 1 h. After one final wash in staining buffer, the cells were resuspended in PBS for flow cytometry and confocal analysis. Stained cells were analyzed by using FACSCalibur (Becton Dickinson) equipped with an argon laser tuned at 488 nm for fluorescence excitation. Confocal images were taken on a Leica TCS inverted scanning microscope equipped with argon (488 nm) and krypton (568 nm) lasers. The specificity of nuclear staining was verified by the monomeric cyanine nucleic acid dye PO-PRO-1 (Molecular Probes).
Recombination End Protection Assay.
The detail protocol will be described elsewhere (D.F. and Y.C., unpublished data). In brief, the isolated nuclei were treated with exonuclease V (Upstate Biotechnology, Lake Placid, NY) and then prepared in agarose plugs for deproteination. The purified DNA molecules were treated with T4 DNA polymerase and subjected to a ligation-mediated PCR for analyses of coding ends.
Results
V(D)J Recombination Induces PARP Activation in scid-ts Cells.
The study of the end processing reaction during V(D)J recombination has been hampered by the rapid kinetics of cleavage, hairpin formation, and religation. In an effort to overcome this limitation and to decipher the end processing events at endogenous antigen receptor gene loci, we developed a scid-ts pre-B cell line that provides the opportunity to analyze each reaction in a piecemeal fashion (21). As described in detail (21, 24), at the permissive temperature (33°C), RAG expression is low, Ig light chain (IgL-chain) loci are inaccessible, and thus a very low level of recombination occurs. At the nonpermissive temperature (39°C), RAG expression is up-regulated and the IgL-chain loci become accessible, resulting in recombination initiation. At this point both signal ends and coding ends are believed to be contained within a synaptic complex held by the RAG proteins. However, the hairpin coding ends of the scid-ts cells appear to be inaccessible to the nicking enzyme as they persist at the nonpermissive temperature. When returned to the permissive temperature (39°C→33°C, postinduction), the hairpin ends are made accessible to a nicking enzyme resulting in the appearance of stable, opened coding ends, which is followed by their religation to form coding joints (24).
Given the large quantity of unresolved ends produced in scid lymphocytes undergoing V(D)J recombination, we investigated whether the poly(ADP-ribosyl)ation system becomes activated in response to the increased numbers of recombination intermediates. To examine endogenous pADPr levels in single intact cells under the different states of V(D)J recombination, we performed immunofluorescence staining and flow cytometry by using the highly specific mAb 10H (23, 25). As shown in Fig. 1A Left, the scid heterozygous counterpart (s/+-ts) displayed no change in pADPr formation under any of the culture conditions. This is not unexpected, given that the ends are continually self-contained within the synaptic complex until they are efficiently resolved. In contrast, the scid-ts cells displayed increased pADPr formation after 2 days of recombination induction, followed by an even more dramatic shift toward higher pADPr levels once the cells were returned to 33°C (Fig. 1A Center). The amount of PARP-1 protein was comparable under various experimental conditions (data not shown). This finding suggests that the coding ends that were released from the synaptic complex induced a stimulation of the poly(ADP-ribosyl)ation system. The increase in fluorescence intensity could be blocked by using the ADP-ribosylation inhibitor 3-AB, which confirmed the specificity of the immunostaining (Fig. 1A Right). The levels of pADPr formed during V(D)J recombination in scid-ts cells were comparable to control cells exposed to γ-irradiation (6 Gy; Fig. 1A Right).
Figure 1.

Increased pADPr formation in response to V(D)J recombination in scid-ts cells. After incubation under various culture conditions as indicated, s/+-ts and scid-ts cells were fixed in 1% paraformaldehyde and pADPr was detected by indirect immunofluoresence by using 10H and alexa-488 goat anti-mouse Abs. (A) FACS analysis. (B) Confocal analysis of scid-ts immunostained cells.
We next analyzed the immunostained cells by confocal microscopy to determine the distribution of pADPr within single cells (Fig. 1B). Consistent with the above results, scid-ts cells at 33°C displayed very low fluorescence (39°C Fig. 1B Inset) and despite an overall increase in fluorescent activity in the cells cultured at 39°C, the cells appeared only slightly speckled. However, once the cells were returned to 33°C, several bright pADPr foci formed in ≈50–60% of the cells analyzed. Prior treatment with 3-AB resulted in suppression of these foci (39°C →33°C Fig. 1B Inset). Therefore, not only did the overall level of poly(ADP-ribosyl)ation increase in scid-ts cells after up-regulation of V(D)J recombination, but this activity appeared to be confined to specific areas within the cell.
Inhibiting ADP-Ribosylation Interferes with Coding End Resolution.
We next wanted to determine whether the high level of localized poly(ADP-ribosyl)ation had any functional relevance to V(D)J recombination. We used a PCR assay to compare the levels of coding joints formed at the κ loci during incubation of the cells with or without 3-AB (Fig. 2A). Wild-type s/+-ts cells immediately formed coding joints at 39°C, and 3-AB even at a concentration of 10 mM had no effect on coding joint formation. In contrast, coding joints in scid-ts cells did not form in abundance until the cultures were returned from 39°C to 33°C, and coding joint formation was inhibited by 3-AB in a dose-dependent manner (Fig. 2A). At lower numbers of PCR cycles (i.e., 20 cycles), the inhibitory effect of 3-AB was already evident at a concentration of 1 mM, which is known to inhibit poly(ADP-ribosyl)ation but not mono-ADP-ribosyl transfer reactions or NAD+ hydrolysis (26) (Fig. 2B). Similar results were obtained by using another ADP-ribosylation inhibitor, 4-amino-1,8-naphthalimide (data not shown). We then determined whether the observed reduction in coding joints was caused by a decrease in recombination cleavage. A standard LM-PCR designed to detect the recombination intermediates, Jκ signal ends (21), revealed that drug treatment did not reduce the level of signal ends (Fig. 2C). Therefore, the inhibitor did not affect V(D)J recombination induction but clearly interfered with coding end resolution. These data indicate a marked difference in the requirement for poly(ADP-ribosyl)ation during V(D)J recombination between s/+-ts and scid-ts cells. In scid-ts cells, coding end resolution facilitated by poly(ADP-ribosyl)ation whereas in s/+-ts cells it does not. Because pADPr foci are absent in s/+-ts cells and are present in the scid-ts cells only during the recombination resolution phase, where nascent opened coding ends are generated, it is conceivable that the observed foci in Fig. 1B may represent the dysregulated recombination complexes. The lack of pADPr foci in s/+-ts cells is consistent with rapid resolution of coding ends and/or containment of the recombination intermediates within the RAG complexes. However, in the scid-ts cells, coding end processing occurs upon RAG down-regulation and thus it is rather unlikely that coding end processing occurs within the RAG complex (24). Therefore, it is probable that once the RAG proteins dissociate from the recombination ends, PARP-1 recognizes and binds to protect the exposed ends, in essence forming a new DNA–protein complex.
Figure 2.

Reduction of scid-ts-coding joints in the presence of the ADP-ribosylation inhibitor 3-AB. DNA was isolated from s/+ and scid-ts cells treated with varying concentrations of 3-AB, as described in the text. (A) Analysis of VJκ-coding joints in s/+-ts and scid-ts cells, using 25 cycles of PCR amplification. (B) Dose-dependent reduction of coding joint formation in scid-ts cells analyzed after 20 PCR cycles. (C) Analysis of Jκ signal ends by LM-PCR.
Effect of Poly(ADP-Ribosyl)ation on the Accessibility of Coding Ends to Processing Enzymes.
The above finding led us to postulate that the poly(ADP-ribosyl)ation event might modify the recombination synaptic complex such that the previously protected coding ends become accessible to the processing and joining machinery required for their resolution. Based on this argument, an inhibition of pADPr formation by 3-AB should conceal the ends and therefore result in a failure of end resolution. To directly test this hypothesis, we used a recombination end protection assay recently developed in our laboratory (D.F. and Y.C., unpublished data) to measure the accessibility of coding ends in intact nuclei to a processing enzyme, exonuclease V. If the substantial number of opened coding ends present in the postinduction of scid-ts cells (21) were contained in a DNA end binding protein complex, they should be resistant to exonuclease V treatment. If, on the other hand, the recombination ends were free from protein complexes, they should be accessible to degradation mediated by exonuclease V digestion. We amplified the coding ends remaining after exonuclease V digestion, which were presumably protected from the digestion. The total amount of input coding ends was revealed by LM-PCR of purified DNA without previous treatment of exonuclease, which serves as a reference for estimating the extent of end protection. A representative analysis is shown in Fig. 3A and the quantitative comparison is given in Fig. 3B. It is clear that in the absence of 3-AB, although the level of total coding ends is high, treating nuclei with exonuclease V greatly reduces the detection of these ends (Fig. 3A, lanes 1 and 3). This finding indicates that recombination coding ends are accessible to the processing machinery when poly(ADP-ribosyl)ation is active. In contrast, the opened coding ends appear to be significantly more protected when PARP activity is inhibited even though the total level of input opened coding ends is reduced (Fig. 3A, lanes 4 and 6 as well as lane 1). These data provide a clear indication for the involvement of poly(ADP-ribosyl)ation in regulating the accessibility of opened coding ends. The reduction of input opened coding ends in 3-AB-treated cells is not caused by a decrease in recombination cleavage because the level of signal ends in these cells is comparable to the cells without 3-AB treatment (Fig. 2C). Instead, the reduction is more likely to result from a decrease in hairpin end opening (unpublished observation).
Figure 3.

Inhibition of poly(ADP-ribosyl)ation increases protection of coding ends to enzymatic degradation. (A) Representative LM-PCR analyses of recombination coding ends in scid-ts cells after recombination induction (39°C→33°C). Nuclei isolated from cultures with and without 3-AB (5 mM) were subjected to exonuclease V (Exo) digestion; the remaining nuclear-coding ends were then revealed by LM-PCR (lanes 6 and 3, respectively). The positive control (K) represents coding ends retrieved from DNA purified by Proteinase K digestion, which reflects the total amount of input-coding ends (lanes 1 and 4). KE is a control for the efficacy of exonuclease digestion, in which LM-PCR was performed on the purified DNA sample that had been digested with exonuclease V (lanes 2 and 5). (B) Increased protection of coding ends in the 3-AB-treated scid-ts cells. Relative protection (percentage) was assessed by using a PhosphorImager and imagequant and is defined as (“Exo treatment” density value/“input” density value) × 100.
Mre11 Foci Colocalize with pADPr Foci.
Based on the biochemical properties of Mre11/Rad50/NBS in end processing reactions, it is possible that these proteins might participate in the formation of protein complexes along with PARP at recombination ends of scid cells. To address this possibility, we immunostained the respective cells with both Mre11 (green) and pADPr (red) Abs to determine the localization of Mre11 in relation to pADPr (Fig. 4). In the scid-ts cells cultured at 39°C to induce recombination (Fig. 4 a–c), no intense Mre11 foci were discernable although there were several areas of diffuse fluorescence, and again there was very little pADPr fluorescence. When the scid-ts cells were returned from 39°C to 33°C (Fig. 4 d–f), very striking foci formed for both Mre11 and pADPr, and these foci were colocalized in ≈40% of the cells. Interestingly, RAG-2 did not colocalize to pADPr foci in the scid-ts cells (data not shown). In comparison, when the s/+-ts cells were simultaneously stained with Mre11 and pADPr Abs, <5% of the cells at 39°C showed double staining. This low frequency may be caused by enhanced recombination activity being induced under these conditions (Fig. 4 g–l). Furthermore, in scid-ts cells exposed to ionizing radiation (Fig. 5A), multiple foci were evident throughout the cell and coincident foci could be observed. Thus, the doubly positive foci observed in the postinduction scid-ts cells displayed characteristics similar to the pattern of the γ-irradiated cells. Therefore, both Mre11 and PARP, apparently, can associate with DNA lesions generated either by genotoxic agents or recombination events.
Figure 4.

Mre11 and pADPr foci colocalize in scid-ts cells. Cells were cultured as described in the text and stained with anti-Mre11 (green) and 10H (red) Abs. Nuclear staining was verified by counterstaining with PO-PRO-1 (data not shown). (a–c) scid-ts cells at 39°C; scid-ts cells at 33°C (Insets). (d–f) scid-ts cells after 39°C→33°C. (g–i) S/+-ts cells at 39°C; s/+-ts cells at 33°C (Insets). (j–l) S/+-ts cells after 39°C→33°C.
Figure 5.

Analysis of Mre11 and pADPr in the scid-ts cells treated with genotoxic agents (ionizing radiation or etoposide). (A) Confocal analysis of Mre11 and pADPr. The scid-ts cells cultured at 33°C exposed to 6 Gy of γ-irradiation or treated with 20 μM etoposide. (B) Apoptosis assessment by flow cytometry. The scid-ts cells that had gone through different culture schemes (33°C, 39°C, or 39°C→33°C) were stained with annexin V-FITC and propidium iodide (PI).
Because apoptotic cells are also known to display intense pADPr staining (27), we performed flow cytometry by using annexin V and propidium iodide to determine whether the high level of polymer staining could be attributed to apoptosis. Although etoposide treatment, used as a positive control for apoptosis induction (27), led to clearly higher numbers of cells positive for annexin V, only a few percentage of wild-type (data not shown) or scid cells were found to stain with annexin V (Fig. 5B). Furthermore, the pADPr immunostaining associated with early apoptosis in etoposide-treated cells did not show colocalization with Mre11 (Fig. 5A). Thus, the pADPr staining observed in the scid-ts cell lines during the course of recombination resolution and its colocalization with Mre11 was not the consequence of an apoptotic process, but reflected an involvement of poly(ADP-ribosyl)ation in DNA-PKcs-independent V(D)J recombination resolution.
Discussion
Here we have demonstrated that a DNA damage pathway involving poly(ADP-ribosyl)ation becomes activated in response to V(D)J recombination in DNA-PK-deficient cells. This activation appears to be important for resolution of recombination intermediates generated in these cells. On the surface, our results might seem to contradict the observation by Morrison et al. (17) that PARP-1 is an anti-recombinogenic factor because PARP-1 deficiency by genetic knockout rescues the block in V(D)J formation present in scid mice. However, knockout cells lack both the PARP-1 protein with its intrinsic DNA strand break-binding function and pADPr formation by PARP-1 (including PARP-1 automodification). The latter is known to reduce the strand break-binding affinity of PARP-1 by electrostatic repulsion. In contrast, ADP-ribosylation inhibitors abrogate only the catalytic function of the enzyme and thus actually preserve its very high DNA-binding affinity (28). Therefore, as demonstrated in Fig. 3, without being poly(ADP-ribosyl)ated, PARP-1 protein or other DNA-binding proteins would remain bound to the recombination ends and block their accessibility for processing and joining enzymes, thereby reducing their resolution. Viewing these two situations together, we propose the hypothesis that in scid cells, the binding of PARP-1 to DNA strand breaks is an obstacle to productive V(D)J recombination. This block can be overcome either by preventing PARP-1 biosynthesis in the first place, by genetic knockout, or by allowing catalytic activity of PARP-1 molecules bound to the strand breaks.
PARP-1 is believed to be one of the primary sensors involved in detection of DNA damage. Histone H2AX appears to be another early indicator of DNA damages as this protein becomes rapidly phosphorylated in response to γ-irradiation (29). It was reported recently that H2AX phosphorylation is mediated primarily by ataxia-telangiectasia mutation (ATM) proteins(ATM) but not DNA-PKcs (30). This phosphorylation has been visualized to occur at the T cell antigen receptor α locus in developing thymocytes (20), implying that a surveillance mechanism might exist to monitor the status of double-strand breaks generated during V(D)J recombination. However, the observance of normal V(D)J recombination in patients with ataxia-telangiectasia suggests that ATM plays a role in sensing but not necessarily in processing recombination ends. Thus, in the absence of functional DNA-PKcs, scid cells may be capable of sensing but not efficiently processing those ends. Poly(ADP-ribosyl)ation may represent a default and nonefficient pathway in protecting and/or processing recombination intermediates. This hypothesis is further substantiated by our finding of pADPr colocalization with the Mre11/Rad50/NBS complex in scid cells. The latter is also implicated in DNA damage recognition. In both irradiated cells and recombination active cells, H2AX phosphorylation was shown to colocalize with the NBS complex (20, 31). It is possible that poly(ADP-ribosyl)ation may complement H2AX in attracting the Mre11/Rad50/NBS complex to aid in processing and repairing of the recombination ends. Alternatively, the recently identified factor Artemis, which has been implicated to participate in opening the hairpin coding ends (32), may be activated by poly(ADP-ribosyl)ation to process scid hairpin coding ends. In conclusion, our results suggest that poly(ADP-ribosyl)ation serves as an important mechanism in resolving recombination-induced DNA lesions and offers a plausible explanation for the occurrence of low levels of productive rearrangements in DNA-PK-deficient cells.
Acknowledgments
We thank T. Sugimura and M. Miwa for 10H hybridoma cells and E. Jacobsen for comments on the manuscript. We greatly appreciate technical support from the Arizona State Uuniveristy W. M. Keck Lab for confocal microscopy. This work was supported by National Institutes of Health Grant CA73857 (to Y.C.) and an Achievement Rewards for College Scientists Fellowship (to M.L.B.).
Abbreviations
- scid
severe combined immunodeficient
- ts
temperature sensitive
- PARP-1
poly(ADP-ribose) polymerase-1
- DNA-PKcs
catalytic subunit of DNA-dependent protein kinase
- 3-AB
3-aminobenzamide
- LM-PCR
ligation-mediated PCR
- RAG
recombination-activating gene
- pADPr
poly(ADP-ribose)
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.McBlane J F, van Gent D C, Ramsden D A, Romeo C, Cuomo C A, Gellert M, Oettinger M A. Cell. 1995;83:387–395. doi: 10.1016/0092-8674(95)90116-7. [DOI] [PubMed] [Google Scholar]
- 2.van Gent D, McBlane J F, Ramsden D A, Sadofsky M J, Hesse J E, Gellert M. Cell. 1995;81:1–20. doi: 10.1016/0092-8674(95)90012-8. [DOI] [PubMed] [Google Scholar]
- 3.Eastman Q M, Leu T M J, Schatz D G. Nature (London) 1996;380:85–88. doi: 10.1038/380085a0. [DOI] [PubMed] [Google Scholar]
- 4.Taccioli G E, Rathbu G, Oltz E, Stamato T, Jeggo P A, Alt F W. Science. 1993;260:207–210. doi: 10.1126/science.8469973. [DOI] [PubMed] [Google Scholar]
- 5.Blunt T, Finnie N J, Taccioli G E, Smith G C, Demengeot J, Gottlieb T M, Mizuta R, Varghese A J, Alt F W, Jeggo P A, et al. Cell. 1995;80:813–823. doi: 10.1016/0092-8674(95)90360-7. [DOI] [PubMed] [Google Scholar]
- 6.Li Z, Otevrel T, Gao Y J, Cheng H L, Seed B, Stamato T D, Taccioli G E, Alt F W. Cell. 1995;83:1079–1089. doi: 10.1016/0092-8674(95)90135-3. [DOI] [PubMed] [Google Scholar]
- 7.Grawunder U, Zimmer D, Fugmann S, Schwarz K, Lieber M R. Mol Cell. 1998;2:477–484. doi: 10.1016/s1097-2765(00)80147-1. [DOI] [PubMed] [Google Scholar]
- 8.Carroll A M, Hardy R R, Bosma M J. J Immunol. 1989;143:1087–1093. [PubMed] [Google Scholar]
- 9.Petrini J H-J, Carroll A M, Bosma M J. Proc Natl Acad Sci USA. 1990;87:3450–3453. doi: 10.1073/pnas.87.9.3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao Y, Chaudhuri J, Zhu C, Davidson L, Alt F W. Immunity. 1998;9:367–376. doi: 10.1016/s1074-7613(00)80619-6. [DOI] [PubMed] [Google Scholar]
- 11.Bogue M A, Jhappan C, Roth D B. Proc Natl Acad Sci USA. 1998;95:15559–15564. doi: 10.1073/pnas.95.26.15559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurimasa A, Ouyang H, Dong L-J, Wang S, Li X, Cordon-Cardo C, Chen D J, Li G C. Proc Natl Acad Sci USA. 1999;96:1403–1408. doi: 10.1073/pnas.96.4.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bürkle A. BioEssays. 2001;23:795–806. doi: 10.1002/bies.1115. [DOI] [PubMed] [Google Scholar]
- 14.D'Amours D, Desnoyers S, D'silva I, Poirier G G. Biochem J. 1999;342:249–268. [PMC free article] [PubMed] [Google Scholar]
- 15.Smith S. Trends Biochem Sci. 2001;26:174–179. doi: 10.1016/s0968-0004(00)01780-1. [DOI] [PubMed] [Google Scholar]
- 16.Wang Z Q, Stingl L, Morrison C, Jantsch M, Los M, Schulze-Osthoff K, Wagner E F. Gene Dev. 1997;11:2347–2358. doi: 10.1101/gad.11.18.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morrison C, Smith G C M, Sting L, Jackson S P, Wagner E F, Wang Z-Q. Nat Genet. 1997;17:479–482. doi: 10.1038/ng1297-479. [DOI] [PubMed] [Google Scholar]
- 18.Nelms B E, Maser B S, MacKay J F, Lagally M G, Petrini J H. Science. 1998;280:590–592. doi: 10.1126/science.280.5363.590. [DOI] [PubMed] [Google Scholar]
- 19.Paull T T, Gellert M. Gene Dev. 1999;13:1276–1288. doi: 10.1101/gad.13.10.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen H T, Bhandoola A, Difilippantonio M J, Zhu J, Brown M J, Tai X, Rogakou E P, Brotz T M, Bonner W M, Ried T, et al. Science. 2000;290:1962–1965. doi: 10.1126/science.290.5498.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang Y, Brown M L. Proc Natl Acad Sci USA. 1999;96:191–196. doi: 10.1073/pnas.96.1.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ogata S, Okumura K, Taguchi H. Biosci Biotechnol Biochem. 2000;64:510–515. doi: 10.1271/bbb.64.510. [DOI] [PubMed] [Google Scholar]
- 23.Bürkle A, Chen G, Küpper J H, Grube K, Zeller W J. Carcinogenesis. 1993;14:559–561. doi: 10.1093/carcin/14.4.559. [DOI] [PubMed] [Google Scholar]
- 24.Brown M L, Chang Y. J Immunol. 2000;164:4135–4142. doi: 10.4049/jimmunol.164.8.4135. [DOI] [PubMed] [Google Scholar]
- 25.Kawamitsu H, Hoshino H, Okada H, Miwa M, Momoi H, Sugimura T. Biochemistry. 1984;23:3771–3777. doi: 10.1021/bi00311a032. [DOI] [PubMed] [Google Scholar]
- 26.Rankin P W, Jacobson E L, Benjamin R C, Moss J, Jacobson M K. J Biol Chem. 1989;264:4312–4317. [PubMed] [Google Scholar]
- 27.Negri C, Donzelli M, Bernardi R, Rossi L, Bürkle A, Scovassi A I. Exp Cell Res. 1997;234:174–177. doi: 10.1006/excr.1997.3591. [DOI] [PubMed] [Google Scholar]
- 28.Shall S. Mol Cell Biochem. 1994;138:71–75. doi: 10.1007/BF00928445. [DOI] [PubMed] [Google Scholar]
- 29.Rogakou E P, Pilch D R, Orr A H, Ivanova V S, Bonner W M. J Biol Chem. 1998;273:25858–25868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 30.Burma S, Chen B P, Murphy M, Kurimasa A, Chen D J. J Biol Chem. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 31.Paull T T, Rogakou E P, Yamazaki V, Kirchgessner C U, Gellert M, Bonner W M. Curr Biol. 2000;10:886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- 32.Moshous D, Callebaut I, de Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, Tezcan I, Sanal O, Bertrand Y, Philippe N, et al. Cell. 2001;105:177–186. doi: 10.1016/s0092-8674(01)00309-9. [DOI] [PubMed] [Google Scholar]