

Summary
Chlamydia spp. are major causes of important human diseases, but dissecting the host–pathogen interactions has been hampered by the lack of bacterial genetics and the difficulty in carrying out forward genetic screens in mammalian hosts. RNA interference (RNAi)-based methodologies for gene inactivation can now be easily carried out in genetically tractable model hosts, such as Drosophila melanogaster, and offer a new approach to identifying host genes required for pathogenesis. We tested whether Chlamydia trachomatis infection of D. melanogaster S2 cells recapitulated critical aspects of mammalian cell infections. As in mammalian cells, C. trachomatis entry was greatly reduced by heparin and cytochalasin D. Inclusions were formed in S2 cells, acquired Golgi-derived sphingolipids, and avoided phagolysosomal fusion. Elementary body (EB) to reticulate body (RB) differentiation was observed, however, no RB to EB development or host cell killing was observed. RNAi-mediated inactivation of Rac, a Rho GTPase recently shown to be required for C. trachomatis entry in mammalian cells, inhibits C. trachomatis infection in S2 cells. We conclude that Drosophila S2 cells faithfully mimic early events in Chlamydia host cell interactions and provides a bona fide system to systematically dissect host functions important in the pathogenesis of obligate intracellular pathogens.
Introduction
Chlamydia species cause a wide range of acute diseases in humans, including sexually transmitted, ocular, and respiratory tract infections (reviewed in Schachter, 1988). Important sequelae of chronic Chlamydia infection include female infertility, blindness, arthritis and possibly atherosclerosis (Campbell and Kuo, 2003). Despite the broad spectrum of disease, all Chlamydia spp. are obligate intracellular pathogens that share a common strategy to survive within the hostile intracellular compartment (Moulder, 1991). They alternate between an extra-cellular spore-like form, the elementary body (EB), and an intracellular metabolically active but non-infectious form, the reticulate body (RB). Chlamydia species can productively infect most cultured cells, suggesting that the receptor(s) is widespread. For some species and serovars, including the more invasive lymphogranuloma venerum strain (LGV) L2, heparan sulphate may act as a bridging molecule for a relatively weak and reversible interaction (Zhang and Stephens, 1992; Gutierrez-Martin et al., 1997; Taraktchoglou et al., 2001; Wuppermann et al., 2001) that is followed by a stronger, more specific binding to an unidentified secondary receptor (Carabeo and Hackstadt, 2001; Fudyk et al., 2002). Internalization is accompanied by induction of a microvillus-like structure over a large portion of the host cell in a process that is dependent upon actin polymerization (Carabeo et al., 2002) and is mediated through Rac (Carabeo et al., 2004). Upon entry, the organism is sequestered in a membrane bound compartment, termed the chlamydial ‘inclusion’, over a period of approximately 1–2 h, during which the primary differentiation process is initiated (Fields and Hackstadt, 2002). Bacterial metabolism and replication commence, and the EB differentiates into the larger, less compact RB form. The inclusion rapidly segregates from the endocytic pathway in a process that may initially depend upon the secretion of previously synthesized proteins that are exported through the type III secretion system (Fields et al., 2003); ultimately, continued avoidance of fusion with endocytic or lysosomal compartments requires bacterial protein synthesis (Scidmore et al., 2003). The inclusion is transported via microtubules to the perigolgi region by a dynein-dependent process (Grieshaber et al., 2003). Interestingly, the inclusion acquires characteristics of the Golgi apparatus or of an exocytic compartment and receives cholesterol and host sphingolipids from the Golgi (Hackstadt et al., 1996; Carabeo et al., 2004). After replicating by binary fission within the ever-enlarging inclusion over a 48–72 h time period, the RB undergoes a second differentiation process back to an infectious EB. This is accompanied by the expression of two histone-like proteins, Hc1 and Hc2, which are involved in DNA compaction and cessation of transcription (Perara et al., 1992; Barry et al., 1993; Brickman et al., 1993; Pedersen et al., 1996a; 1996b). At this late stage in development (40–60 h) the host cell lyses, possibly via a toxin-B related protein (Belland et al., 2001), to release mature EBs that then infect neighbouring cells.
The study of Chlamydia pathogenesis has been hampered by the failure to stably introduce DNA into this obligate intracellular pathogen and by the inability to grow this bacterium ex vivo. Modern cell biological approaches and reverse genetics have facilitated the identification of some host proteins that are involved in the successful intracellular survival of this pathogen, but much remains to be learned. Genetically tractable non-mammalian models, such as Drosophila melanogaster, are being developed as model hosts for studying host–pathogen interactions, including Listeria monocytogenes, Pseudomonas aeruginosa and Mycobacterium marinum (Rahme et al., 2000; D’Argenio et al., 2001; Baldini et al., 2002; Dionne and Schneider, 2002; Cheng and Portnoy, 2003; Dionne et al., 2003; Lau et al., 2003; Mansfield et al., 2003; Vodovar et al., 2004). Drosophila has recently become an even more attractive model host because of the ease and availability of using RNA interference (RNAi) to inactivate gene expression and the lack of redundancy in the genome compared with mammals. RNAi is an evolutionarily conserved process in which gene expression is suppressed at the post-transcriptional level by the introduction of homologous small double-stranded RNAs (dsRNAs) (Elbashir et al., 2001). While complete gene inactivation is rarely obtained, there is usually sufficient loss of function to result in phenotypic changes. RNAi-based forward genetic screens in Drosophila S2 cells, a cell line derived from phagocytic haematopoietic cells which recapitulates key aspects of innate immunity (Echalier, 1997), have been used with great success to identify new genes involved in cell division, cell motility, phagocytosis and recognition of Gram-positive and Gram-negative bacteria (Gottar et al., 2002; Ramet et al., 2002; Kiger et al., 2003; Rogers et al., 2003; Boutros et al., 2004; Innocenti et al., 2004).
In this study, we have established S2 cells as a valid in vitro model to study early aspects of Chlamydia infections. We have examined C. trachomatis infection of Drosophila S2 cells for key processes that have been observed during Chlamydia infection of mammalian cells and demonstrate that infection of S2 cells closely mimics important initial steps of mammalian infections. These studies set the stage to use this novel system to identify host genes important in the pathogenesis of Chlamydia infections. These methods can be extended to the study of other obligate and facultative intracellular parasites.
Results
S2 cells can be efficiently infected by diverse Chlamydia trachomatis serovars
To determine whether Drosophila can serve as a model host for Chlamydia infection, we assessed the ability of Chlamydia to form inclusions in S2 cells. S2 cells are propagated in vitro at temperatures of 25–30°C; at higher temperatures they rapidly undergo apoptosis (Echalier, 1997). S2 cells were infected with C. trachomatis serovars L2, D, E and K for 1 h and incubated at 28°C. At various times after infection, the Chlamydia-infected S2 cells were plated for 1 h onto concanavalin A (ConA)-treated cover slips, which allows the normally round and nonadherent S2 cells to spread out and efficiently adhere (Rogers et al., 2003). The infected cells were then fixed and stained with DAPI (which stains the DNA of the host nucleus and of Chlamydia) and with an antibody to the Chlamydia major outer membrane protein (MOMP) for examination by immunofluoresence microscopy (IF) (Fig. 1A). As a result of the large number of lysosomes and other membrane bound compartments in this macrophage-like cells line (for example see Fig. 6), visualization of the inclusions by phase microscopy was often difficult.
Fig. 1.
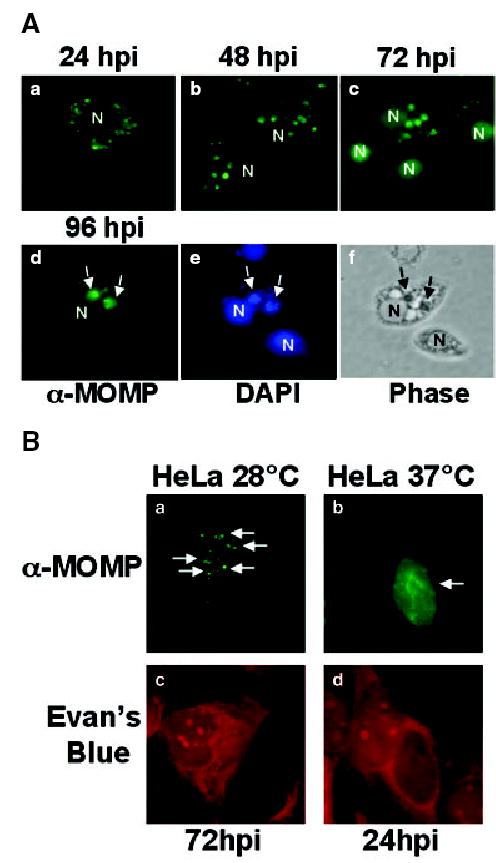
A. L2 inclusion maturation in S2 cells. S2 cells were infected with L2 for 1 h, and at the indicated times post infection, cells were fixed and stained with an antibody to MOMP (panels a–d) to localize the Chlamydia and with DAPI (panel e) to localize the inclusion (arrow) or the host cell nucleus (N). The corresponding phase image of a single field of infected cells at 96 hpi is included (panel f). The inclusions increased in size as a function of time. All micrographs are shown at 1000× final magnification and are representative of multiple fields that were examined.
B. L2 inclusion maturation in HeLa Cells. HeLa cells grown at 28°C (panels a and c) or 37°C (panels b and d) were infected with L2 for 1 h. At the indicated times post infection, cells were fixed and stained with an antibody to MOMP (a and b) to visualize Chlamydia and counterstained with Evan’s blue (c and d) to visualize the HeLa cells. Note the presence of multiple small inclusions spread throughout the cytoplasm of a HeLa cell grown at 28°C compared with the single large inclusion present in a HeLa cell grown at 37°C. Arrows are pointing to inclusions. All micrographs are shown at 1000× final magnification and are representative of multiple fields that were examined.
Fig. 6.
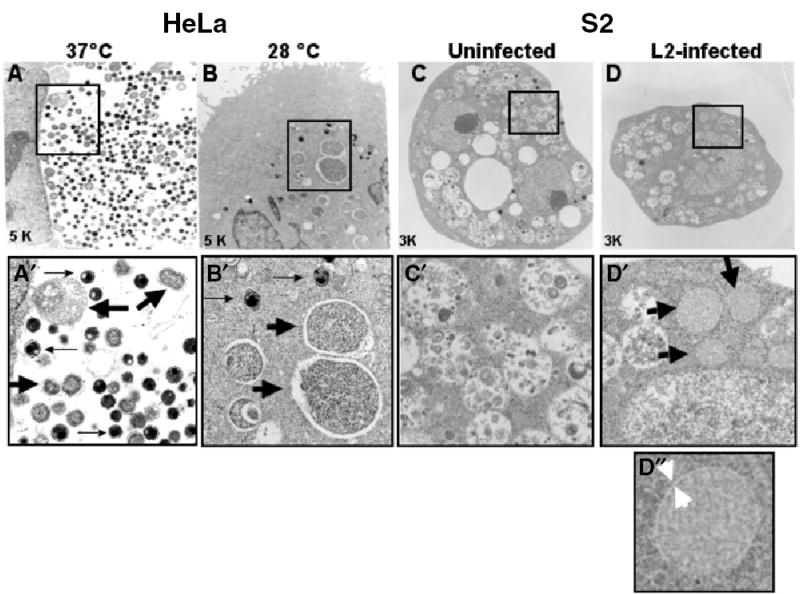
Transmission electron microscopy (TEM) of L2 inclusions in HeLa and S2 cells. HeLa and S2 cells were infected with L2 for 1 h at the indicated temperature and examined by TEM at the indicated times post infection. The middle panels (A′–D′) represent an enlargement of the boxed area in the corresponding upper panels (A–D). In HeLa cells infected with L2 at 37°C for 24 h (A and A′), EBs (thin arrows), RBs (thick arrows), and intermediate forms are easily identified in a single large inclusion. In HeLa cells infected with L2 at 28°C for 24 h (B and B′), multiple inclusions containing single RBs (thick arrow) or EBs (thin arrow) are present. In S2 cells infected with L2 at 60 hpi (D and D′), multiple inclusions containing single RB-like structures are present (thick arrow) whereas these structures are absent from untreated samples (C and C′). Note that there are many other vacuolar-like structures visible in S2 cells. The bottom panel shows an enlargement of a single RB from S2 cells (D″). Thick white arrowheads point to a possible inclusion membrane.
Infection of S2 cells with L2 led to the formation of multiple small inclusions of variable sizes (Fig. 1A) that increased in number in a dose dependent manner (data not shown). Inclusions were easily detectable as early as 24 h post infection (hpi) (Fig. 1A, panel a). At this time, the inclusions were spread throughout the cell, although they were specifically excluded from the nucleus. By 48–72 hpi (Fig. 1A, panels b and c), many of the inclusions aggregated to discrete regions of the cell, indicating that the inclusions were trafficking. At 96 hpi (Fig. 1A, panels d–f), many cells contained two or three large inclusions. Although the inclusions appeared to mature over time, there was significant heterogeneity with respect to inclusion number and size within an infected population. Inclusions could be detected up to 1 week after infection without loss of MOMP staining (data not shown). Inclusion formation was greatly reduced when the infected cells were treated with doxycycline (data not shown), a well-established inhibitor of chlamydial protein synthesis (Stamm and Holmes, 1990); therefore the development of inclusions in the S2 cells was dependent upon chlamydial protein synthesis similar to that observed in mammalian cells. The genital-associated serovars D, E and K infected S2 cells with similar kinetics and inclusion morphology to that seen with LGV L2 (data not shown). Unless otherwise noted, we used serovar L2 for the remainder of the experiments described. Because the L2 inclusions inside S2 cells were small, a high MOI was necessary in order to accurately visualize inclusions.
Chlamydia trachomatis infection of mammalian cells at 37°C at high MOIs typically results in the formation of a single large inclusion (Fig. 1B), unlike S2 cells which contain multiple inclusions (Fig. 1A). To determine whether this inclusion morphology was resulting from growth at 28°C, we examined inclusion morphology in HeLa cells infected at 28°C. As shown in Fig. 1B, multiple inclusions were also observed in HeLa cells grown at 28°C. These results are consistent with the studies of Fields et al. and van Ooij et al. demonstrating that inclusion fusion does not occur in cells grown at temperatures below 32°C (van Ooij et al., 1998; Fields et al., 2002).
L2 entry into S2 cells can be blocked by excess heparin and requires an intact actin cytoskeleton
Initial attachment of both C. pneumoniae and some biovars of C. trachomatis (including L2) to mammalian cells is thought to involve electrostatic interactions mediated by heparan sulphate-like molecules, as binding is inhibited by excess heparan sulphate (Zhang and Stephens, 1992; Gutierrez-Martin et al., 1997; Taraktchoglou et al., 2001; Wuppermann et al., 2001). As shown in Fig. 2, incubation of S2 cells with heparin 1 h before and during the 1 h infection significantly decreased inclusion formation as evidenced by the lack of MOMP staining of L2-infected S2 cells at 48 hpi. Treatment with heparin had no effect on S2 cell number or viability (Fig. 2 and data not shown).
Fig. 2.
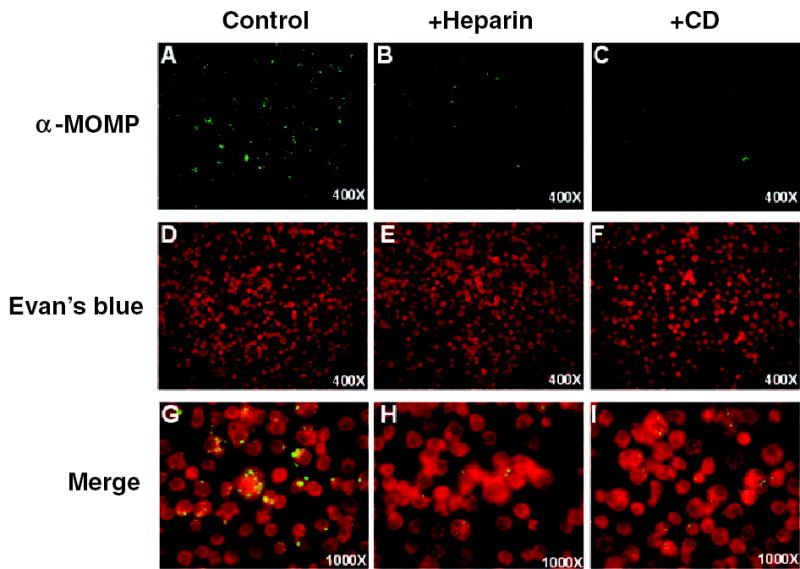
Heparin and cytochalasin D inhibit L2 Infection in S2 cells. S2 cells were pretreated with heparin or cytochalasin D for 1 h, infected with L2 for 1 h in the presence of drug, and incubated for 48 h. Cells were fixed and stained with anti-MOMP antibody to visualize the bacteria (AC; 400× final magnification) and counterstained with Evan’s blue to visualize S2 cells numbers (D–F); 400× final magnification). A merged set of images for each sample is shown at 1000× final magnification to allow a more detailed visualization of the inclusions (G–I).
Chlamydia entry into mammalian cells is dependent upon actin polymerization, and uptake of EBs can be inhibited by cytochalasin D, an actin-depolymerizing agent (Schramm and Wyrick, 1995; Carabeo et al., 2002). Likewise, preincubation of S2 cells with cytochalasin D for 1 h before and during the 1 h infection decreased Chlamydia infection of S2 cells (Fig. 2A–C) without affecting S2 number (Fig. 2D–F) or viability (data not shown). In both heparin and cytochalasin D treatments, there was a reduction in the number of infected cells and a reduction in the size of inclusions in infected cells (Fig. 2). Thus, entry into S2 cells faithfully mimics two important characteristics seen in mammalian cells, initial attachment via heparin sulphate and internalization via the actin cytoskeleton. These findings suggest that S2 cells will be useful for identifying the host cell receptor and entry pathways.
Internalized Chlamydia do not fuse with lysosomes and acquire Golgi-derived sphingolipids
Once inside mammalian cells, Chlamydia modify the properties of the inclusion to prevent fusion with lysosomes in a process that requires ongoing bacterial protein synthesis (Scidmore et al., 2003). Lamp-1 is a well-established marker for lysosomes. Using S2 cells that express Drosophila Lamp-1 (DmLamp1) fused to GFP, we examined whether C. trachomatis inclusions avoided fusion with lysosomes in these cells and whether chlamydial inhibition of phagolysosomal fusion required de novo bacterial synthesis as it does in mammalian cells. S2 cells expressing GFP-DmLamp1 were infected with L2 for 1 h in the presence or absence of chloramphenicol, a well established inhibitor of bacterial protein synthesis. At 48 hpi, cells were fixed and stained with the MOMP antibody to visualize inclusions. As shown in Fig. 3B and E, S2 cells contain many lysosomal vesicles; however, the GFP-DmLamp1 containing compartments were distinct and non-overlapping with the MOMP-staining L2 inclusions (Fig. 3A–C). No lysosomal fusion was detected as long as 5 days post infection (data not shown). In contrast, when bacterial synthesis was inhibited with chloramphenicol throughout the 48 h incubation, the L2 inclusions localized to GFP-DmLamp1-containing lysosomes (Fig. 3D–F). Thus, C. trachomatis inclusions avoided phagolysosomal fusion in S2 cells in a process that required active bacterial protein synthesis similar to what is observed in mammalian cells.
Fig. 3.
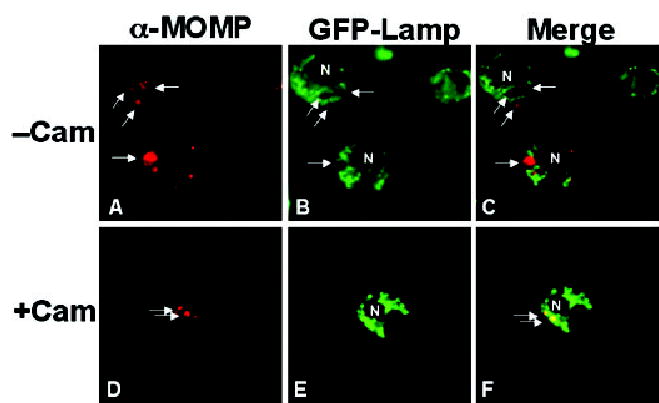
L2-containing inclusions do not fuse with lysosomes in S2 cells. S2 cells expressing GFP-Lamp (which labels lysosomes) were infected with L2 for 1 h. At 48 hpi, the samples were fixed, and stained with anti-MOMP. In some samples, chloramphenicol (Cam) was present throughout the infection and subsequent 48 h incubation (D–F). A and D show the MOMP staining; B and E show the GFP-lamp staining; and C and F show the merged panels. In the absence of Cam treatment, the inclusion is distinct from the GFP-lamp staining lysosomes. In the presence of Cam, the inclusion and the GFP-Lamp staining lysosomes overlap, demonstrating fusion of the inclusion and lysosomes. The arrows point to selected inclusions. N, host nucleus. All micrographs are 1000× final magnification. Samples shown are representatives of multiple fields examined.
Once inside mammalian cells, C. trachomatis quickly segregates from the endocytic pathway and becomes fusogenic with exocytic vesicles containing sphingomyelin and cholesterol en route from the Golgi apparatus to the plasma membrane (Hackstadt et al., 1996; Carabeo et al., 2004). Fluorescently labelled ceramide analogues, such as C6-NBD ceramide, have been useful markers for studying sphingolipid transport to the Chlamydia inclusion in mammalian cells (Hackstadt et al., 1995; Hackstadt et al., 1996; Wolf and Hackstadt, 2001). Like endogenous ceramide, the analogue C6-NBD ceramide is processed to sphingomyelin and glucosylceramide within the Golgi apparatus before transport to the plasma membrane (Lipsky and Pagano, 1985). However, in Chlamydia-infected mammalian cells, the fluorescent lipid is incorporated into both the inclusion membrane and intracellular Chlamydia cells (Hackstadt et al., 1995; Hackstadt et al., 1996; Wolf and Hackstadt, 2001).
Using fluorescently labelled C6-NBD ceramide, we examined whether L2-infected S2 cells acquired sphingolipids from S2 cells. S2 and HeLa cells were infected with L2 and incubated for 72 or 24 hpi, respectively, and subsequently labelled with FITC-conjugated C6-NBD ceramide. Samples were counterstained with DAPI to visualize Chlamydia nuclei. It is worth noting that host nuclei are also stained with DAPI but will not be labelled with FITC-conjugated C6-NBD ceramide whereas the bacterial nuclei will be labelled with both DAPI and FITC-conjugated C6-NBD ceramide. At 24 hpi, inclusions inside HeLa cells incorporated C6-NBD-sphingomyelin as evidenced by the overlap of FITC-conjugated C6-NBD ceramide and DAPI staining (Fig. 4E and F). This result is consistent with previous studies (Hackstadt et al., 1996). We found that at 48 hpi (data not shown) and 72 hpi (Fig. 4C and D), C6-NBD-sphingomyelin was incorporated into Chlamydia inclusions inside S2 cells as shown by the overlap of FITC-conjugated C6-NBD ceramide and DAPI staining of Chlamydia DNA. It was difficult to visualize incorporation of C6-NBD-sphingomyelin at times before 48 hpi resulting from the small size of the inclusions. Together, these results clearly show that Chlamydia was growing and viable in S2 cells and suggests that Chlamydia inclusions were quickly segregated from the endocytic pathway in these cells. It further indicates that the Chlamydia requirement for host cell sphingolipids can be met by growth in S2 cells at 28°C and that the host cell molecules that are required for these events can be identified by studying Chlamydia infected S2 cells.
Fig. 4.
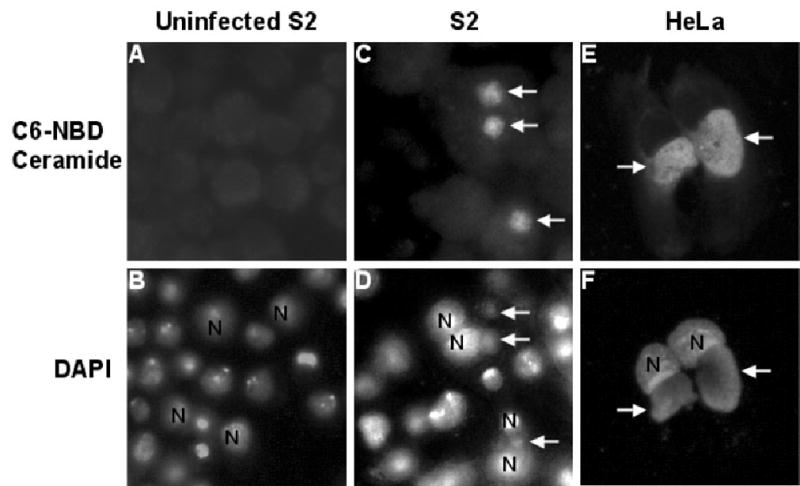
L2 inclusions in S2 cells acquire sphingomyelin from the host. S2 (A–D) and HeLa cells (E and F) were infected with L2 for 1 h and then incubated for 72 h (A–D) or 24 h (E and F). The cells were labelled with C6-NBD ceramide (upper panels) and DAPI (lower panels) to visualize the inclusion and the host nuclei as described in the Experimental procedures. The arrows point to inclusions and the host nucleus is labelled with N. Inclusions in both S2 cells and in HeLa cells acquired C6-NBD ceramide. All micrographs are shown at 1000× final magnification.
Elementary body (EB) to RB differentiation occurs
To test whether the EBs differentiated into RBs in S2 cells, we examined whether we could detect expression of an RB-specific protein in L2-infected S2 cells. IncG is an inclusion membrane protein that is synthesized only after EB to RB conversion (Scidmore-Carlson et al., 1999). It has been shown to interact with mammalian 14-3-3β, although its exact role in pathogenesis is unclear (Scidmore and Hackstadt, 2001). The cellular localization of IncG in Chlamydia-infected S2 or HeLa cells was determined by IF microscopy with an antibody to IncG. Samples were counterstained with DAPI to visualize Chlamydia nuclei. As shown in Fig. 5E and F, IncG was expressed in the inclusion of Chlamydia-infected HeLa cells at 24 hpi (as evidenced by the overlap of FITC-conjugated C6-NBD ceramide and DAPI staining) and was enriched at the inclusion membrane in one hemisphere of the RB, consistent with previously published data (Scidmore-Carlson et al., 1999). Similarly, IncG was clearly detectable in the inclusion of L2-infected S2 cells at 48 hpi (Fig. 5C and D); however, the IncG staining pattern did not appear as a rim around the inclusion but rather appeared to stain individual RBs (Fig. 5C). This staining pattern provides support for the notion that inclusion fusion was not occurring efficiently in S2 cells (see below). These results demonstrate that RB formation occurs in S2 cells, as IncG is an RB-specific protein (Scidmore and Hackstadt, 2001).
Fig. 5.
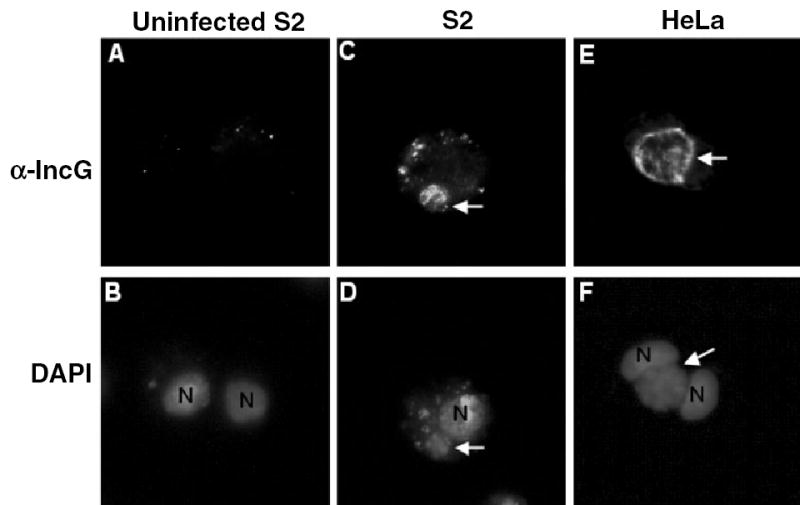
IncG, an RB-specific protein, is expressed in the inclusions of L2-infected S2 cells. S2 and HeLa cells were infected with L2 for 1 h, and then fixed and stained with an IncG monoclonal antibody (upper panels) and DAPI (lower panels) at 48 (S2) or 24 h (HeLa at 37°C) post infection to visualize the inclusion and the host nuclei.
A and B. Uninfected S2 cells.
C–D. S2 cells at 48 hpi.
E and F. HeLa cells at 24 hpi.
Arrows point to inclusions that express IncG, demonstrating that EB to RB differentiation has occurred. N, host nucleus. All micrographs are 1000× final magnification. Samples shown are representatives of multiple fields examined.
In order to more closely examine the morphology of the C. trachomatis developmental forms present in S2 cells, L2-infected cells were examined by transmission electron microscopy (TEM). For comparison, we examined L2-infected HeLa cells grown at 28°C and 37°C. As shown in Fig. 6A and A′, a single large inclusion was present in HeLa cells infected with L2 for 24 h at 37°C that contained EBs, RBs and the various intermediate forms. In contrast, HeLa cells infected with L2 for 24 h at 28°C had multiple inclusions that mostly contained individual RBs (Fig. 6B and B′), consistent with earlier reports that vacuolar fusion is inhibited at lower temperatures (van Ooij et al., 1998). Several of the RB-like structures in HeLa cells at 28°C were somewhat enlarged compared with 37°C, suggesting that they could represent aberrant or persistent forms. As shown in Fig. 6D and D′, inclusions containing single RB-like structures were observed in S2 cells grown at 28°C for 60 hpi. These RB-like structures were distinct from other vacuolar structures present in untreated (Fig. 6C and C′) and treated cells. Together, these results suggest that bacterial protein synthesis is initiated and that EB to RB differentiation occurred during Chlamydia infection of S2 cells. Although it is possible that RBs may be free in the cytoplasm, the EM studies suggest that the RBs are enclosed in a membrane bound compartment (Fig. 6D′).
Late cycle events do not occur in S2 cells
Our results thus far suggest that early events were recapitulated in C. trachomatis infection of S2 cells. To determine if the entire intracellular life cycle occurs, we assayed whether infectious progeny were formed. S2 cells were infected with serovar L2 and incubated for 5 days. At 24 h intervals, the cells were harvested, lysed in Triton X-100, and replated on HeLa cells grown under standard conditions (37°C) for 48 h. Infectious progeny were not obtained from infected S2 cells up to 5 days post infection (data not shown). To determine whether the lack of progeny formation was a result of low growth temperatures, we also tested progeny formation in L2-infected HeLa cells grown at 28°C. Viable progeny were also not obtained in these cells (data not shown). Examination by immunofluorescence microscopy of the HeLa cells infected with the putative progeny stained with an anti-MOMP antibody revealed a speckled, extracellular staining pattern at both early and late times after infection, suggestive that only RBs were obtained (data not shown). Similar results were obtained with infection by serovars D, E and K (data not shown).
Because we were not able to detect production of infectious progeny, we next examined whether the inhibition of EB transition was resulting from altered expression of the late cycle histone-like protein, Hc2. This protein is important for condensation of chromatin during the final RB to EB transition (Barry et al., 1993; Grieshaber et al., 2004). S2 cells were infected with L2 and incubated for up to 96 hpi. At various times post infection, lysates from infected S2 cells were prepared and immunoblotted with antibodies to Hc2. Mock infected cells and EB lysates were included as specificity controls for antibodies. As a control for normal Chlamydia protein expression, lysates from Chlamydia-infected HeLa cells grown at 37°C were prepared and immunoblotted in parallel (Fig. 7). As expected Hc2 expression was greatly increased at 24 hpi in L2-infected HeLa cells and was present in EB lysates (Fig. 7). In contrast, we were unable to detect an increase in expression of Hc2 at any time point following infection of S2 cells with L2 at 28°C. Lysates were immunoblotted with antibodies to Drosophila β-tubulin and human GAPDH as loading controls for S2 cells and HeLa cells respectively. These samples were also immunoblotted with an antibody to MOMP, as production of this protein increases during RB conversion and bacterial replication in infected HeLa cells. In infected S2 cells, there was an increase in MOMP expression from two to 18 hpi, however, no further increase in MOMP expression was observed thereafter (Fig. 7). Together, these results indicate that RB conversion does occur, however, the terminal differentiation step into EBs does not take place in S2 cells. These results also suggest that C. trachomatis is not efficiently replicating in S2 cells.
Fig. 7.
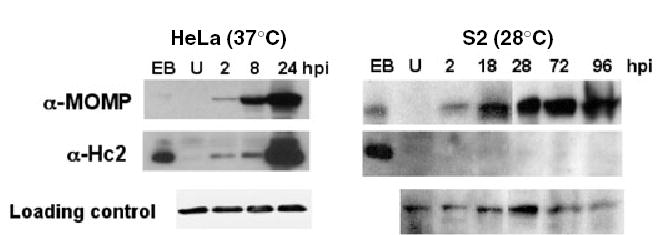
L2-infected S2 cells do not produce the late EB-specific protein Hc2. HeLa cells grown at 37°C or S2 cells grown at 28°C were infected with L2 for 1 h, incubated for the indicated times, and lysates were immunoblotted with antibodies to MOMP, Hc2, GAPDH (loading control for HeLa cells) or β-tubulin (loading control for S2 cells). Elementary body lysates (EB) were included as a positive control. Expression of the late cycle Chlamydia protein, Hc2, did not increase in S2 cells at any time point following infection up to 96 hpi whereas expression of Hc2 was greatly increased by 24 hpi in infected HeLa cells. All samples were run on the same gels and exposed for the same amount of time.
Although RB to EB differentiation was not detected, we tested whether infection of S2 cells resulted in host cell death, a late event that occurs during L2 infection of HeLa cells. S2 cells grown at 28°C were infected with increasing doses of L2 for 1 h and further incubated for 5 days post infection. For comparison, HeLa cells grown at 28°C or 37°C were infected in parallel. Quantification of cell death in HeLa cells was measured by crystal violet staining, a vital dye taken up by living cells. As shown in Fig. 8A, cell death increased in HeLa cells grown at 37°C in a time-and dose-dependent manner, whereas no significant cell death was observed in HeLa cells grown at 28°C (Fig. 8B). As S2 cells are only semi-adherent, we measured cell viability by trypan blue, which is only taken up by damaged and dying cells. The data are plotted as the percentage of viable cells. As shown in Fig. 8C, minimal increase in cell death over background was observed in infected-S2 cells compared with untreated, even up to 5 days post infection. Taken together, these results demonstrate that L2 infection of S2 cells does not result in host cell death. We conclude that the failure to complete the life cycle is a consequence of growth at low temperature. Similarly, Chlamydia-infected HeLa cells grown at 28°C did not exhibit cell death. However, we cannot rule out the possibility that S2 cells may lack other factors required for completion of the Chlamydia intracellular life cycle.
Fig. 8.
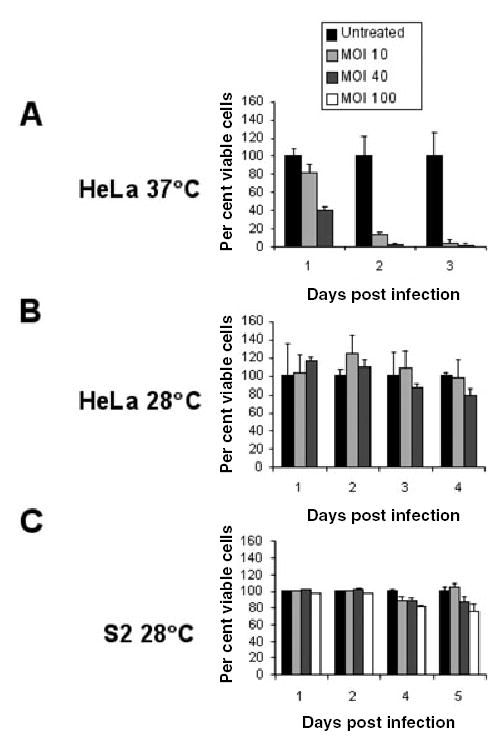
L2 infection of S2 cells does not result in host cell death. HeLa cells grown at 37°C or 28°C and S2 cells were infected with increasing doses of L2 for 1 h and then subsequently incubated for 3, 4 or 5 days respectively. Cell viability for infected HeLa cells was quantified by crystal violet staining and data (means ± SD) were plotted as a percentage of viable cells compared with untreated controls. Cell death for infected S2 cells was quantified by trypan blue staining and data (means ± SD) were plotted as the percentage of viable cells compared with untreated controls. Little cell death was observed in infected S2 cells and HeLa cells grown at 28°C compared with untreated controls. All experiments were performed in triplicate.
RNAi can be used to study the requirement for host genes in Chlamydia infection of S2 cells
A major advantage to using Drosophila S2 cells as a model for Chlamydia infections is the opportunity to utilize RNAi-mediated gene inactivation for large-scale forward genetic screens. To test whether this technology could be employed to identify host genes required for Chlamydia infections, we used RNAi to inactivate a target gene known to be required for Chlamydia infections in mammalian cells. Recently, Hackstadt and coworkers demonstrate that the Rac GTPase, but not Rho1 or Cdc42 GTPases is required for C. trachomatis invasion of mammalian cells (Carabeo et al., 2004). To inhibit expression of the Rho family GTPases, we treated Drosophila S2 cells with dsRNA specific for Rho1, Rac1/2 and Cdc42 individually for 4 days and subsequently infected the cells with L2 for 1 h. Forty-eight hours post infection, the cells were fixed, stained with an antibody to MOMP, and examined by IF. As shown in the upper panels of Fig. 9A, RNAi-mediated inactivation in S2 cells of Rac1/2, but not Rho1 or Cdc42, greatly reduced L2 infection. These data were quantified by counting the number of cells containing inclusions and were plotted as a percentage of the total number of cells. As shown in Fig. 9B, depletion of Rac1/2 resulted in a 5-fold decrease in infectivity (P < 0.001 compared with control cells, one-way anova).
Fig. 9.

A. L2 infection of S2 cells is Rac dependent. S2 cells were treated with dsRNA against Rho1, Rac1/2 and Cdc42 for 4 days and then subsequently infected with L2 for 1 h. At 48 hpi, the S2 cells were fixed and stained with an antibody to MOMP to visualize the inclusion (upper panels) and counterstained with Evan’s blue to visualize the cells (middle panels). At the time of infection, a portion of the RNA-treated cells (lower panels) was removed, plated onto coverslips, fixed, and stained with Texas red-phalloidin (to visualize the actin cytoskeleton) and with DAPI (to visualize the nuclei). RNAi-mediated depletion of Rho1 resulted in enlarged cells, whereas RNAi-mediated depletion of Rac1/2 and Cdc42 caused clear alterations in the actin cytoskeleton. Only depletion of Rac1/2 resulted in a decrease in Chlamydia infection. The upper and middle panel micrographs are 400× final magnification and the lower panel micrographs are 1000× final magnification.
B. Quantification of inclusion formation in RNAi-treated S2 cells. Values (mean ± SD) are shown as percentage of cells with inclusions/total cells. Approximately, 1200 cells from each sample were counted. *P < 0.001 compared with control RNAi-treated cells (anova).
C. Efficiency of Rho1 depletion. Lysates were prepared from control S2 cells (U) or S2 cells exposed to dsRNA specific to Rho1 or Cdc42 for 4 days and immunoblotted with antibodies to Drosophila Rho1 or β-tubulin. All samples were run on the same gel and exposed for the same amount of time.
We used two approaches, microscopic examination and Western blot analysis, to assess the efficacy of RNAi-mediated depletion of the target proteins. As previously reported (Rogers et al., 2003), RNAi-mediated depletion of Rac1/2 or Cdc42 results in gross alterations in the actin cytoskeleton. To examine cytoskeletal changes, RNAi-treated S2 cells were stained with fluorescently conjugated-phalloidin, which binds actin. S2 cells do not form stress fibres; instead, they exhibit a radially symmetrical actin cytoskeleton (Rogers et al., 2003). RNAi-mediated inactivation of Rac1/2 and Cdc42 clearly resulted in similar severe alterations in the actin cytoskeleton (Fig. 9A, lower panels). However, only Rac1/2 depletion decreased Chlamydia infectivity, indicating that this decrease was not a non-specific effect resulting from gross changes in host cell morphology.
Depletion of Rho1 results in more subtle changes in the host cell architecture, with a slight increase in cell size (Rogers et al., 2003). We observed similar modest changes in cell and nuclear size (Fig. 9A). To further assess the efficiency of Rho1 depletion, we examined Rho1 protein levels in RNA-treated cells by Western blot analysis with a Drosophila-specific antibody to Rho1 (Fig. 9C). As a control, cells were treated with dsRNA specific for Cdc42; this treatment showed no diminution of Rho1 protein levels. It was not possible to directly quantify Cdc42 or Rac1/2 protein levels as antibodies to these Drosophila proteins are not available.
Discussion
In this work, we demonstrate that C. trachomatis infection of Drosophila S2 cells recapitulates key steps in the early infection of mammalian cells, including entry, inclusion formation, inhibition of phagolysosomal fusion, and acquisition of Golgi-derived sphingolipids. S2 cells could be efficiently infected in a dose-dependent manner with serovars characteristic of many different human Chlamydia infections, including the more invasive LGV serovar as well as several genital serovars (D, E and K). Infection was dependent upon bacterial protein synthesis, inhibited by heparin, required an intact actin cytoskeleton, and dependent upon the small GTPase, Rac. Our finding that bacterial entry was dependent on Rac but not dependent on the closely related Rho GTPase family members, Rho1 or Cdc42, suggests that the uptake pathway resembled that of mammalian cells (Carabeo et al., 2004). It was Chlamydia-specific and did not represent uptake by a more general phagocytic process.
Based on several criteria, we conclude that EB to RB differentiation occurred in S2 infections. Reticulate body (RB)-like forms were visualized by TEM, and expression of IncG, an RB-specific protein, was detected in L2-infected S2 cells. Furthermore, treatment of L2-infected S2 cells with penicillin resulted in enlarged RBs (unpubl. data), similar to the changes reported in mammalian cells (Matsumoto and Manire, 1970; Kramer and Gordon, 1971). The fact that IncG was produced and localized to the inclusion suggests that Chlamydia was metabolically active and modified the inclusion as it does during mammalian infection.
In contrast to mammalian infection where multiple Chlamydia inclusions fuse to form one large inclusion, fusion of inclusions in infected S2 cells appeared to be inefficient. In mammalian cells, it has been shown that IncA is required for fusion. In cells grown at temperatures below 32°C, IncA is not properly localized and inclusion fusion does not occur (van Ooij et al., 1998; Fields et al., 2002). Thus, temperature may play an important role in preventing inclusion fusion in S2 cells. In addition or alternatively, the macrophage-like nature of the S2 cells could inhibit fusion, as Chlamydia infection of human monocytes at 37°C results in multiple inclusions that are remarkably diverse in their sizes (Manor and Sarov, 1986; Airenne et al., 1999).
While early events of Chlamydia infections were recapitulated in S2 cells, the later steps of the life cycle were not observed. Several findings suggest that there was minimal RB to EB differentiation. First, expression of Hc2, a late cycle specific protein, did not increase at any time up to 4 days post infection. Second, no infectious progeny were obtained. Third, no host cell killing was detected. Similar phenomena were observed in L2-infected HeLa cells grown at 28°C. The lack of infectious progeny was not a result of bacterial killing by the S2 cells because L2 was still present in cells up to 5 days post infection, and no phagolysosomal fusion was observed, even at these late times.
The simplest explanation for the incomplete life cycle and lack of host cell killing observed in C. trachomatis-infected S2 cells is that bacterial replication is inefficient at these lower temperatures in any cell line, regardless of origin. Alternatively, eukaryotic cells grown at 28°C may lack host factors that are essential for completion of the C. trachomatis intracellular life cycle. In addition, the macrophage-like properties of S2 cells may prevent completion of the developmental cycle, as has been observed in human monocytes (Manor and Sarov, 1986; Airenne et al., 1999). Finally, it is possible that C. trachomatis infection of S2 cells results in persistent infection, as seen in penicillin-exposed, tryptophan-starved or interferon-gamma-treated mammalian cells (Morrison, 2003). Persistent growth is characterized by specific changes in RB morphology, gene expression, limited replication and a failure to convert to EBs and produce infectious progeny (Moulder et al., 1980; Morrison, 2003), all features that are in common with infection of S2 cells. Persistent infection of monocytes/macrophages is significant because it may enable Chlamydia to remain in these cells for long periods of time and thus allow for distribution of Chlamydia from the primary site of infection to other organs. Although S2 cells do not produce IFN-γ, they do synthesize antimicrobial peptides that are important in mediating the innate response to pathogens (Hoffmann, 2003). Therefore, it is possible that this restriction in S2 cells may be an active process via host–pathogen interactions in S2 cells similar to human macrophages. This possibility is currently under investigation.
It is not surprising that there are differences in bacterial infection of non-mammalian model systems, such as Drosophila, as compared with their mammalian systems. For example, the facultative intracellular pathogen, L. monocytogenes, displays a slower growth rate in S2 cells and is able to escape from its intracellular vacuole even in the absence of vacuole acidification (Cheng and Portnoy, 2003). Nonetheless, Listeria infection of S2 cells has provided a means to study new aspects of listeriolysin-O-dependent vacuolar escape.
In summary, we have shown that Chlamydia infection of Drosophila S2 cells closely resembles early critical events in mammalian infections. While RNAi-mediated gene inactivation in Drosophila has begun to be used to identify genes important in cell shape and cell division (Somma et al., 2002; Kiger et al., 2003), the use of this approach to identify host genes important in pathogenesis of infectious disease agents is new. As a proof of principle, we have demonstrated that Rac1/2 depletion, a host protein necessary for Chlamydia infection of human epithelial cells, also decreases infection in S2 cells. The stage is now set to use genome-wide RNAi-mediated gene inactivation in S2 cells to carry out forward genetic screens. The conservation of the innate immune processes between D. melanogaster and mammalian cells make this approach particularly useful to study host–pathogen interactions. It promises to open new avenues to elucidate host factors important for microbial pathogenesis, particularly for obligate intracellular pathogens are not genetically tractable, such as Chlamydia.
Experimental procedures
Reagents
HeLa 229 cells and L929 cells were obtained from ATCC and passaged as previously described (van Ooij et al., 1997). S2 wild type and GFP-Lamp1 expressing cells were kind gifts from Dr Ron Vale (UCSF). Cholesterol, MβCD, heparin and ConA were obtained from Sigma-Aldrich. Texas red-conjugated phalloidin was obtained from Molecular Probes. Antibodies were obtained from the following sources: mouse anti-Chlamydia FITC conjugate (Meridian Diagnostics), goat anti-C. trachomatis MOMP (Cortex Biochem), mouse anti-GAPDH (Chemicon), goat anti-Drosophila tubulin (Santa Cruz), mouse anti-Drosophila RhoI (P1D9; Developmental Studies Hybridoma Bank), rabbit anti-Hc2 and rabbit anti-IncG were kind gifts from the Hackstadt laboratory, rabbit anti-goat IgG HRP (Calbiochem), goat anti-rabbit IgG HRP (Amersham Biosciences), goat anti-mouse IgG HRP (Amersham Biosciences) and donkey anti-goat Alexa 594 (Molecular Probes).
Cell culture and C. trachomatis propagation
HeLa cells were routinely cultured in minimal essential medium (MEM) supplemented with 10% fetal bovine serum (FBS). S2 cells were cultured at 28°C in Schneider’s Medium supplemented with 10% FBS. Chlamydia trachomatis serovar LGV L2 was propagated in L929 cells. Chlamydia trachomatis serovars D, E and K were routinely propagated in HeLa cells. Chlamydia trachomatis EBs were harvested from infected cells and purified using a renografin step-gradient essentially as described (Caldwell et al., 1981). Alternatively, crude lysates obtained from infected cells were used for infection. Similar results were obtained with both crude preparations of C. trachomatis and renografin purified EBs.
Infection of Drosophila S2 cells
For infection with C. trachomatis serovars, S2 cells were infected with an MOI of 100, unless otherwise indicated. Thirty minutes before and during infection, S2 cells were treated with 50 μM MβCD-cholesterol and incubated at 28°C with centrifugation for 5 min at 1000 r.p.m. (Sorvall RT6000B). After 1 h of infection, bacteria were removed, cells were rinsed with phosphate buffered saline (PBS), fresh media supplemented with 1 mg ml−1 heparin was added, and cells were incubated for the indicated times. For binding and internalization experiments, 1 mg ml−1 heparin or 2.5 μg ml−1 cytochalasin D were added to S2 cells 30 min before and during infection. At various times following infection, S2 cells were replated onto ConA-coated 12 mm cover slips and allowed to adhere for 30 min S2 cells were fixed in ice-cold Methanol for 5 min, stained with anti-Chlamydia antibody (Merifluor) for 1 h, counterstained with Evan’s blue to visualize the cells, and examined by immunofluorescence microscopy. To assess the disruption of the actin cytoskeleton by cytochalasin D, cells were fixed in 4% paraformaldehyde, permeabilized with 1% saponin, and stained with Phalloidin-Texas Red (Molecular Probes). To determine if infection was dependent on bacterial protein synthesis, infected cells were incubated in the presence/absence of 10 μg ml−1 doxycycline. All experiments were performed in triplicate, and a minimum of 300 infected cells was counted per sample. Images for all immunofluorescent studies were acquired with a CCD camera (Nikon) using a 40× or 100× objective lens mounted on a Nikon TE2000 inverted microscope driven by Simple PCI software (Compix). A 10× ocular lens was used, making the total magnification for all images either 400× or 1000×. Images were processed with Adobe Photoshop CS.
Lysosomal fusion studies
S2 cells expressing GFP-Lamp were seeded in 24 well plates. Cells were then infected with L2 for 1 h as described above and further incubated in the absence or presence of 150 μg ml−1 chloramphenicol. At various times post infection, cells were replated onto Con-A coated cover slips, fixed in 4% paraformaldehyde for 30 min at room temperature, permeabilized with 1% saponin, and blocked for 1 h in 1% fish skin gelatin/2% FBS. Samples were stained with goat anti-MOMP (1:1000) for 1 h and subsequently stained with donkey anti-goat Alexa 594 (1:1000). Cover slips were then mounted in mounting media and visualized by immunofluorescence microscopy.
Labelling with FITC-C6-NBD-ceramide labelling
FITC-C6-NBD-ceramide (Molecular Probes) was complexed with 0.034% defatted bovine serum albumin (dfBSA) in MEM as per manufacturer’s protocol to yield complexes ~5 μM in both dfBSA and C6-NBD-ceramide. Chlamydia trachomatis L2-infected S2 and HeLa cells were incubated with the dfBSA/NBD-Cer complex at 4°C for 30 min in the dark. Cells were rinsed in PBS and incubated with MEM/0.34% dfBSA to ‘back exchange’ excess probe from plasma membrane. Cells on cover slips were rinsed in PBS, fixed in 4% paraformaldehyde for 30 min, mounted in mounting medium containing DAPI (Vectashield), and visualized by fluorescent microscopy.
IncG labelling
HeLa cells grown on 12 mm cover slips were infected with L2 for 24 h at 37°C and S2 cells were infected with L2 for 48 h. At the end of the infection, S2 cells were replated onto ConA-coated cover slips. Both HeLa and S2 cells were fixed with 4% paraformaldehyde for 30 min at room temperature, permeabilized with 1% saponin, and blocked for 1 h in 1% fish skin gelatin/2% FBS. Cells were stained with rabbit anti-IncG (1:2000) for 1 h and subsequently stained with goat anti-rabbit Alexa 488 (1:1000). Cover slips were then mounted in mounting media with DAPI to stain host and bacterial nuclei and visualized by immunofluorescence microscopy.
Electron microscopy
HeLa cells were seeded into 24 well plates on 12 mm cover slips and allowed to adhere either at 37°C or 28°C overnight. S2 cells were seeded without cover slips into 24 well plates and allowed to adhere at 28°C for 30 min Subsequently, HeLa and S2 cells were left untreated or infected with serovars L2, D and E. After 12 h and 24 h (for HeLa) and 60 h (for S2) incubation, S2 cells were replated onto ConA coated cover slips and both HeLa and S2 cells were fixed with 2% glutaraldehyde in 0.1 M phosphate buffer (pH 7.2) for 30 min in the cold and processed as described in (Pascopella et al., 1995).
Western blot analysis
HeLa and S2 cells were infected with L2 as described below. At various times post infection, cells were harvested, resuspended in PBS, and lysed by passage through a 22-gauge needle. For RNAi samples, S2 cells were incubated with RNA for 4 days and lysates were subsequently prepared as described above. Lysates were diluted two-fold with 2× SDS Sample Buffer containing 100 mM DTT, boiled for 5 mins, and subjected to SDS-PAGE on 12% gels. Gels were transferred to Immobilon (Millipore) by semidry blotting and blocked with a 5% solution of skim milk powder. Blots were probed with anti-MOMP and anti-Hc2 antibodies to detect expression of Chlamydia-specific proteins or probed with anti-Drosophila RhoI antibody to detect RNAi-depletion of Rho1 protein. Blots were also probed with anti-GAPDH (HeLa) or antibeta tubulin (Drosophila) antibodies as loading controls. Proteins were detected by ECL (Amersham Biosciences) according to the manufacturer’s protocol.
RNAi-mediated gene inactivation
S2 cells were plated in 96 well microplates with 50 000 cells in 200 μl Schneider’s Drosophila medium (Gibco) supplemented with 10% heat inactivated FBS, penicillin, streptomycin. DsRNA was generated as previously described (Foley and O’Farrell, 2004) and added to each well at a final concentration of 10 μg ml−1. Cells were cultured for 4 days at 28°C. dsRNA treated cells were replated into 96 well plates, infected with L2 for 1 h with centrifugation for 5 min, and then incubated for an additional 48 h. After 96 h, an aliquot of cells was removed and stained with phalloidin-Texas red (1:500) to visualize the actin cytoskeleton after RNAi-mediated depletion of Rho1, Rac and Cdc42. At 48 hpi, infected cells were replated onto 96 well glass bottom plates (Greiner) that had been coated with ConA and allowed to adhere for 30 min Cells were then fixed with methanol for 5 min, stained with anti-Chlamydia antibody (Meridian) for 1 h, and visualized by immunofluorescence microscopy.
Quantification of viable progeny and host cell viability
S2 and HeLa cells were grown at 28°C (for S2 and HeLa) or 37°C (for HeLa), were infected for 1 h with different doses of serovars L2, D, E and K, and incubated for 5 days. At various time points, the infected host cells were removed with a cell scraper. To quantify viable progeny, the host cells were pelleted in a micro-centrifuge for 10 min at 17 000× g. The host cells were washed once with medium, pelleted again, and lysed by drawing them sequentially through a 22-gauge and a 27-gauge needle. Two different aliquots of the lysate were diluted in DMEM and incubated for 1 h with 2 × 104 HeLa cells that had been incubated overnight on cover slips. The fraction of HeLa cells that contained inclusions was determined 18–24 h later to determine the relative amount of infectious progeny. To quantify viability of HeLa cells, infected cells were stained with crystal violet staining solution (0.2% crystal violet, 20% methanol) and absorbance at OD620 was measured. To quantify host cell viability of S2 cells, a portion of infected cells were removed and viability assessed by microscopic examination for exclusion of the vital stain trypan blue. All experiments were performed in triplicate, and a minimum of 300 infected cells was counted per sample.
Statistical analysis
The software program Instat was used for statistical analysis of data.
Acknowledgments
We thank members of the Engel, Vale, Davis, Portnoy and O’Farrell labs for advice and encouragement. We kindly acknowledge the kind gift of reagents from Drs Stephen Rogers and Ron Vale (UCSF) and Dr Ted Hackstadt (NIH). We thank Nafisa Ghori (Stanford University) for processing of samples for TEM. This work was supported by Grants to JNE from the NIH (R01 AI42806) and the Sandler Family Foundation. During a portion of this work, C.E. was supported by a postdoctoral fellowship from the American Lung Association.
References
- Airenne S, Surcel HM, Alakarppa H, Laitinen K, Paavonen J, Saikku P, Laurila A. Chlamydia pneumoniae infection in human monocytes. Infect Immun. 1999;67:1445–1449. doi: 10.1128/iai.67.3.1445-1449.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldini RL, Lau GW, Rahme LG. Use of plant and insect hosts to model bacterial pathogenesis. Methods Enzymol. 2002;358:3–13. doi: 10.1016/s0076-6879(02)58077-0. [DOI] [PubMed] [Google Scholar]
- Barry CE, Brickman TJ, Hackstadt T. Hc1-mediated effects on DNA structure: a potential regulator of chlamydial development. Mol Microbiol. 1993;9:273–283. doi: 10.1111/j.1365-2958.1993.tb01689.x. [DOI] [PubMed] [Google Scholar]
- Belland RJ, Scidmore MA, Crane DD, Hogan DM, Whitmire W, McClarty G, Caldwell HD. Chlamydia trachomatis cytotoxicity associated with complete and partial cytotoxin genes. Proc Natl Acad Sci USA. 2001;98:13984–13989. doi: 10.1073/pnas.241377698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros M, Kiger AA, Armknecht S, Kerr K, Hild M, Koch B, et al. Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science. 2004;303:832–835. doi: 10.1126/science.1091266. [DOI] [PubMed] [Google Scholar]
- Brickman TJ, Barry CE, III, Hackstadt T. Molecular cloning and expression of hctB encoding a strain-variant chlamydial histone-like protein with DNA-binding activity. J Bacteriol. 1993;175:4274–4281. doi: 10.1128/jb.175.14.4274-4281.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell HD, Kromhout J, Schachter J. Purification and partial characterization of the major outer membrane protein of Chlamydia trachomatis. Infect Immun. 1981;31:1161–1176. doi: 10.1128/iai.31.3.1161-1176.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell LA, Kuo CC. Chlamydia pneumoniae and atherosclerosis. Semin Respir Infect. 2003;18:48–54. doi: 10.1053/srin.2003.50006. [DOI] [PubMed] [Google Scholar]
- Carabeo RA, Hackstadt T. Isolation and characterization of a mutant Chinese hamster ovary cell line that is resistant to Chlamydia trachomatis infection at a novel step in the attachment process. Infect Immun. 2001;69:5899–5904. doi: 10.1128/IAI.69.9.5899-5904.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carabeo RA, Grieshaber SS, Fischer E, Hackstadt T. Chlamydia trachomatis induces remodeling of the actin cytoskeleton during attachment and entry into HeLa cells. Infect Immun. 2002;70:3793–3803. doi: 10.1128/IAI.70.7.3793-3803.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carabeo RA, Grieshaber SS, Hasenkrug A, Dooley C, Hackstadt T. Requirement for the Rac GTPase in Chlamydia trachomatis invasion of non-phagocytic cells. Traffic. 2004;5:418–425. doi: 10.1111/j.1398-9219.2004.00184.x. [DOI] [PubMed] [Google Scholar]
- Cheng LW, Portnoy DA. Drosophila S2 cells: an alternative infection model for Listeria monocytogenes. Cell Microbiol. 2003;5:875–885. doi: 10.1046/j.1462-5822.2003.00327.x. [DOI] [PubMed] [Google Scholar]
- D’Argenio DA, Gallagher LA, Berg CA, Manoil C. Drosophila as a model host for Pseudomonas aeruginosa infection. J Bacteriol. 2001;183:1466–1471. doi: 10.1128/JB.183.4.1466-1471.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dionne MS, Schneider DS. Screening the fruitfly immune system. Genome Biol. 2002;3:REVIEWS 1010. doi: 10.1186/gb-2002-3-4-reviews1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dionne MS, Ghori N, Schneider DS. Drosophila melanogaster is a genetically tractable model host for Mycobacterium marinum. Infect Immun. 2003;71:3540–3550. doi: 10.1128/IAI.71.6.3540-3550.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echalier, G. (1997) Drosophila Cells in Culture New York: Academic Press.
- Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields KA, Hackstadt T. The Chlamydial inclusion: escape from the endocytic pathway. Annu Rev Cell Dev Biol. 2002;18:221–245. doi: 10.1146/annurev.cellbio.18.012502.105845. [DOI] [PubMed] [Google Scholar]
- Fields KA, Fischer E, Hackstadt T. Inhibition of fusion of Chlamydia trachomatis inclusions at 32 degrees C correlates with restricted export of IncA. Infect Immun. 2002;70:3816–3823. doi: 10.1128/IAI.70.7.3816-3823.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields KA, Mead DJ, Dooley CA, Hackstadt T. Chlamydia trachomatis type III secretion: evidence for a functional apparatus during early-cycle development. Mol Microbiol. 2003;48:671–683. doi: 10.1046/j.1365-2958.2003.03462.x. [DOI] [PubMed] [Google Scholar]
- Foley E, O’Farrell PH. Functional dissection of an innate immune response by a genome-wide RNAi screen. PLos Biol. 2004;2:E203. doi: 10.1371/journal.pbio.0020203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fudyk T, Olinger L, Stephens RS. Selection of mutant cell lines resistant to infection by Chlamydia trachomatis and Chlamydia pneumoniae. Infect Immun. 2002;70:6444–6447. doi: 10.1128/IAI.70.11.6444-6447.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottar M, Gobert V, Michel T, Belvin M, Duyk G, Hoffmann JA, et al. The Drosophila immune response against Gram-negative bacteria is mediated by a peptidoglycan recognition protein. Nature. 2002;416:640–644. doi: 10.1038/nature734. [DOI] [PubMed] [Google Scholar]
- Grieshaber NA, Fischer ER, Mead DJ, Dooley CA, Hackstadt T. Chlamydial histone–DNA interactions are disrupted by a metabolite in the methylerythritol phosphate pathway of isoprenoid biosynthesis. Proc Natl Acad Sci USA. 2004;101:7451–7456. doi: 10.1073/pnas.0400754101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieshaber SS, Grieshaber NA, Hackstadt T. Chlamydia trachomatis uses host cell dynein to traffic to the microtubule-organizing center in a p50 dynamitin-independent process. J Cell Sci. 2003;116:3793–3802. doi: 10.1242/jcs.00695. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Martin CB, Ojcius DM, Hsia R, Hellio R, Bavoil PM, Dautry-Varsat A. Heparin-mediated inhibition of Chlamydia psittaci adherence to HeLa cells. Microb Pathog. 1997;22:47–57. doi: 10.1006/mpat.1996.0090. [DOI] [PubMed] [Google Scholar]
- Hackstadt T, Rockey D, Heinzen R, Scidmore M. Chlamydia trachomatis interrupts an exocytic pathway to acquire endogenously synthesized sphingomyelin in transit from the Golgi apparatus to the plasma membrane. EMBO J. 1996;15:964–977. [PMC free article] [PubMed] [Google Scholar]
- Hackstadt T, Scidmore MA, Rockey DD. Lipid metabolism in Chlamydia trachomatis-infected cells: directed trafficking of Golgi-derived sphingolipids to the chlamydial inclusion. Proc Natl Acad Sci USA. 1995;92:4877–4881. doi: 10.1073/pnas.92.11.4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann JA. The immune response of Drosophila. Nature. 2003;426:33–38. doi: 10.1038/nature02021. [DOI] [PubMed] [Google Scholar]
- Innocenti M, Zucconi A, Disanza A, Frittoli E, Areces LB, Steffen A, et al. Abi1 is essential for the formation and activation of a WAVE2 signalling complex. Nat Cell Biol. 2004;6:319–327. doi: 10.1038/ncb1105. [DOI] [PubMed] [Google Scholar]
- Kiger A, Baum B, Jones S, Jones M, Coulson A, Echeverri C, Perrimon N. A functional genomic analysis of cell morphology using RNA interference. J Biol. 2003;2:27. doi: 10.1186/1475-4924-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer MJ, Gordon FB. Ultrastructural analysis of the effect of penicillin and chlortetracyline on the development of a genital tract Chlamydia. Infect Immun. 1971;3:333–341. doi: 10.1128/iai.3.2.333-341.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau GW, Goumnerov BC, Walendziewicz CL, Hewitson J, Xiao W, Mahajan-Miklos S, et al. The Drosophila melanogaster toll pathway participates in resistance to infection by the gram-negative human pathogen Pseudomonas aeruginosa. Infect Immun. 2003;71:4059–4066. doi: 10.1128/IAI.71.7.4059-4066.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipsky NG, Pagano RE. Intracellular translocation of fluorescent sphingolipids in cultured fibroblasts: endogenously synthesized sphingomyelin and glucocerebroside analogues pass through the Golgi apparatus en route to the plasma membrane. J Cell Biol. 1985;100:27–34. doi: 10.1083/jcb.100.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manor E, Sarov I. Fate of Chlamydia trachomatis in human monocytes and monocyte-derived macrophages. Infect Immun. 1986;54:90–95. doi: 10.1128/iai.54.1.90-95.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfield BE, Dionne MS, Schneider DS, Freitag NE. Exploration of host–pathogen interactions using Listeria monocytogenes and Drosophila melanogaster. Cell Microbiol. 2003;5:901–911. doi: 10.1046/j.1462-5822.2003.00329.x. [DOI] [PubMed] [Google Scholar]
- Matsumoto A, Manire GP. Electron microscopic observations on the effects of penicillin on the mophology of Chlamydia psittaci. J Bacteriol. 1970;101:278–285. doi: 10.1128/jb.101.1.278-285.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison RP. New insights into a persistent problem – chlamydial infections. J Clin Invest. 2003;111:1647–1649. doi: 10.1172/JCI18770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder JW. Interaction of chlamydiae and host cells in vitro. Microbiol Rev. 1991;55:143–190. doi: 10.1128/mr.55.1.143-190.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder JW, Levy NJ, Schulman LP. Persistent infection of mouse fibroblasts (L cells) with Chlamydia psittaci: evidence for a cryptic chlamydial form. Infect Immun. 1980;30:874–883. doi: 10.1128/iai.30.3.874-883.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ooij C, Apodaca G, Engel J. Characterization of the Chlamydia trachomatis vacuole and its interaction with the host endocytic pathway in HeLa cells. Infect Immun. 1997;65:758–766. doi: 10.1128/iai.65.2.758-766.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ooij C, Homola E, Kincaid E, Engel J. Fusion of vacuoles containing Chlamydia trachomatis is inhibited at low temperature and requires bacterial protein synthesis. Infect Immun. 1998;66:5364–5371. doi: 10.1128/iai.66.11.5364-5371.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascopella L, Raupach B, Ghori N, Monack D, Falkow S, Small PL. Host restriction phenotypes of Salmonella typhi and Salmonella gallinarum. Infect Immun. 1995;63:4329–4335. doi: 10.1128/iai.63.11.4329-4335.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen LB, Birkelund S, Christiansen G. Purification of recombinant Chlamydia trachomatis histone H1-like protein Hc2, and comparative functional analysis of Hc2 and Hc1. Mol Microbiol. 1996a;20:295–311. doi: 10.1111/j.1365-2958.1996.tb02618.x. [DOI] [PubMed] [Google Scholar]
- Pedersen LB, Birkelund S, Holm A, Ostergaard S, Christiansen G. The 18-kilodalton Chlamydia trachomatis histone H1-like protein (Hc1) contains a potential N-terminal dimerization site and a C-terminal nucleic acid-binding domain. J Bacteriol. 1996b;178:994–1002. doi: 10.1128/jb.178.4.994-1002.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perara E, Ganem D, Engel JN. A developmentally regulated chlamydial gene with apparent homology to eukaryotic histone H1. Proc Natl Acad Sci USA. 1992;89:2125–2129. doi: 10.1073/pnas.89.6.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahme LG, Ausubel FM, Cao H, Drenkard E, Goumnerov BC, Lau GW, et al. Plants and animals share functionally common bacterial virulence factors. Proc Natl Acad Sci USA. 2000;97:8815–8821. doi: 10.1073/pnas.97.16.8815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramet M, Manfruelli P, Pearson A, Mathey-Prevot B, Ezekowitz RA. Functional genomic analysis of phagocytosis and identification of a Drosophila receptor for E. coli. Nature. 2002;416:644–648. doi: 10.1038/nature735. [DOI] [PubMed] [Google Scholar]
- Rogers SL, Wiedemann U, Stuurman N, Vale RD. Molecular requirements for actin-based lamella formation in Drosophila S2 cells. J Cell Biol. 2003;162:1079–1088. doi: 10.1083/jcb.200303023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachter J. The intracellular life of Chlamydia. Curr Top Microbiol Immunol. 1988;138:109–139. [PubMed] [Google Scholar]
- Schramm N, Wyrick PB. Cytoskeletal requirements in Chlamydia trachomatis infection of host cells. Infect Immun. 1995;63:324–332. doi: 10.1128/iai.63.1.324-332.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scidmore MA, Hackstadt T. Mammalian 14-3-3beta associates with the Chlamydia trachomatis inclusion membrane via its interaction with IncG. Mol Microbiol. 2001;39:1638–1650. doi: 10.1046/j.1365-2958.2001.02355.x. [DOI] [PubMed] [Google Scholar]
- Scidmore MA, Fischer ER, Hackstadt T. Restricted fusion of Chlamydia trachomatis vesicles with endocytic compartments during the initial stages of infection. Infect Immun. 2003;71:973–984. doi: 10.1128/IAI.71.2.973-984.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scidmore-Carlson MA, Shaw EI, Dooley CA, Fischer ER, Hackstadt T. Identification and characterization of a Chlamydia trachomatis early operon encoding four novel inclusion membrane proteins. Mol Microbiol. 1999;33:753–765. doi: 10.1046/j.1365-2958.1999.01523.x. [DOI] [PubMed] [Google Scholar]
- Somma MP, Fasulo B, Cenci G, Cundari E, Gatti M. Molecular dissection of cytokinesis by RNA interference in Drosophila cultured cells. Mol Biol Cell. 2002;13:2448–2460. doi: 10.1091/mbc.01-12-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamm, W.E., and Holmes, K.K. (1990) Chlamydia trachomatis infections of the adult. In Sexually Transmitted Diseases Holmes, K., Mardh, P.-A., Sparling, P.F., Weisner, P.J., Cates, W., Lemon, S.M., and Stamm, W.E. (eds). New York: McGraw-Hill, pp. 181–194.
- Taraktchoglou M, Pacey AA, Turnbull JE, Eley A. Infectivity of Chlamydia trachomatis serovar LGV but not E is dependent on host cell heparan sulfate. Infect Immun. 2001;69:968–976. doi: 10.1128/IAI.69.2.968-976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodovar N, Acosta C, Lemaitre B, Boccard F. Drosophila: a polyvalent model to decipher host–pathogen interactions. Trends Microbiol. 2004;12:235–242. doi: 10.1016/j.tim.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Wolf K, Hackstadt T. Sphingomyelin trafficking in Chlamydia pneumoniae-infected cells. Cell Microbiol. 2001;3:145–152. doi: 10.1046/j.1462-5822.2001.00098.x. [DOI] [PubMed] [Google Scholar]
- Wuppermann FN, Hegemann JH, Jantos CA. Heparan sulfate-like glycosaminoglycan is a cellular receptor for Chlamydia pneumoniae. J Infect Dis. 2001;184:181–187. doi: 10.1086/322009. [DOI] [PubMed] [Google Scholar]
- Zhang JP, Stephens RS. Mechanism of Chlamydia trachomatis attachment to eukaryotic host cells. Cell. 1992;69:861–869. doi: 10.1016/0092-8674(92)90296-o. [DOI] [PubMed] [Google Scholar]