

Abstract
Purpose
Oxidative stress from reactive oxygen species (ROS) has been implicated in many diseases, including age-related macular degeneration (AMD), in which the retinal pigment epithelium (RPE) is considered a primary target. Because manganese superoxide dismutase (SOD2), localized in mitochondria, is known to be a key enzyme that protects the cells against oxidative stress, this study was undertaken to examine oxidation-induced apoptosis in cultured RPE cells with various levels of SOD2.
Methods
Primary cultures of RPE cells were established from wild-type (WT), heterozygous Sod2-knockout mouse (HET) and hemizygous Sod2 mice with overexpression of the enzyme (HEMI). Purity of the RPE cell cultures was verified by immunostaining with antibody to RPE65 and quantified by flow cytometry. Oxidative stress was induced in RPE cells by exposing them to H2O2 (0 –500 μM) for 1 hour and reculturing them in normal medium for various times (0 –24 hours). Apoptosis in the RPE was examined by TUNEL staining and quantified by cell-death– detection ELISA. Mitochondrial transmembrane potential (MTP) was measured by a cationic dye, and cytochrome c leakage from mitochondria was analyzed by Western blot analysis.
Results
More than 95% of the cells in each culture were RPE65 positive, and the relative SOD2 levels in HET, WT, and HEMI cells were 0.6, 1.0, and 3.4, respectively. H2O2-induced apoptotic cell death was both dose and time dependent, and apoptosis in these cells was related to the cellular SOD2 level. Disruption of MTP and release of cytochrome c were observed to occur before apoptotic cell death, and they correlated with cellular SOD2.
Conclusions
The results demonstrate a critical role of SOD2 in protection against oxidative challenge. Cells from HET mice showed greater apoptotic cell death, whereas in those from HEMI mice, cell death cell death induced by oxidative injury was suppressed.
It has been shown that oxidative stress from reactive oxygen species (ROS) is one of the key factors in the pathogenesis of aging associated diseases, including age-related macular degeneration (AMD) and cataracts.1–4 The retinal pigment epithelium (RPE) is considered a primary target in AMD, because RPE cell death is observed in the early phase of the disease.5–7 Since the RPE plays a role in transporting selective molecules between the choroidal blood and the neural retina, which forms the outer blood–retinal barrier, dysfunction or death of the RPE cells may induce degeneration of photoreceptors. Several factors may contribute to the pathogenesis of AMD, all of which may involve the RPE: a decrease in the number of RPE cells in the macular area, accumulation of degenerated substance (drusen) in the inner layer of Bruch’s membrane, and leakage at the Bruch’s membrane of the macula, which may result in subretinal neovascularization. Although oxidative stress has been thought to be one of the major factors involved in RPE cell death in AMD, the mechanisms are still unclear.
Under normal physiological conditions, metabolism of oxygen by aerobic organisms generates ROS, which could lead to cellular and/or mitochondrial damage. The damaging effect of ROS is prevented by endogenous antioxidant compounds and enzymes that are present in various cells. Under conditions of a diminution of the antioxidant systems or enzyme function during normal aging, the ability of the various cells to protect against oxidative challenge is compromised, eventually leading to apoptotic cell death. Mitochondria are known to be one of the vulnerable components in the cell against oxidative damage, because nearly 4% of the oxygen consumed by the electron transport chain in mitochondria is converted to superoxide anions.8,9 These ROS are removed by manganese superoxide dismutase (SOD2). Oxidative insult to the mitochondria can lead to perturbation of its metabolism and redox status, which alters mitochondrial membrane permeability transition and induces cytochrome c leakage from the mitochondria. There is increasing evidence that leakage of cytochrome c from the mitochondrial inner membrane compartment triggers a series of apoptosis signal transduction processes, resulting in apoptotic cell death.
ROS generated in mitochondria is normally regulated by antioxidant enzymes such as SOD2 and phospholipid hydroperoxide glutathione peroxidase (PHGPx), which protect the mitochondria from oxidative insult. SOD2 is localized in the mitochondrial matrix and catalyzes the dismutation of two superoxide radicals, to yield hydrogen peroxide and oxygen.10 There are several studies in which the deficiency of SOD2 has been implicated in apoptotic cell death.11–15
Transgenic and knockout mutant mice with different levels of SOD2 have served as useful models for investigating the functional role of this enzyme in protecting against oxidation-induced cell death. Experiments with knockout mice that lack this enzyme provide insights into gene function and the pathophysiology of various diseases related to oxidative stress. Two Sod2-knockout mouse models having different disruptions in the Sod2 gene have been reported.16,17 In both of these knockout models, the homozygous (Sod2−/−) mutants display embryonic or neonatal lethality, and thus, SOD2 has been considered a critical enzyme for aerobic life. However, the Sod2-knockout mice survive for only a short time, and it has not been possible to study the ocular manifestations resulting from the total absence of the Sod2 protein. Heterozygous Sod2-knockout mice (HET) appeared relatively normal, and no phenotype has been reported, although the activities of this enzyme in the mitochondria were reduced in all the tissues studied. Sod2 activity in HET is roughly 50% of wild-type (WT) mice, but activity of other antioxidant enzymes such as glutathione peroxidase and copper/zinc superoxide dismutase (SOD1) is not affected.18 Although the partial reduction of this enzyme induces mitochondrial oxidative damage and altered mitochondrial function, there is no significant oxidative damage in cytosolic protein or nuclear DNA.18 Thus, the heterozygous knockout mouse that shows mitochondrial oxidative damage could serve as a useful model for studying the functional role of this enzyme in apoptotic cell death. We have also studied the role of this enzyme in Sod2 transgenic mice (HEMI) in which the enzyme was overexpressed. Specific activity of SOD2 in the lungs of these animals increased by 170% compared to WT control animals and provides antioxidant defense in the lung.19,20 We have reported that SOD2 overexpression in human lens epithelial cell culture suppresses apoptotic cell death induced by photochemical oxidative stress.21 These studies demonstrated increased cytochrome c leakage from the mitochondria and greater apoptosis in SOD2-deficient cells when compared with the cells overexpressing the enzyme.
In this study, we investigated the protective role of SOD2 against H2O2-induced oxidative stress in RPE cells containing various levels of the enzyme. The primary cultures were derived from WT and mutant mice, partial knockout mice (HET), and transgenic mice that overexpress the enzyme (HEMI).
Materials and Methods
Animals
Sod2 heterozygous knockout mice were obtained from the Jackson Laboratory (Bar Harbor, ME; stock no. 002973, strain B6.129S7-Sod2tm1Leb) and were fed standard laboratory chow and water ad libitum. To minimize genetic variations, HET and WT mice were initially derived from breeding two heterozygous knockout mice. The Sod2 HEMI mice were first generated by microinjecting the human SOD2 expression vector into fertilized mouse eggs isolated from mating of (C57BL/6 X C3H; hereafter called B6C3 F1) male and female mice, as described previously.19 Experimental animals were generated by breeding of hemizygous Sod2 transgenic mice with WT B6C3 F1 mice. PCR was used for genotyping the offspring, as described previously.21 All animals used were maintained and treated in accordance with ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Cell Culture
Mouse RPE cells were isolated from eyes obtained from 1-year-old HET, WT, and HEMI mice. In brief, the mice were killed by CO2 asphyxia, eyes were enucleated, and the anterior segment, lens, and neuroretina were removed. Eyecups were treated for 30 minutes with papain (5 U/mL) to isolate the RPE from Bruch’s membrane. After incubation, the RPE was removed by trituration with a plastic pipette and cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen-Gibco, Grand Island, NY) containing 15% fetal bovine serum (FBS; Invitrogen-Gibco) and 50 μg/mL of gentamicin in culture plates (Falcon; BD Biosciences, Bedford, MA). Mouse eyecups became soft and flat when treated with papain, because both the inside and outside of the eyecups were in contact with the enzyme solution. Preliminary studies showed that the harvested RPE cells were often contaminated with fibroblasts from the sclera. To avoid fibroblast contamination of the RPE, the early stage of the initial culture was carefully monitored, and all possible fibroblast growth (which appear earlier than RPE cell growth) was ablated with a red-hot platinum loop that burned out all the fibroblast colonies, and cell culturing continued. When RPE cells became confluent they were subcultured by splitting in a 1:3 ratio. After three passages, the cells were frozen in liquid N2 and later used for the various experiments.
Purity of the cells was verified by immunostaining with antibody to RPE65, a specific marker for RPE, and the SOD2 level was determined by Western blot analysis, as described below. These cells were cultured in DMEM with 15% FBS at 37°C in a 5% CO2 environment. After the cells were confluent, they were trypsinized and recultured to adjust the number of cells in each group, as desired.
RPE65 Immunohistochemistry and Flow Cytometry
The purity of the cell culture was established by an immunocytochemical method, with an antibody against RPE65, and quantified by flow cytometry. The primary cell cultures (2 × 104/well) from WT, HET, and HEMI mice were cultured on eight-well chambered slides (Nunc, Roskilde, Denmark) for 24 hours and then stained with RPE65 antibody and analyzed by fluorescence microscopy. Briefly, the cultured cells were fixed with 4% methanol-free formaldehyde for 30 minutes at room temperature and washed with PBS. They were then blocked with a blocking solution (20% sheep serum and 0.5% Triton X-100 in PBS) for 30 minutes at room temperature and incubated overnight at 4°C with primary antibody to RPE65 (kindly provided by Debra Thompson, Kellogg Eye Center) diluted in wash solution (2% sheep serum and 0.2% Triton X-100 in PBS). The cells were then rinsed again with wash solution and incubated for 1 hour at room temperature with tetramethylrhodamine isomer R (TRITC)-conjugated secondary antibody (Molecular Probe, Eugene, OR), which was diluted in wash solution (1: 250).
For quantification, the stained cells were analyzed by flow cytometry, as follows. Cells (5 × 105) were trypsinized and collected by centrifugation at 1000g for 5 minutes and then fixed with 4% methanol-free formaldehyde for 30 minutes at room temperature. They were then incubated in PBS containing 0.25% Triton X-100 for 5 minutes at 4°C to permeabilize the cell membrane. After the cells were washed with PBS, they were incubated with primary antibody to RPE65 or without the antibody, as a negative control, in wash solution for 1 hour at room temperature. The cells were washed two times and then incubated for 30 minutes at room temperature with TRITC-conjugated secondary antibody diluted in wash solution (1:100). The cells were analyzed by flow cytometry (FACS; BD Biosciences, San Jose, CA), with FITC signal detector [FL2]).
Analysis of SOD2 Protein Expression
SOD2 levels in these cells were measured by Western blot analysis. Trypsinized cultures were washed with PBS and centrifuged at 1000g for 5 minutes. Collected cells were dissolved in 0.5% Triton buffer containing proteinase inhibitor (Roche Diagnostics, Mannheim, Germany) and incubated for 10 minutes on ice. After incubation, each sample was centrifuged at 10,000g for 10 minutes at 4°C, to remove the debris. Supernatants were assayed for protein and all samples adjusted to the same protein concentration and mixed with lithium dodecyl sulfate sample buffer (Invitrogen, Carlsbad, CA) and 2-mercaptoethanol, according to Invitrogen protocols. The samples were subjected to SDS/PAGE followed by Western blot analysis using a protein expression assay (Western Breeze; Invitrogen, Carlsbad, CA). Electrophoresed proteins were transferred from gel onto nitrocellulose membrane (Trans-Blot transfer medium; Bio-Rad, Hercules, CA) and incubated with 0.1 μg/mL primary antibody against SOD2 (Anti MnSOD; product no. SOD-111; Stressgen, Victoria, British Columbia, Canada) overnight at 4°C and then incubated for 30 minutes at room temperature with alkaline-phosphatase–conjugated secondary antibody. The staining reaction was quantified by densitometry with a computerized image analysis program (NIH Image, ver. 1.63; available by ftp at zippy.nimh.nih.gov/ or at http://rsb.info.nih.gov/nih-image; developed by Wayne Rasband, National Institutes of Health, Bethesda, MD).
H2O2-Induced Oxidative Stress
Primary cultures were incubated in serum-free DMEM containing different levels of H2O2 (0–500 μM) for 1 hour, and then the media were replaced with DMEM with 15% FBS. After 0, 4, 8, 16, and 24 hours of reculturing, these cells were used for experiments involving apoptosis and mitochondrial transmembrane potential.
Analysis of Apoptosis
The cells were cultured on eight-well chambered slides (2 × 104/well) for 24 hours and then exposed to H2O2, as described earlier. They were fixed with 4% methanol-free formaldehyde and subjected to terminal deoxynucleotidyltransferase dUTP nick-end labeling (TUNEL) staining using a cell-death–detection kit (In Situ Cell Death Detection Fluorescein kit; Roche Applied Science, Penzberg, Germany). For quantification of apoptosis under different experimental conditions, apoptotic cell death was determined by a cell-death–detection ELISA kit (Roche Applied Science), which is based on a quantitative sandwich-enzyme immunoassay using antibody binding to DNA and histones H1, H2A, H2B, H3, and H4 and photometric measurement at 405 nm with a microplate reader (Spectra Max 190; Molecular Devices, Sunnyvale, CA).
Measurement of Mitochondrial Transmembrane Potential
After exposure to H2O2 as previously described, the cells on the slide were examined for change in mitochondrial transmembrane potential (MTP) by using a cationic dye (MitoCapture; BioVision, Mountain View, CA), and detected with fluorescence microscopy. The cultured cells on the chamber slide after exposure to H2O2 were incubated with prewarmed cationic dye solution (MitoCapture; BioVision) for 15 minutes at 37°C and then immediately observed under a fluorescence microscope. The assay is based on the aggregation of the dye and its fluorescence in the mitochondria. In the undamaged mitochondria, the aggregated dye appears as red fluorescence, whereas in the apoptotic cell with altered mitochondrial transmembrane potential, the dye remains as monomers in the cytoplasm with diffuse green fluorescence.
Measurement of Cytochrome c Leakage from Mitochondria
After 8 hours of reculturing after exposure to 300 μM of H2O2 for 1 hour, the RPE cells (5 × 107) were collected by centrifugation at 600g for 5 minutes at 4°C and washed with ice-cold PBS. The cells were assayed with the cytochrome c–releasing apoptosis kit (BioVision). Briefly, they were homogenized with the cytosol extraction buffer provided in the kit and then centrifuged at 700g for 10 minutes at 4°C to remove the debris. The supernatant was then centrifuged at 10,000g for 30 minutes at 4°C. The pellet was used as the mitochondrial fraction, and the supernatant was collected as the cytosolic fraction. These fractions were analyzed for cytochrome c by Western-blot as described earlier, with the cytochrome c antibody provided in the kit.
Results
Purity of RPE Cell Cultures
In this study, first it was important to establish the purity of the RPE cultures. Preliminary studies indicated considerable contamination with fibroblast cells, probably derived from the sclera. However, ablation of the fibroblast colonies, which appeared first in the initial cultures, with a hot platinum wire was highly successful. The purity of the RPE cells was verified by immunostaining with antibody to RPE65 and quantified by flow cytometry. RPE65-positive stain was observed in nearly all the cells (Fig. 1A). Quantification with flow cytometry (Fig. 1B) showed that nearly 100% of the cells in each of the cell cultures were RPE65 positive. Incubation without RPE65 antibody, as negative control, showed less than 5% of the cells were stained. Thus, we concluded that the RPE cell cultures derived from WT and the two mutant mice are not contaminated with other cell types.
Figure 1.
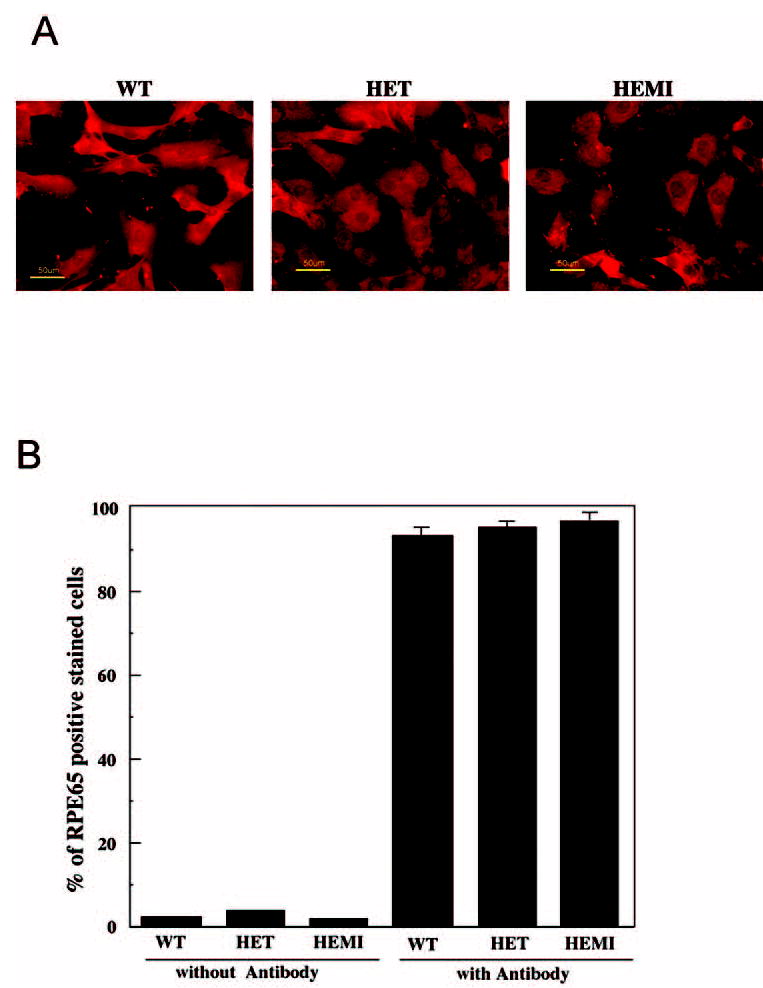
Purity of the RPE cell culture. (A) Immunocytochemistry of cultured RPE derived from mice with different levels of SOD2. The cells were stained with antibody to RPE65 and visualized by fluorescence microscope. Nearly all the cells are stained with the antibody. (B) Flow cytometric analysis of antibody-stained cells. Nearly 100% of the cells were RPE65 positive. In the absence of the antibody, less than 3% of the cells showed staining. Data are expressed as the mean ± SEM (n = 3).
Levels of SOD2 Protein in RPE Cells
Expression of SOD2 in the RPE cells from the three groups of mice (WT, HET, and HEMI) was detected by Western blot analysis with antibody against SOD2. A 25-kDa band was observed in each of the samples (Fig. 2A). Density scanning of these immunoblots showed that the relative expression of SOD2 in RPE cells from HET was approximately 60% of the endogenous level in the control cells (WT), whereas the expression in RPE cells from HEMI was approximately 3.4 times that of the control cells (Fig. 2B).
Figure 2.
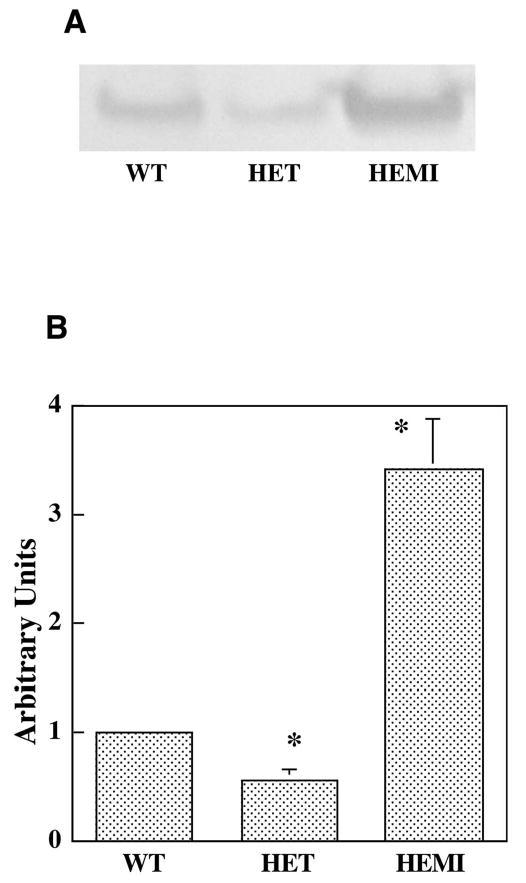
Differential expression of SOD2 in RPE cells. (A) Extracts containing 10 μg protein from each of the cell cultures were subjected to SDS-PAGE followed by Western-blot analysis with antibody to SOD2. (B) The enzyme level was quantified by integrated density scanning of the immunoblot membrane. The relative SOD2 levels in WT, HET, and HEMI mice were 1.0, 0.6, and 3.4, respectively. Data are expressed as the mean ± SEM (n = 4). *P < 0.05 compared with those of WT.
Effect of Oxidative Stress on Apoptosis in RPE Cell Culture
The normal phagocytosis of outer segments of photoreceptor by RPE is known to accumulate H2O2, which is detoxified by antioxidant enzymes. We therefore assessed the protective effect of SOD2 against oxidation-induced apoptosis after exposure to H2O2. To detect apoptosis, we analyzed by fluorescence microscopy the occurrence of DNA fragmentation in the cells (Fig. 3). The number of TUNEL-positive cells increased with H2O2 treatment in a dose- and time-dependent manner (data not shown). Figure 3 shows that when RPE cells were exposed to a 300-μM concentration of H2O2 for 1 hour and then recultured for 16 hours, apoptotic cell death increased compared with that in untreated control cells (without H2O2). The number of TUNEL-positive RPE cells from HET mice was much greater than that in cells derived from WT or HEMI mice. Furthermore, in cells derived from HEMI mice, in which the SOD2 level was nearly 3.4 times that of WT, apoptosis was suppressed. It is notable that the least number of TUNEL-positive cells was present in the cells derived from the HEMI mice. To obtain further confirmation of peroxide-induced apoptosis, additional experiments were performed for quantitative assessment. RPE cells from the three groups of mice were exposed to different doses of H2O2 for 1 hour and recultured for various times, and apoptotic cell death was measured by cell-death– detection ELISA kit (Figs. 4, 5). H2O2-induced apoptosis was both dose and time dependent. Figure 4 shows cell death as a function of the concentration of H2O2 ranging from 0 to 500 μM. The cell death in SOD2-deficient cells (HET) was significantly greater than in those from WT and HEMI mice at all concentrations of H2O2 higher than 200 μM. The time dependency of H2O2-induced apoptosis was also related to the cellular level of SOD2 (Fig. 5). The cells overexpressing SOD (HEMI) had a greater protective effect against apoptosis induced by H2O2 than did those from WT or HET mice.
Figure 3.
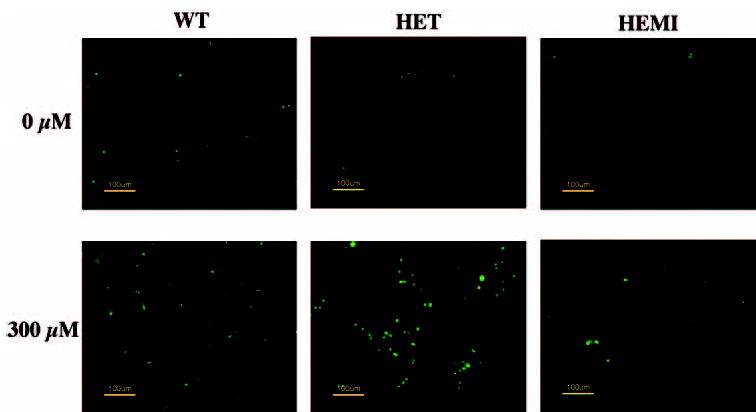
The effect of oxidative stress on apoptosis in RPE cells derived from mice with different levels of SOD2. Cell cultures were exposed to 300 μM H2O2 for 1 hour and recultured for 16 hours in a normal medium, and apoptosis was determined by TUNEL staining. H2O2-treated cells from HET mice showed more TUNEL-positive cells than did those from the WT or HEMI mice, whereas apoptotic cell death was suppressed in the cells from the HEMI mice.
Figure 4.
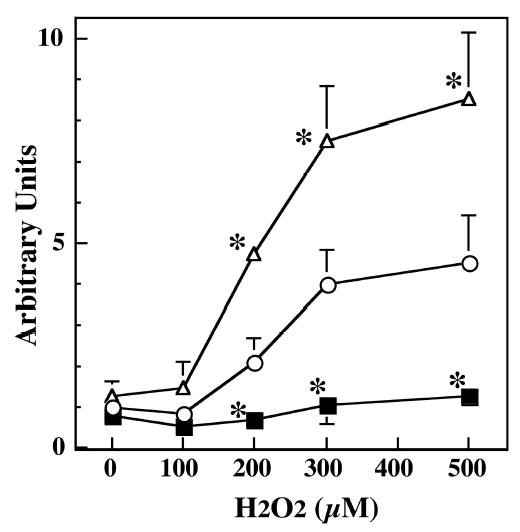
Dose-dependent effect of H2O2 exposure on apoptosis in RPE with different levels of SOD2. An equal number of RPE cells derived from WT (○), HET (▵), and HEMI (▪) mice were exposed to various concentrations of H2O2 for 1 hour and recultured in normal medium for 16 hours, and apoptosis was determined by a cell-death–detection ELISA kit. Cells from HET mice showed greater apoptosis than did those from WT or HEMI mice at all levels of H2O2 exposure. A higher level of SOD2 (HEMI) protected the RPE cells from apoptosis. Data are expressed as the mean ± SEM (n = 4 to 7). *P < 0.05 compared with those of WT.
Figure 5.
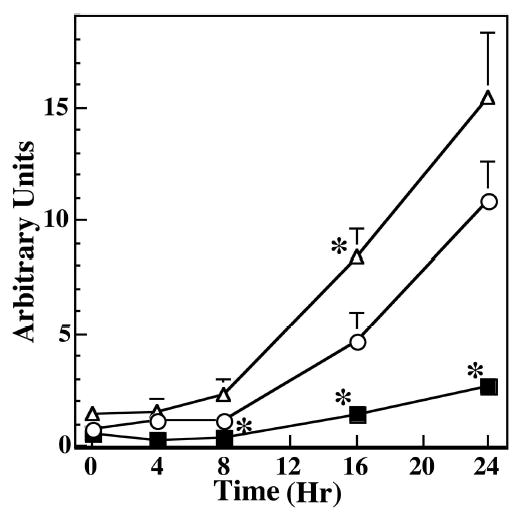
Time course of the effect of H2O2 exposure on apoptosis in RPE with different levels of SOD2. An equal number of RPE cells derived from WT (○), HET (▵), and HEMI (▪) mice were exposed to 300 μM H2O2 for 1 hour and cultured in normal medium for the different periods indicated. Apoptosis was determined by a cell-death–detection ELISA kit. Apoptotic cell death increased progressively as a function of time. Also, greater apoptosis was noted in HET cells than in WT and HEMI cells. Apoptotic cell death in HEMI mice was well suppressed until 16 hours of incubation. Data are expressed as the mean ± SEM (n = 4–7). *P < 0.05 compared with those of WT.
Effect of Oxidative Stress on MTP
Altered MTP is known to be an early event in the apoptosis signaling process.22–24 We therefore investigated the relationship between mitochondrial damage and apoptosis when cells were treated with H2O2. The cells were exposed to 300 μM of H2O2 for 1 hour and then recultured for 4, 8, and 16 hours. Disruptions of the MTP in these cells increased in a time-dependent manner, and the extent of the mitochondrial damage was related to the level of SOD2. Fig. 6 shows mitochondrial membrane damage (green fluorescence) in cells derived from WT and HET mice first noted after 4 hours of incubation. At this time point, no apoptotic cell death was observed (Fig. 5). Loss of membrane potential after 4 and 8 hours was much greater in HET than WT mice, whereas the MTP in cells derived from HEMI mice was still preserved until 8 hours of incubation. Thus, deficiency of SOD2 results in greater disruption of the MTP, whereas overexpression of the enzyme protects against mitochondrial membrane damage. Furthermore, altered MTP was observed before apoptosis was detected.
Figure 6.
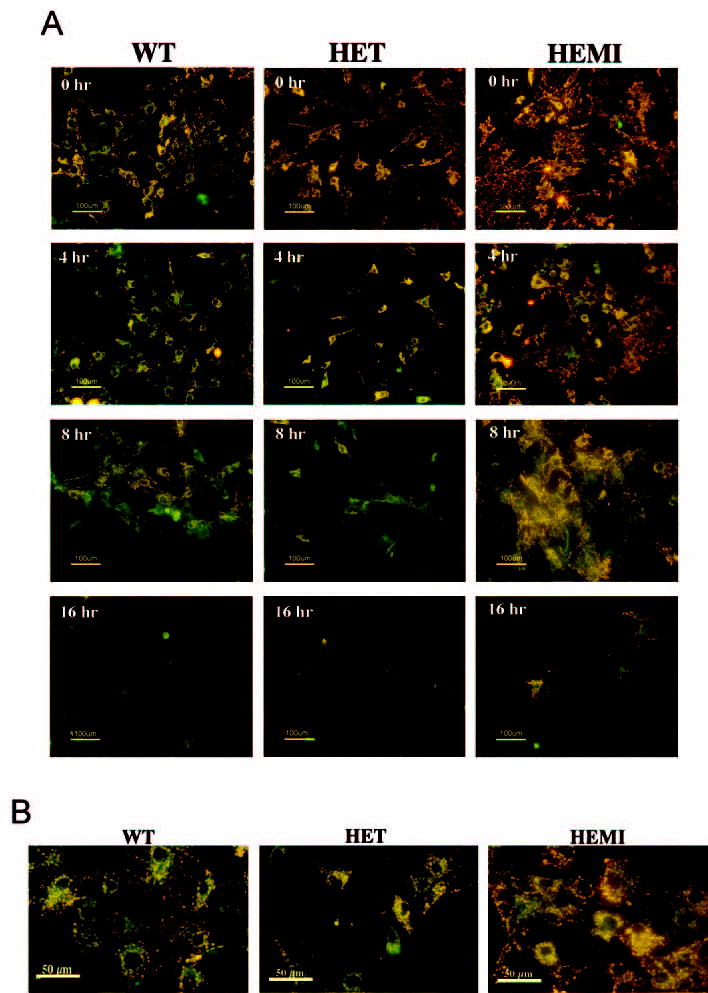
Effect of H2O2 on mitochondrial transmembrane potential in RPE cells with various levels of SOD2. (A) An equal number of cells from each of the cultures were exposed to 300 μM of H2O2 for 1 hour and then recultured for 4, 8, and 16 hours in normal medium. The cells were then treated with a cationic dye and observed under a fluorescence microscope. The decreased fluorescence (orange) began to appear at 4 hours in the WT and HET cells, indicative of the dispersion of MTP. This was more marked in cells from the HET mice than in those from the WT and HEMI mice. The images are representative of one experiment after 4, 8, and 16 hours of reculture in three separate experiments. (B) Higher magnification of the stained cells with exposure to 300 μM of H2O2 for 1 hour and reculture for 4 hours.
Effect of Oxidative Stress on Cytochrome c Leakage
The leakage of cytochrome c from mitochondria into the cytoplasm is known to activate caspases and initiate apoptosis.22,25 We therefore examined leakage of cytochrome c from the three groups of the RPE cell cultures derived from WT, HET, and HEMI after oxidative insult. The cells were exposed to 300 μM H2O2 for 1 hour and then incubated for 8 hours, and the cytochrome c in the cytoplasm was determined, as described in the Methods section. As seen in Figure 7, there was a marked increase in cytochrome c leakage in H2O2-treated RPE cells compared with that in the untreated control cells (shown only for WT). Moreover, there was a greater cytochrome c leakage in H2O2-treated cells derived from HET mice compared with those of WT and HEMI mice. The relative amounts of cytochrome c in the cytosolic fractions of H2O2-treated RPE cells from HET, WT, and HEMI mice were 1.4, 1.0, and 0.6 respectively (cytochrome c leakage in non-H2O2-treated cells was similar in the three groups; data not shown). Thus, the leakage of cytochrome c was directly related to the SOD2 levels in the RPE cell cultures.
Figure 7.
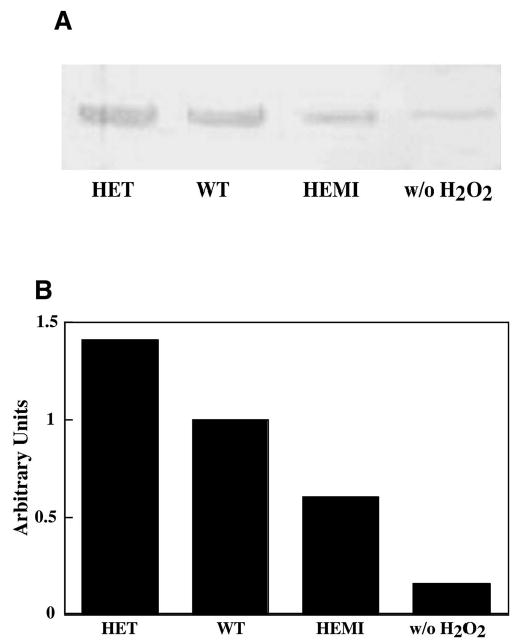
Effect of H2O2 on cytochrome c leakage from mitochondria in RPE cells with various levels of SOD2. Cells from each of the cultures were exposed to 300 μM of H2O2 for 1 hour and then recultured for 8 hours in normal medium. (A) Cytosolic fractions containing 10 μg protein were subjected to SDS-PAGE followed by Western-blot analysis with antibody to cytochrome c. (B) The enzyme level was quantified by integrated density scanning of the immunoblot membrane. The relative levels of cytochrome c in the cytosol from RPE derived from the HET, WT, and HEMI mice were 1.4, 1.0, and 0.6 respectively.
Discussion
This study demonstrates the critical role of the mitochondrial enzyme SOD2 in protecting RPE cells in culture against H2O2-induced apoptotic cell death. The protective effect of SOD2 against oxidation-induced apoptosis was directly related to the cellular level of the enzyme. The cells derived from heterozygous mice (partial knockout, +/−), with SOD2 levels of 60% compared with WT controls, showed a greater number of TUNEL-positive cells. In contrast, the cells derived from transgenic (hemizygous mice) that overexpressed, having an SOD2 level nearly 3.4 times that of WT mice, were resistant to the cytotoxic effect of H2O2. Apoptosis in these cells was suppressed or prevented when exposed to similar concentrations of H2O2. The studies also show that RPE cells exposed to H2O2 undergo alterations in mitochondrial transmembrane potential and induce cytochrome c leakage from the mitochondria, an event that precedes apoptotic cell death. These changes in the mitochondrial damage and cytochrome c leakage, in response to oxidative stress, are also associated with cellular levels of SOD2 in RPE cells established from the mutant and WT control mice.
Our findings, which show the protective role of SOD2 against oxidative stress, are consistent with a number of observations that suggest that mitochondrial damage might be involved in RPE cell death.4,26–28 In a cultured human RPE cell line, it was reported that oxidative challenge with t-butyl hydroperoxide (t-BHP) results in apoptotic cell death by triggering the mitochondrial permeability transition accompanied by mitochondrial swelling and release of intermembrane protein, including leakage of cytochrome c.26 It has also been reported that mitochondrial DNA deletions accumulated in the aging retina may contribute to the changes in macular function observed in aging and age-related maculopathy.29 As mitochondria are organelles that are vulnerable to oxidative stress, increased intracellular oxidative challenge in the aging process could be a cause of mitochondrial damage. Although multiple processes are involved in the pathogenesis of various retinal-degeneration diseases, alteration of mitochondrial function appears to play an important role in triggering apoptosis in RPE cells.
For a model, we used cells with different levels of SOD2 protein, which was varied by gene manipulation of the mice, because this protein is well known to be an important enzyme in maintaining mitochondrial function against oxidative stress.30 In an earlier study on human lens epithelial cells, in which SOD2 was up- and downregulated with plasmids containing sense and antisense vectors for cDNA for SOD2, it was demonstrated that the cells with higher enzyme levels were more resistant to the cytotoxic effect of H2O2, O2−, and UVB radiation as evidenced by decreased apoptosis.21,31 Furthermore, SOD2-deficient cells revealed dramatic mitochondrial damage, increased cytochrome c leakage, and activation of caspase 3 and enhanced apoptotic cell death.21
Although the functional role of SOD2 in protecting against oxidant-induced damage has been studied in animals or cells derived from knockout animals, this approach was not possible in the current investigation because the homozygous (Sod2−/−) mice null for the enzyme survive only for a short postnatal period (3 days in our hands). In previous reports16,17 Sod2-knockout mice survived for 5 to 18 days. In one study, it was reported that treatment of these mice with SOD2 mimetic compound briefly extended the lifespan of these animals, which exhibited progressive thinning of the retina with early effects on photoreceptors.32 Postnatal reduction of SOD2 expression in mouse eyes using ribozyme targeted against SOD2 mRNA to reduce expression of SOD2 was found to increase ROS generation from mitochondria leading to thinning of retinal layer and loss of axons and myelin in the optic nerve and ganglion cells.33 The aforementioned studies and those described in this communication clearly indicate the importance of SOD2 in the retina in protection against mitochondrial oxidative stress.
Because of our inability to breed Sod2-knockout mice, we studied the role of this enzyme in apoptotic cell death in RPE cells that were derived from mutant mice with different levels of SOD2. It was thought that this model, with heterozygous mice with roughly half the enzyme of the WT, might be analogous to decreased antioxidant status during the aging process and might give some insight into the ocular manifestation of SOD2 deficiency. There is little information regarding the ocular disease in partial Sod2-knockout mice because of the absence of a phenotype in this model. Although the animals grow and reproduce normally, when subjected to oxidative stress by arterial ischemia and reperfusion injury, there is exacerbation of cerebral infarction caused by overgenerated O2− in the mitochondrial compartment.34,35 There is another report that chronic mitochondrial oxidative stress in the Sod2 heterozygous mouse results in the age-related decline of mitochondrial function and induction of premature apoptosis in the liver.36 Similar mitochondrial decline was also observed in the mice with normal SOD2 levels, but it occurred later in life.36 Thus, it is possible that similar changes may occur in the ocular tissues in Sod2 heterozygous mouse during the aging process, leading to pathologic changes. In the present investigation, RPE cells derived from heterozygous mice had a normal growth rate in culture (data not shown), and there were no differences in mitochondrial transmembrane potential or apoptosis, unless the cells were subjected to oxidative stress by H2O2 exposure. Thus, under normal physiological conditions in vivo, the RPE cells with a partial deficiency of SOD2 (60% of that in WT) may be able to withstand the oxidative stress generated by ROS under normal metabolic conditions of the retina.
The RPE is known to play an important role in phagocytosis of the photoreceptor outer segment with the generation of H2O2.37,38 A decrease in SOD2 (and other antioxidants) may compromise the ability of the cells to remove or detoxify the continuous oxidative challenge from H2O2 and other ROS. The retina is particularly susceptible to oxidative insult, because of the high concentration of polyunsaturated fatty acid, such as docosahexaenoic acid (DHA) in the membrane of photoreceptor/pigment epithelium complex. DHA has been considered a potential target of lipid peroxidation induced by ROS in retina.39,40 Yet another source of oxidative challenge to the retina is the continuous generation of O2− radicals, since nearly 4% of oxygen consumed by the electron transport chain in mitochondria is thought to be converted to superoxide anions, which must be removed by SOD2 through the dismutation of O2− to H2O2. The retina is known to be one of the highest oxygen-consuming tissues of the human body because it requires a large amount of metabolic energy for visual processes. Because the RPE is located between the choroidal plexus and neural retina, which forms the outer blood–retinal barrier, it may be exposed to relatively high oxygen concentration in the retina and may be targeted by a large amount of ROS under physiological conditions. In the retina, deficiency of SOD2 enzyme in the mitochondria could increase generation of ROS and lead to perturbation of mitochondrial metabolism and the redox status in the cells. O2− is a major source of ROS produced by mitochondria, and these ROS possibly induce decline of mitochondrial function and increase levels of oxidative products, which positively correlate to aging. To the extent that similar changes may occur in the human retina during aging, we hypothesize that an accumulation of oxidative stress from ROS during the aging process may be one of the factors that contributes to AMD, a leading cause of legal blindness in the developed world. We therefore suggest that partial knockout mice and mice with overexpression of SOD2 protein could serve as a useful model for studying mitochondria-based AMD.
In the present investigation, H2O2 was used to induce oxidative challenge in cultured RPE cells. The mechanism by which SOD2 protects the cells against H2O2-induced oxidative stress is not clear, because this enzyme detoxifies O2− radicals rather than H2O2 directly. A possible explanation is that H2O2 can serve as a substrate for the iron-mediated (Fe2+) Fenton reaction and can generate O2−. Therefore, SOD2 may protect the cells by removing the O2− generated from the increased concentration of H2O2. Alternatively, O2− can reduce Fe3+ to form Fe2+, which can then participate in the Fenton reaction.31
In conclusion, we have shown that SOD2 plays a critical role in apoptosis in RPE cells in vitro and may have a similar function in vivo. We hypothesize that a decrease of this enzyme below some critical level during the aging process may be a significant contributory factor in AMD.
Acknowledgments
The authors thank Austra Liepa for the breeding and long-term care of the mutant mice.
Footnotes
Disclosure: E. Kasahara, None; L.-R. Lin, None; Y.-S. Ho, None; V.N. Reddy, None
Supported by National Eye Institute Grants EY00484 (VNR), Vision Core Grant EY07003, and National and Heart, Lung, and Blood Institute Grant HL56421 (Y-SH).
References
- 1.Ames BN, Shigenaga MK, Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci USA. 1993;90:7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cai J, Nelson KC, Wu M, et al. Oxidative damage and protection of the RPE. Prog Retin Eye Res. 2000;19:205–221. doi: 10.1016/s1350-9462(99)00009-9. [DOI] [PubMed] [Google Scholar]
- 3.Reddy VN, Giblin FJ, Lin LR, et al. Glutathione peroxidase-1 deficiency leads to increased nuclear light scattering, membrane damage, and cataract formation in gene-knockout mice. Invest Ophthalmol Vis Sci. 2001;42:3247–3255. [PubMed] [Google Scholar]
- 4.Liang FQ, Godley BF. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: a possible mechanism for RPE aging and age-related macular degeneration. Exp Eye Res. 2003;76:397–403. doi: 10.1016/s0014-4835(03)00023-x. [DOI] [PubMed] [Google Scholar]
- 5.Green WR, McDonnell PJ, Yeo JH. Pathologic features of senile macular degeneration. Ophthalmology. 1985;92:615–627. [PubMed] [Google Scholar]
- 6.Young RW. Pathophysiology of age-related macular degeneration. Surv Ophthalmol. 1987;31:291–306. doi: 10.1016/0039-6257(87)90115-9. [DOI] [PubMed] [Google Scholar]
- 7.Spraul CW, Lang GE, Grossniklaus HE. Morphometric analysis of the choroid, Bruch’s membrane, and retinal pigment epithelium in eyes with age-related macular degeneration. Invest Ophthalmol Vis Sci. 1996;37:2724–2735. [PubMed] [Google Scholar]
- 8.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 9.Wallace DC. Mitochondrial genetics: a paradigm for aging and degenerative diseases? Science. 1992;256:628–632. doi: 10.1126/science.1533953. [DOI] [PubMed] [Google Scholar]
- 10.Ho YS, Crapo JD. Isolation and characterization of complementary DNAs encoding human manganese-containing superoxide dismutase. FEBS Lett. 1988;229:256–260. doi: 10.1016/0014-5793(88)81136-0. [DOI] [PubMed] [Google Scholar]
- 11.Karbowski M, Kurono C, Wozniak M, et al. Free radical-induced megamitochondria formation and apoptosis. Free Radic Biol Med. 1999;26:396–409. doi: 10.1016/s0891-5849(98)00209-3. [DOI] [PubMed] [Google Scholar]
- 12.Liu L, Trimarchi JR, Keefe DL. Involvement of mitochondria in oxidative stress-induced cell death in mouse zygotes. Biol Reprod. 2000;62:1745–1753. doi: 10.1095/biolreprod62.6.1745. [DOI] [PubMed] [Google Scholar]
- 13.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 14.Jonas EA, Hoit D, Hickman JA, et al. Modulation of synaptic transmission by the BCL-2 family protein BCL-xL. J Neurosci. 2003;23:8423–8431. doi: 10.1523/JNEUROSCI.23-23-08423.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parone P, Priault M, James D, et al. Apoptosis: bombarding the mitochondria. Essays Biochem. 2003;39:41–51. doi: 10.1042/bse0390041. [DOI] [PubMed] [Google Scholar]
- 16.Li Y, Huang TT, Carlson EJ, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 17.Lebovitz RM, Zhang H, Vogel H, et al. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci USA. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams MD, Van Remmen H, Conrad CC, et al. Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J Biol Chem. 1998;273:28510–28515. doi: 10.1074/jbc.273.43.28510. [DOI] [PubMed] [Google Scholar]
- 19.Ho YS, Vincent R, Dey MS, et al. Transgenic models for the study of lung antioxidant defense: enhanced manganese-containing superoxide dismutase activity gives partial protection to B6C3 hybrid mice exposed to hyperoxia. Am J Respir Cell Mol Biol. 1998;18:538–547. doi: 10.1165/ajrcmb.18.4.2959. [DOI] [PubMed] [Google Scholar]
- 20.Chen Z, Siu B, Ho YS, et al. Overexpression of MnSOD protects against myocardial ischemia/reperfusion injury in transgenic mice. J Mol Cell Cardiol. 1998;30:2281–2289. doi: 10.1006/jmcc.1998.0789. [DOI] [PubMed] [Google Scholar]
- 21.Reddy VN, Kasahara E, Hiraoka M, et al. Effects of variation in superoxide dismutases (SOD) on oxidative stress and apoptosis in lens epithelium. Exp Eye Res. 2004;79:859–868. doi: 10.1016/j.exer.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 22.Yang J, Liu X, Bhalla K, et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 23.Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 24.Jiang X, Wang X. cytochrome C-mediated apoptosis. Annu Rev Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- 25.Liu X, Kim CN, Yang J, et al. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 26.Cai J, Wu M, Nelson KC, et al. Oxidant-induced apoptosis in cultured human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1999;40:959–966. [PubMed] [Google Scholar]
- 27.Suter M, Reme C, Grimm C, et al. Age-related macular degeneration. The lipofusion component N-retinyl-N-retinylidene ethanolamine detaches proapoptotic proteins from mitochondria and induces apoptosis in mammalian retinal pigment epithelial cells. J Biol Chem. 2000;275:39625–39630. doi: 10.1074/jbc.M007049200. [DOI] [PubMed] [Google Scholar]
- 28.King A, Gottlieb E, Brooks DG, et al. Mitochondria-derived reactive oxygen species mediate blue light-induced death of retinal pigment epithelial cells. Photochem Photobiol. 2004;79:470–475. doi: 10.1562/le-03-17.1. [DOI] [PubMed] [Google Scholar]
- 29.Barron MJ, Johnson MA, Andrews RM, et al. Mitochondrial abnormalities in ageing macular photoreceptors. Invest Ophthalmol Vis Sci. 2001;42:3016–3022. [PubMed] [Google Scholar]
- 30.Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 31.Matsui H, Lin LR, Ho YS, et al. The effect of up- and downregulation of MnSOD enzyme on oxidative stress in human lens epithelial cells. Invest Ophthalmol Vis Sci. 2003;44:3467–3475. doi: 10.1167/iovs.02-0830. [DOI] [PubMed] [Google Scholar]
- 32.Sandbach JM, Coscun PE, Grossniklaus HE, et al. Ocular pathology in mitochondrial superoxide dismutase (Sod2)-deficient mice. Invest Ophthalmol Vis Sci. 2001;42:2173–2178. [PubMed] [Google Scholar]
- 33.Qi X, Lewin AS, Hauswirth WW, et al. Optic neuropathy induced by reductions in mitochondrial superoxide dismutase. Invest Ophthalmol Vis Sci. 2003;44:1088–1096. doi: 10.1167/iovs.02-0864. [DOI] [PubMed] [Google Scholar]
- 34.Murakami K, Kondo T, Kawase M, et al. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J Neurosci. 1998;18:205–213. doi: 10.1523/JNEUROSCI.18-01-00205.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fujimura M, Morita-Fujimura Y, Kawase M, et al. Manganese superoxide dismutase mediates the early release of mitochondrial cytochrome C and subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. J Neurosci. 1999;19:3414–3422. doi: 10.1523/JNEUROSCI.19-09-03414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kokoszka JE, Coskun P, Esposito LA, et al. Increased mitochondrial oxidative stress in the Sod2 (+/−) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc Natl Acad Sci USA. 2001;98:2278–2283. doi: 10.1073/pnas.051627098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tate DJ, Jr, Miceli MV, Newsome DA. Phagocytosis and H2O2 induce catalase and metallothionein gene expression in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1995;36:1271–1279. [PubMed] [Google Scholar]
- 38.Tate DJ, Miceli MV, Newsome DA. Expression of metallothionein isoforms in human chorioretinal complex. Curr Eye Res. 2002;24:12–25. doi: 10.1076/ceyr.24.1.12.5426. [DOI] [PubMed] [Google Scholar]
- 39.Organisciak DT, Darrow RM, Jiang YL, et al. Retinal light damage in rats with altered levels of rod outer segment docosahexaenoate. Invest Ophthalmol Vis Sci. 1996;37:2243–2257. [PubMed] [Google Scholar]
- 40.Mukherjee PK, Marcheselli VL, Serhan CN, et al. Neuroprotectin D1: a docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc Natl Acad Sci USA. 2004;101:8491–8496. doi: 10.1073/pnas.0402531101. [DOI] [PMC free article] [PubMed] [Google Scholar]