

Abstract
Extracellular adenosine has been widely implicated in adaptive responses to hypoxia. The generation of extracellular adenosine involves phosphohydrolysis of adenine nucleotide intermediates, and is regulated by the terminal enzymatic step catalyzed by ecto-5′-nucleotidase (CD73). Guided by previous work indicating that hypoxia-induced vascular leakage is, at least in part, controlled by adenosine, we generated mice with a targeted disruption of the third coding exon of Cd73 to test the hypothesis that CD73-generated extracellular adenosine functions in an innate protective pathway for hypoxia-induced vascular leakage. Cd73 −/− mice bred and gained weight normally, and appeared to have an intact immune system. However, vascular leakage was significantly increased in multiple organs, and after subjection to normobaric hypoxia (8% O2), Cd73 −/− mice manifested fulminant vascular leakage, particularly prevalent in the lung. Histological examination of lungs from hypoxic Cd73 −/− mice revealed perivascular interstitial edema associated with inflammatory infiltrates surrounding larger pulmonary vessels. Vascular leakage secondary to hypoxia was reversed in part by adenosine receptor agonists or reconstitution with soluble 5′-nucleotidase. Together, our studies identify CD73 as a critical mediator of vascular leakage in vivo.
Keywords: adenosine, inflammation, edema, endothelium, knockout
Introduction
Acute increases in vascular leakage to macromolecules closely coincide with tissue injury of many etiologies, and can result in fluid loss, edema, and organ dysfunction (1–3). Macromolecule transit across blood vessels is tightly controlled, whereby relatively low macromolecular permeability is essential for maintenance of a physiologically optimal equilibrium between intravascular and extravascular compartments (4, 5). The normal vasculature is generally quite impermeable to large macromolecules, and the predominant barrier (∼90%) to movement of macromolecules across a blood vessel wall is presented by the endothelium (2, 6). Passage of macromolecules across a cellular monolayer can occur via either a paracellular route (i.e., between cells) or a transcellular route (i.e., through cells). In conditions of vascular leakage, macromolecules such as albumin (molecular mass ∼70 kD) appear to cross the cell monolayer by passing between adjacent endothelial cells (i.e., paracellular), although some degree of transcellular passage may also occur (7, 8). Endothelial macromolecular permeability is inversely related to macromolecule size, and such permeability is also dependent on the tissue of origin. For example, endothelial cells in the cerebral circulation (i.e., blood–brain barrier) demonstrate an exceptionally low permeability (9, 10). Endothelial permeability may increase markedly upon exposure to a variety of inflammatory compounds (e.g., histamine, thrombin, reactive oxygen species, leukotrienes, bacterial endotoxins) or adverse conditions (e.g., hypoxia, ischemia; references 2, 11).
Previous studies indicated that extracellular nucleotide metabolites, predominantly adenosine, may trigger an endogenous protective mechanism during hypoxia and ischemia (12–15). Ecto-5′-nucleotidase (5′-NT, CD73) is a membrane-bound glycoprotein that functions to hydrolyze extracellular nucleoside monophosphates into bioactive nucleoside intermediates (14). Surface-localized CD73 converts AMP into adenosine, which in turn can activate transmembrane adenosine receptors or can be internalized through dipyridamole-sensitive carriers (14). Adenosine generated by CD73 expressed on barrier cell types (e.g., endothelia, epithelia) has been shown to result in such diverse endpoints as regulation of endothelial permeability (16), attenuation of neutrophil adhesion (17), and stimulation of epithelial electrogenic chloride secretion (responsible for mucosal hydration; reference 18).
Endothelial cells of many origins express CD73 constitutively. The primary function attributed to endothelial CD73 has been catabolism of extracellular nucleotides, although CD73 may also mediate lymphocyte binding under some circumstances (19). Rather little is known about the regulation of endothelial CD73 expression, and whether this molecule contributes to the regulation of endothelial permeability. Given that adenosine receptor activation (via adenosine liberated from CD73) elevates intracellular cAMP, and that elevated cAMP in endothelia promotes barrier function (2, 20), we considered the possibility that endothelial CD73 functions to regulate permeability. In support of this hypothesis, it has been shown that CD73 expression is regulated by hypoxia (21–24), primarily by hypoxia-inducible factor-1 (21). However, there is no direct in vivo evidence implicating CD73 in vascular barrier function.
In the present studies, we sought to determine whether CD73 functionally regulates murine vascular leakage under basal conditions or during hypoxia. Initial studies using the selective CD73 inhibitor α,β-methylene ADP (APCP; reference 25) revealed that inhibition of CD73 significantly increases both basal and hypoxia-induced vascular leakage. To more selectively address this issue, we generated Cd73 −/− mice and used these mice to examine vascular leakage. Such studies identified defects in vascular barrier regulation, particularly in the lung. Treatment of Cd73 −/− mice with the adenosine receptor agonist 5′-(N-ethylcarboxamido)-adenosine (NECA) or reconstitution with purified 5′-NT resulted in partial reversal of these defects. These studies identify CD73 as a critical regulator of vascular leakage in vivo.
Materials and Methods
Generation of Cd73-deficient Mice.
P1 bacteriophage clones containing SV129 Cd73 genomic DNA were purchased from Genome Systems. An 8.9-kb BamHI restriction fragment containing exon 3 was subcloned into Pzero (Invitrogen) to generate PzeroBamHI 81 (see Fig. 1 A). A targeting vector was constructed by ligating a 1-kb fragment (short arm) amplified by PCR from intronic sequence upstream of exon 3 and a 4.2-kb EcoRI fragment (long arm) downstream of exon 3 into pBSneo, a modified version of pBluescript II KS (+/−) with a neomycin resistance cassette directionally ligated into BamHI and EcoRI sites (a gift from T. Mak, Ontario Cancer Institute, Toronto, Canada). The 1-kb fragment contained engineered SacI sites to permit ligation into the SacI site of the multicloning site of pBSneo. Both Cd73 genomic fragments were in the antisense orientation with respect to neo. Targeting vector DNA was CsCl purified before electroporation into embryonic stem (ES) cells.
Figure 1.

Restriction maps of CD73 genomic DNA, targeting vector, and recombined allele. (A) CD73 genomic DNA, including exon 3 and intronic sequences used to construct the short (1 kb) and long arms (4.2 kb) of the targeting vector. (B) CD73 targeting vector showing the antisense orientation of the CD73 sequences relative to the neo cassette. (C) Recombined, gene-targeted CD73 allele. The flanking probe used for Southern blots is shown, as are the positions of the PCR primers (arrows) used for screening the ES cell clones. (D) Southern blot of genomic DNA from Cd73 +/+, Cd73 −/+, and Cd73 −/− mice. Genomic DNA was purified from mouse tails, digested with BamHI, and subjected to Southern blotting by conventional methods. The probe was from intronic sequence 3′ to the long arm as shown in C. The wild-type BamHI fragment is 8.9 kb, whereas that from the gene-targeted mice is 7.4 kb. (E) Northern blot of kidney RNA from Cd73 +/+ and Cd73 −/− mice. Triplicate Northern blots were performed with 15 μg of kidney RNA in each lane. Blots were hybridized to the following probes: exon 2 (5′ probe), exon 3, or exon 4 (3′ probe). Ethidium bromide staining was used to confirm equal RNA loading.
XhoI-linearized targeting vector was electroporated into CJ7 ES cells (a gift from T. Sato, University of Texas Southwestern Medical Center, Dallas, TX) at a ratio of 30 μg vector DNA/5 × 106 ES cells. G418 was added to the media at 300 μg/ml after 24 h of culture. Clones were picked after 7–9 d, expanded, and frozen according to standard protocols. Clones were screened by PCR to detect the presence of the targeted allele (see Fig. 1 C). The forward PCR primer, 5′-AAGGAGGGGTGCATCTTGCTATTC-3′, was from intronic sequence 5′ to that comprising the short arm of the targeting vector and the reverse primer, 5′-CCAGCTCATTCCTCCCACTCATG-3′, was from within the neo cassette. Two independent clones containing a gene-targeted allele were generated. Gene targeting was confirmed by Southern blotting with BamHI-digested genomic DNA and a probe from intronic sequence 3′ to the long arm (see Fig. 1 D). The gene-targeted allele generated a 7.4-kb band that was easily distinguished from the 8.9-kb wild-type band. Blastocyst injections were performed at the University of Cincinnati Gene-Targeted Mouse Service with both ES cell clones. Chimeras were obtained from both of them, but germline transmission was obtained with only one, 4G11. A single female Cd73 +/− mouse was obtained and bred to C57BL/6 males. Her offspring were genotyped by PCR on tail DNA to detect the neo cassette. Cd73 +/− mice were bred to each other to obtain Cd73 −/− mice and control Cd73 +/+ and Cd73 +/− littermates. Cd73 +/− mice were backcrossed to C57BL/6 mice for six generations for use in hypoxia experiments.
Immunofluorescent Staining.
CD73 expression on hematopoietic cells was evaluated by immunofluorescent staining with purified monoclonal antibody TY/23 (26) followed by PE-goat anti–rat IgG (BD Biosciences). Rat IgG2a was used as a negative control. Immunofluorescence was evaluated with a FACSCalibur and CELLQuest software (Becton Dickinson).
Ecto-5′-NT Enzyme Assays.
Ecto-5′-NT enzyme activity was evaluated by measuring the conversion of [14C]IMP to [14C]inosine as described previously (27). APCP (Sigma-Aldrich) was used as a specific inhibitor of CD73 (25). Thus, ecto-5′-NT enzyme activity was calculated as the portion of the total IMP-hydrolyzing activity that could be inhibited by APCP. The results were expressed in nmoles IMP hydrolyzed/h/mg protein.
In subsets of experiments, CD73 activity was measured by quantifying the conversion of etheno-AMP (E-AMP) to etheno-adenosine (E-Ado) by reverse phase HPLC as described previously (16). E-AMP and E-Ado were eluted with a 0–50% methanol/H2O gradient mobile phase (2 ml/min over 10 min). Absorbance was monitored at 260 nm, and ultraviolet absorption spectra were obtained at chromatographic peaks. CD73 activity was expressed as percent E-AMP conversion in this time frame. This methodology of assaying phosphohydrolysis was validated between samples using conversion of [14C-IMP] to [14C-inosine] (see previous paragraph; unpublished data).
In Vivo Hypoxia Model.
Total organ vascular leakage was assessed by intravascular administration of Evan's blue (Sigma-Aldrich) as described previously (23). In brief, Evan's blue (0.2 ml of 0.5% in PBS) was injected intravenously into Cd73 −/− or littermate control Cd73 +/+ animals that were exposed to normobaric hypoxia (8% O2, 92% N2) or room temperature air for 4 h (n = 4–6 animals per condition). After experimental exposure, the animals were killed, and the colon, skeletal muscle (gluteus maximus), kidney, brain, heart, liver, and lungs were harvested. Evan's blue concentrations in organs were quantified after formamide extraction (55°C for 2 h) by measuring absorbances at 610 nm with subtraction of reference absorbances at 450 nm. This protocol was in accordance with National Institutes of Health (NIH) guidelines for use of live animals and was approved by the Institutional Animal Care and Use Committee at Brigham and Women's Hospital.
In subsets of experiments, mice were reconstituted with 5′-NT purified from Crotalus atrox venom (Sigma-Aldrich). Pilot dosing experiments revealed that 5′-NT could be used at concentrations as high as 500 U/kg i.p. without deleterious effects. After administration of 5′-NT, animals were subjected to normoxia or hypoxia, and examined for vascular leakage using Evan's blue as described before.
In other experiments, mice were administered the adenosine A2A receptor antagonist 4-(2-[7-amino-2-(2-furyl)[1,2,4]triazolo[2,3-a][1,3,5]triazin-5-ylamino]ethyl)phenol (ZM 241385; Tocris Cookson Inc.; dosage of 1 mg/kg i.p. plus 1 mg/kg s.c.), adenosine A2B receptor antagonist N-(4-cyanophenyl)-2-[4-(2,3,6,7-tetrahydro-2,6-dioxo-1,3-dipropyl-1H-purin-8-yl)-phenoxy]acetamide (MRS1754; a gift from K. Jacobson, Molecular Recognition Section, NIH, Bethesda, MD; dosage of 1 mg/kg i.p. plus 1 mg/kg s.c.) or the nonmetabolizable adenosine analogue NECA (Sigma-Aldrich; dosage of 0.1 mg/kg i.p. plus 0.1 mg/kg s.c. based on previous work [28]). After administration of drug, animals were subjected to normoxia or hypoxia, as indicated, and examined for vascular leakage using Evan's blue as described before. For assessment of pulmonary edema, lungs were collected, weighed, and dried by speed-vac. Weight differences before and after drying were used to calculate lung water content.
Data Analysis.
Permeability data were compared by two-factor analysis of variance (ANOVA), or by Student's t test where appropriate. Values are expressed as the mean ± SD from at least three separate experiments.
Results
Generation of Cd73-deficient Mice.
Previous studies implicated extracellular adenosine in a protective pathway regulating vascular leakage in multiple organs during hypoxia/ischemia, both in vitro and in vivo (16, 23). To directly address the role of CD73 in generating extracellular adenosine, we generated a Cd73-deficient mouse line. Our strategy was to replace exon 3 with a neomycin resistance cassette (Fig. 1), as our earlier work showed that exon 3 contained two crucial histidine residues required for ecto-5′-NT enzyme activity (29). Using conventional gene targeting methodology, a single Cd73 +/− female was obtained. She was bred to C57BL/6 males to obtain additional Cd73 −/+ mice, and these were bred to each other to obtain Cd73 −/−, −/+, and +/+ mice. Offspring were obtained in the expected 1:2:1 ratio.
CD73 Expression in Cd73 Gene-targeted Mice.
CD73 expression was assessed in gene-targeted mice by the three following methods: Northern blotting, immunofluorescent staining of lymphoid cells, and ecto-5′-NT enzyme assays on kidney extracts. Fig. 1 E shows a Northern blot of kidney RNA from Cd73 +/+ and Cd73 −/− mice. Three probes were used. The exon 3 probe showed an absence of RNA containing this exon from Cd73 −/− mice, as expected. However, probes from both the 5′ and 3′ sides of exon 3 revealed low level expression (∼10% of wild type) of an altered transcript, from which the neo cassette may have been removed by splicing. Even though such splicing should not produce a frame shift, we believe it is unlikely that this transcript could lead to synthesis of a stable protein, as exon 3 contains two out of the four histidine residues that interact with the two Zn+2 ions in each CD73 molecule (29) and, thus, is likely crucial for maintaining the three-dimensional structure of the protein. A preliminary Western blotting experiment with our goat anti-CD73 antibody (30) failed to reveal a smaller form of CD73 in kidney extracts from CD73 gene-targeted mice as would be expected if the 65 amino acids in exon 3 had been deleted (unpublished data).
Cells from blood, thymus, spleen, lymph nodes, and bone marrow from Cd73 +/+ and Cd73 −/− mice were stained for CD73 expression with monoclonal antibody TY/23 (Fig. 2). The results demonstrated that Cd73 −/− mice lacked detectable cell surface expression of CD73 protein on any of their hematopoietic cells. The results of the immunofluorescent staining were confirmed with ecto-5′-NT enzyme assays on extracts of colon, lung, heart, skeletal muscle, brain, liver, and kidney (Table I). As expected, enzyme activity was nearly undetectable in tissue extracts from Cd73 −/− mice. Interestingly, these assays also revealed a 46-fold variation in CD73 5′-NT enzyme activity in different tissues from wild-type animals (e.g., compare colon vs. skeletal muscle in Table I). Table I also shows the levels of nucleotide hydrolyzing enzyme activities in each tissue that cannot be attributed to CD73 (i.e., nucleotidase and/or phosphatase activity that is not inhibitable by APCP). Such residual enzyme activity was not distiguishable between wild-type and CD73-deficient mice (not depicted) and varied by >20-fold in different tissues (Table I), with colon having the highest level and skeletal muscle the lowest (undetectable) level.
Figure 2.

CD73 expression and function on leukocytes from Cd73 +/+ and Cd73 −/− mice. CD73 expression was evaluated on leukocytes from lymph node, spleen, peripheral blood, bone marrow, and thymus from Cd73 +/+ (left) and Cd73 −/− (right) mice with monoclonal antibody TY/23 + PE–goat anti–rat IgG. Staining with an isotype-matched control antibody is shown in the shaded histograms.
Table I.
Comparison of 5′-NT Levels in Various Tissues
| Tissue activity |
APCP-inhibitable 5′-NT activity |
Non-APCP 5′-NT |
|
|---|---|---|---|
| Cd73 +/+ | Cd73 −/− | Pooleda | |
|
|
nmol/h/mg ± SEM
|
nmol/h/mg ± SEM
|
|
| Colon | 1,523 ± 116 | 9 ± 3b | 237 ± 41 |
| Lung | 248 ± 56 | 14 ± 4b | 73 ± 4 |
| Heart | 171 ± 64 | 1 ± 0.1b | 33 ± 5 |
| Muscle | 33 ± 5 | 6 ± 0.9b | undetectable |
| Brain | 891 ± 128 | 10 ± 6b | 89 ± 16 |
| Liver | 347 ± 26 | 1 ± 0.1b | 40 ± 4 |
| Kidney | 799 ± 158 | 6 ± 4b | 90 ± 5 |
Non-APCP inhibitable 5′-NT activity, pooled from both Cd73 +/+ and Cd73 −/− animals.
Indicates significantly different than wild type (P < 0.001) for n = 7–9 mice per group.
Evaluation of the Immune System in Cd73−/− Mice.
CD73 expression increases during lymphocyte development in both mice (26) and humans (31). Furthermore, CD73 expression is markedly reduced on the lymphocytes of patients with a variety of immunodeficiency diseases (31). These observations suggested that CD73 might be important either for lymphocyte maturation or for the function of mature lymphocytes. Therefore, Cd73 +/+ and Cd73 −/− mice were examined for the cellularity and composition of the lymphoid organs at various ages up to 1 yr. No abnormalities were found (unpublished data). Lymphocyte function was also evaluated. Lymphocytes from Cd73 −/− mice showed normal proliferative responses to anti-CD3 and LPS and made normal antibody responses to immunization with ovalbumin (unpublished data). No tumors were observed. In short, Cd73 −/− mice appear to have a normal immune system and are healthy when housed in a specific pathogen-free animal facility.
Vascular Leakage in Cd73−/− Mice.
Based on previous findings indicating that extracellular adenosine is critical for control of vascular leakage in vitro and in vivo (16, 23), we hypothesized that CD73 deficiency would significantly influence vascular leakage in vivo. To this end, we used normobaric hypoxia as a stimulus for vascular leakage in multiple organs (23). During the initial period of hypoxia (∼30 min), Cd73 −/− mice were more lethargic and less compliant than their wild-type littermates. Nonetheless, all wild-type and Cd73 −/− mice recovered and survived the 4-h period of hypoxia. Comparative analysis of basal permeability (i.e., in normoxia) using Evan's blue revealed that, in three out of seven organs studied (lung, liver, and skeletal muscle), vascular leakage was significantly increased in Cd73 −/− mice (Fig. 3 A, P < 0.025). Consistent with previous reports (23, 32), normobaric hypoxia (8% O2, 92% N2) significantly increased leakage of highly vascular organs of wild-type mice, including the colon, lung, liver, muscle, heart, and kidney (Fig. 3 B, P < 0.025 by ANOVA). Furthermore, under hypoxic conditions, vascular leakage was increased (compared with Cd73 +/+) in all Cd73 −/− organs examined (P < 0.01 by ANOVA), with the notable exception of the brain. Changes in overall vascular leakage (Evan's blue extravasation) between Cd73 +/+ and Cd73 −/− mice under both normoxic and hypoxic conditions were also evident in open abdominal images taken at necropsy (Fig. 3 C). These findings support the hypothesis that CD73 is a critical determinant for permeability changes associated with adenine nucleotide metabolism, particularly in the posthypoxic vasculature.
Figure 3.

Vascular leakage in Cd73-deficient mice in vivo. Cd73 −/− mice (black bars) and age-, weight-, and gender-matched littermate controls (white bars) were administered intravenous Evan's blue (0.2 ml of 0.5% in PBS per mouse) and exposed to room temperature air (A) or normobaric hypoxia (B, 8% O2, 92% N2) for 4 h. Animals were killed, and the colon (Co), lung (Lg), liver (Lv), muscle (Mu), heart (Ht), kidney (Kd), and brain (Br) were harvested. Evan's blue concentrations in organs were quantified as described in Materials and Methods. Data are expressed as mean ± SD Evan's blue OD/50 mg wet tissue and are pooled from four to six animals per condition, where * indicates P < 0.025 between Cd73 +/+ and Cd73 −/− mice and # indicates P < 0.025 between hypoxia and normoxia. (C) Images of abdominal dissections from wild-type and Cd73 −/− mice subjected to normoxia and hypoxia for 4 h.
Influence of the CD73 Inhibitor APCP on Vascular Leakage.
To verify that CD73 catalyzes a crucial metabolic step for maintenance of vascular barrier during hypoxia, we assessed the consequences of the highly selective CD73 inhibitor APCP (20 mg/kg i.p.) on Evan's blue accumulation in vivo in multiple organs from wild-type mice subjected to normoxia (room temperature air) or normobaric hypoxia (8% O2, 92% N2) for 4 h. This protocol resulted in an 81.2 ± 8.5% decrease in circulating (serum) 5′-NT enzyme activity after 4 h (Fig. 4 C, P < 0.01). As shown in Fig. 4 A, permeability of the lung and colonic vasculature to Evan's blue was increased in normoxic mice preexposed to APCP (P < 0.05 compared with vehicle controls), suggesting that CD73 contributes to basal barrier function in these organs. APCP also increased vascular leakage in colon, lung, liver, heart, and kidney of mice subjected to normobaric hypoxia (Fig. 4 B, P < 0.025 by ANOVA compared with vehicle controls). Moreover, the APCP-mediated increases in permeability were amplified compared with those observed under normoxic conditions (P < 0.025 by ANOVA).
Figure 4.

Influence of CD73 inhibition on vascular leakage in vivo. Age-, weight-, and gender-matched mice were administered APCP (20 mg/kg i.p.) or an equal volume of PBS followed by intravenous Evan's blue solution (0.2 ml of 0.5% in PBS per mouse) and exposed to room temperature air (A) or to normobaric hypoxia (B, 8% O2, 92% N2) for 4 h. Animals were killed and the colon (Co), lung (Lg), liver (Lv), muscle (Mu), heart (Ht), kidney (Kd), and brain (Br) were harvested. Evan's blue concentrations in organs were quantified as described in Materials and Methods. Data are expressed as mean ± SD Evan's blue OD/50 mg wet tissue and are pooled from four to six animals per condition where * indicates P < 0.05 in comparisons between APCP and PBS, and # indicates P < 0.025 between hypoxia and normoxia. (C) HPLC analysis of CD73 5′-NT enzyme activity (conversion of 1 mM E-AMP to E-Ado) in serum harvested at 4 h from normoxic animals administered −APCP (top) or +APCP (bottom).
These results were further confirmed by a loss of APCP response in Cd73 −/− mice. Indeed, administration of APCP to hypoxic Cd73 −/− mice resulted in no significant differences in Evan's blue permeability compared with hypoxic Cd73 −/− animals administered PBS; i.e., values for Evan's blue accumulation in the colon were 1.7 ± 0.4 and 1.9 ± 0.5 OD U/50 mg tissue for PBS and APCP, respectively (p-value was NS). Paired experiments in wild-type animals revealed a prominent influence by APCP; values for Evan's blue accumulation in the colon were 0.7 ± 0.2 and 2.2 ± 0.7 OD U/50 mg tissue for animals administered PBS and APCP, respectively (P < 0.01). Similar results were observed in the lung, liver, and kidney (unpublished data). Together, these results verify our findings in Cd73 −/− mice and indicate that inhibition of CD73 results in increased vascular leakage that is significantly amplified in hypoxia.
Reconstitution of Cd73−/− Mice with 5′-NT.
As proof of principle for the assertion that CD73 plays an important role in regulation of vascular leakage during hypoxia, Cd73 −/− mice were reconstituted with 5′-NT purified from C. atrox venom (500 U/kg i.p.) and subjected to normoxia or normobaric hypoxia. This dose of 5′-NT resulted in a 4.1 ± 0.8-fold increase in plasma 5′-NT activity (P < 0.025; unpublished data). As shown in Fig. 5, this treatment significantly enhanced vascular barrier function in all animals tested (P < 0.05 by ANOVA; Fig. 5, A–C for most organs compared with vehicle control). Moreover, exogenous 5′-NT dramatically rescued increased vascular leakage observed in Cd73 −/− animals exposed to hypoxia in all tissues examined except brain (Fig. 5 D, P < 0.025 by ANOVA). Together, these studies provide strong evidence that CD73 is a critical control point for vascular barrier regulation, particularly during episodes of hypoxia.
Figure 5.

Vascular leakage and reconstitution of Cd73 −/− mice with 5′-NT. Cd73 −/− mice (B and D) and age-, weight-, and gender-matched littermate controls (A and C) were administered 5′-NT purified from C. atrox venom (500 U/kg i.p.; black bars) or PBS (white bars) followed by intravenous Evan's blue solution (0.2 ml of 0.5% in PBS per mouse) and exposed to room temperature air (A and B) or normobaric hypoxia (C and D, 8% O2, 92% N2) for 4 h. Animals were killed and the colon (Co), lung (Lg), liver (Lv), muscle (Mu), heart (Ht), kidney (Kd), and brain (Br) were harvested. Evan's blue concentrations in organs were quantified as described in Materials and Methods. Data are expressed as mean ± SD Evan's blue OD/50 mg wet tissue and are pooled from four to six animals per condition where, in comparisons between 5′-NT and PBS, * indicates P < 0.05 and # indicates P < 0.025.
Influence of Exogenous Adenosine Receptor Agonists on Vascular Leakage.
Our results and those of others suggest that CD73 promotes vascular barrier function by generating adenosine to engage vascular adenosine receptors. Initially, we determined the relative importance of individual adenosine A2 receptors on vascular leakage, using the lung as a model organ. As shown in Fig. 6, administration of either MRS 1754 (selective antagonist for the A2B receptor, dosage of 1 mg/kg i.p. plus 1 mg/kg s.c.) or ZM 241385 (selective antagonist for the A2A receptor, dosage of 1 mg/kg i.p. plus 1 mg/kg s.c.) significantly increased both pulmonary vascular leakage (measured as Evan's blue extravasation) and pulmonary edema (measured as lung water content) in normoxia, and particularly in hypoxia. Such results indicate that both A2A and A2B receptors contribute to pulmonary vascular leakage in hypoxia. Based on these findings, we determined whether the general adenosine receptor agonist NECA could rescue defects in vascular leakage observed in Cd73 −/− mice during hypoxia. As shown in Fig. 7, administration of NECA (0.1 mg/kg i.p. plus 0.1 mg/kg s.c.) decreased vascular leakage in both hypoxia and normoxia, particularly in the colon, lung, and liver. This difference was notably more pronounced in Cd73 −/− mice subjected to hypoxia (P < 0.025 by ANOVA), thus supporting our hypothesis that CD73-generated extracellular adenosine promotes barrier function during hypoxia, likely through activation of A2A and A2B receptors. We acknowledge that ZM241385 can also bind to the A2B receptor at high concentrations; thus, it is possible that the A2B receptor is the predominant receptor in adenosine-mediated protection from vascular leakage in our studies.
Figure 6.
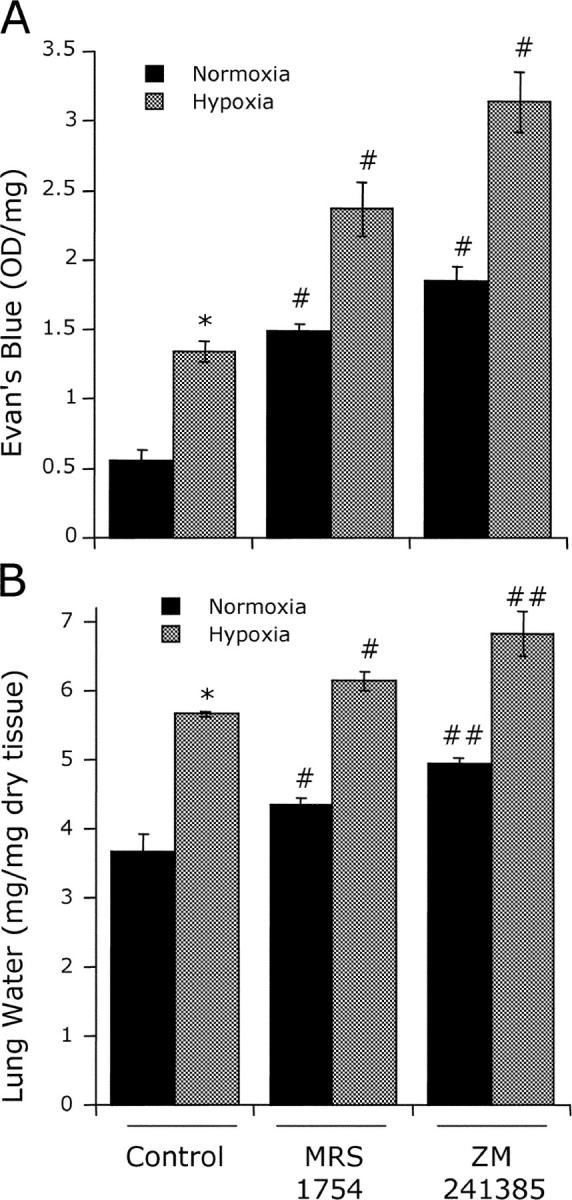
Influence of adenosine receptor antagonists on hypoxia-induced vascular leakage. Wild-type mice were administered either PBS (Control), the A2B receptor antagonist MRS1754 (1 mg/kg i.p. plus 1 mg/kg s.c.) or the A2A receptor antagonist ZM241385 (1 mg/kg i.p. plus 1 mg/kg s.c.) followed by intravenous Evan's blue solution (0.2 ml of 0.5% in PBS per mouse) and exposed to room air (black bars) or normobaric hypoxia (gray bars, 8% O2, 92% N2) for 4 h. Animals were killed, and lungs were harvested. (A) Evan's blue concentrations in organs were quantified as described in Materials and Methods. Data are expressed as mean ± SD Evan's blue OD/50 mg wet tissue and are pooled from four animals per condition where * indicates P < 0.01 between normoxia and hypoxia and # indicates P < 0.01 between treatment and control. (B) Assessment of lung water content. Data are expressed as mean ± SD mg H2O/mg dry tissue, and are pooled from four animals per condition where, in comparisons between hypoxia and normoxia, * indicates P < 0.01, # indicates P < 0.05 between treatment and control, and ## indicates P < 0.025 between treatment and control.
Figure 7.

Influence of the adenosine receptor agonist NECA on vascular leakage of Cd73 −/− mice. Cd73 −/− mice (B and D) and age-, weight-, and gender-matched littermate controls (A and C) were administered the adenosine analogue NECA (0.1 mg/kg i.p. plus 0.1 mg/kg s.c., black bars) or PBS (white bars) followed by intravenous Evan's blue solution (0.2 ml of 0.5% in PBS per mouse) and exposed to room temperature air (A and B) or normobaric hypoxia (C and D, 8% O2, 92% N2) for 4 h. Animals were killed, and the colon (Co), lung (Lg), liver (Lv), muscle (Mu), heart (Ht), kidney (Kd), and brain (Br) were harvested. Evan's blue concentrations in organs were quantified as described in Materials and Methods. Data are expressed as mean ± SD Evan's blue OD/50 mg wet tissue and are pooled from four to six animals per condition where * indicates P < 0.025 and # indicates P < 0.01 between NECA and PBS.
Histology of Cd73−/− Lungs.
Based on the findings of increased pulmonary vascular leakage in APCP-treated animals, as well as significant defects in pulmonary vascular barrier in Cd73 −/− mice, we assessed the pulmonary vascular architecture in Cd73 −/− mice. To do this, wild-type or Cd73 −/− mice were subjected to normoxia or hypoxia as described before. Whole lungs were fixed with 10% formalin at total lung capacity with a constant applied transpulmonary pressure of 25 cm H2O, sectioned, and stained. As shown in Fig. 8 (A and B), histologic sections from wild-type and Cd73 −/− mice, respectively, kept under normoxic conditions revealed normal pulmonary architecture with the capillary endothelium lining the intertwining network of anastomotic capillaries and a normal basement membrane overlying the interstitium. Similar histologic examination of wild-type animals subjected to hypoxia revealed subtle pulmonary edema predominantly in the perivascular regions surrounding major conducting vessels, particularly pulmonary arteries (Fig. 8 D). A small degree of inflammation, characterized by the presence of inflammatory cells, was also evident in wild-type animals subjected to hypoxia. However, lung sections from Cd73 −/− animals subjected to a 4-h period of hypoxia were very revealing (Fig. 8 C). Consistent with our findings of a large degree of vascular leakage, lungs from hypoxic Cd73 −/− animals exhibited perivascular edema surrounding the major pulmonary blood vessels. Perivascular cuffing was also noted with occasional large foci of inflammatory infiltrates (17). These lesions were associated with occasional disruption of epithelial linings (Fig. 8 C, block arrow), although no significant protein leakage into the alveolar space or distal lung was evident. Given the perivascular localization of edema and the absence of frank pulmonary edema, these histological lesions appear to be associated with a hypoxia-induced alteration in large vessel barrier function, with preference for pulmonary arteries.
Figure 8.
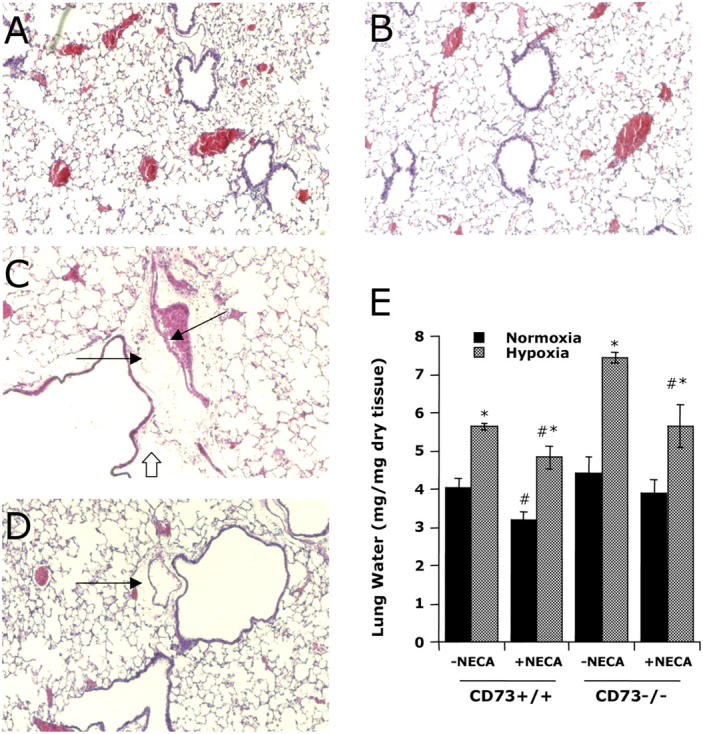
Characterization of lungs from Cd73 −/− mice. Wild-type (A and D) or Cd73 −/− (B and C) mice were subjected to normoxia (A and B) or hypoxia (C and D). Whole lungs were fixed with 10% formalin at total lung capacity, sectioned, and stained with hematoxylin and eosin. (A) A representative image of a wild-type control animal (magnification, 100). (C) Perivascular interstitial edema (line arrows) and epithelial disruption (block arrow) in Cd73 −/− hypoxic mice (magnification, 100). (D) Subtle perivascular interstitial edema (line arrow) associated with wild-type hypoxic mice (magnification, 100). (E) Assessment of lung water content in normoxia (black bars) and hypoxia (gray bars) in the presence and absence of NECA administration (0.1 mg/mg i.p. plus 0.1 mg/kg s.c.). Data are expressed as mean ± SD mg H2O/mg dry tissue and are pooled from three to four animals per condition where, in comparisons between hypoxia and normoxia, * indicates P < 0.025 and, in comparisons between NECA and PBS, # indicates P < 0.025.
To confirm the aforementioned observations, we objectively compared lung water content (wet:dry ratio) in Cd73 +/+ and Cd73 −/− mice subjected to hypoxia in the presence and absence of NECA. As shown in Fig. 8 E, hypoxia increased lung water content by 38 ± 6% in Cd73 +/− mice (P < 0.05). Similar analysis revealed a more pronounced increase (69 ± 7%, P < 0.01) in Cd73 −/− lungs, thereby confirming our histologic findings of pulmonary edema. In animals pretreated with NECA (Fig. 8 E), adenosine receptor activation partially rescued the hypoxia-induced pulmonary edema in both Cd73 +/+ (16 ± 3% decrease, P < 0.05) and Cd73 −/− lungs (32 ± 6% decrease, P < 0.025). Interestingly, NECA alone significantly decreased lung water content in normoxic wild-type animals (29 ± 5% decrease, P < 0.05), but not in Cd73 −/− animals (p-value was NS).
Discussion
Adenosine exerts autocrine and paracrine actions on most cell types. Pathophysiologic conditions of hypoxia/ischemia result in the elevation of extracellular adenosine, with phosphohydrolysis of AMP by CD73 representing the major pathway of extracellular adenosine formation. In the present studies, we explored the in vivo relevance of CD73 in physiologic and pathophysiologic regulation of vascular leakage. These studies revealed that, whereas Cd73 −/− mice are viable and appear to have a functionally intact immune system, significant vascular leakage was observed in multiple organs, particularly when mice were subjected to hypoxic stress.
Interstitial adenosine is critical for adaptation to hypoxia. For example, extracellular adenosine production in the myocardium by CD73 (14) has been demonstrated to contribute to cardiac preconditioning from brief periods of ischemia (12, 14, 33, 34), and CD73 activity increases in response to hypoxia/ischemia (21–24), attributable to a variety of acute activation pathways. Recent studies provide direct evidence that CD73 is transcriptionally induced by hypoxia in pheochromocytoma cells in vitro (22) and in epithelia and endothelia in vivo and in vitro (21, 23). Mechanisms of CD73 induction involve both hypoxia-inducible factor-1–mediated regulation of promoter activity (21) as well as feed-forward regulation of CD73 by adenosine (35). Thus, previous work strongly implicated CD73 as part of an innate protective response in hypoxic/ischemic tissue through the generation of extracellular adenosine, and provided the rationale for us to examine the role of CD73 in vascular function in vivo.
Previous studies suggested that CD73 is a key component of a protective pathway to maintain barrier function in epithelia (21) and endothelia (23). For example, administration of the selective ecto-5′-NT inhibitor APCP to mice by gavage significantly increased intestinal epithelial permeability (21). Furthermore, the barrier-promoting function of ATP released from activated neutrophils in vitro was found to be dependent on ATP hydrolysis to adenosine via the coordinated action of CD39 (ecto-apyrase) and CD73 (23). Here, we provide several new lines of evidence from in vivo studies to support our hypothesis that CD73 functions to protect barrier function in multiple organs. Initial findings of increased vascular leakage (Evan's blue extravasation) that was enhanced by hypoxia in Cd73 −/− animals fortify the developing idea of CD73 as an important adaptive enzyme during hypoxia. Additional support for this hypothesis was provided by the observation that inhibition of CD73 using APCP in wild-type animals resulted in increased vascular leakage in highly vascularized tissues. Finally, Cd73 −/− animals were afforded a degree of protection by reconstitution with purified C. atrox 5′-NT, implying that CD73 may provide a potential target for therapeutic development. Together, these new findings implicate extracellular adenosine as a key control point for regulation of vascular leakage in multiple tissues.
It is important to note that we observed quite significant differences in vascular leakage between different tissues and with various treatments (e.g., APCP and NECA). The factors responsible for these tissue- and treatment-specific differences in vascular leakage are complex and incompletely understood. Clearly, the level of CD73 5′-NT enzyme activity in a given tissue is not the crucial factor. The colon has the highest expression of the tissues examined, yet shows only a modest increase in vascular leakage in hypoxic Cd73 −/− mice. Alternately, 5′-NT activity is very low in skeletal muscle (∼2% of that in colon), yet muscle tissue shows an increase in vascular leakage comparable to that observed in other tissues such as kidney with as much as 24-fold higher 5′-NT enzyme activity. One factor may be the level of AMP-hydrolyzing activity attributed to enzymes other than CD73 (i.e., other nucleotidases and/or phosphatases). Of the tissues we studied, colon had the highest level of nucleotide-hydrolyzing activity that could not be inhibited by APCP, whereas skeletal muscle had the lowest (undetectable) level (Table I). The differences in vascular leakage observed under hypoxic conditions for Cd73 −/− mice versus wild-type mice treated with APCP may reflect limited access of APCP to some tissues and/or the difference between Cd73 −/− mice that are Cd73-deficient since birth as compared with Cd73 +/+ mice that become Cd73 deficient acutely after treatment with APCP. Another confounding difference could be variations in adenosine receptor expression in the various tissues studied.
Although the present studies are the first to define a physiological role for CD73 in barrier function in vivo, these results also raise a number of important questions. First, it will be important to determine the exact source of extracellular nucleotides, particularly during hypoxia and inflammation. Neutrophils are known to accumulate at sites of hypoxia and inflammation and are demonstrated sources of adenine nucleotides (in the form of 5′-AMP [18] and ATP [23]). Moreover, nucleotides released can be metabolized to active intermediates (e.g., adenosine) via biochemical crosstalk pathways involving two different cell types (16–18). Activated platelets comprise an additional reservoir of extracellular adenine nucleotides, particularly ATP. Given the recent evidence implicating ATP/ADP phosphohydrolysis by CD39 during hypoxia and ischemia (23, 36), it is possible that these types of circulating cells provide a readily available substrate pool for vascular barrier regulation by CD73. Second, we do not know which adenosine receptors predominate in this vascular response. Four subtypes of G protein–coupled adenosine receptors exist, designated A1, A2A, A22B, and A3 (14). These receptors are classified according to utilization of pertussis toxin–sensitive pathways (A1 and A3) or adenylate cyclase activation pathways (A2A and A2B). Our evidence indicates that adenosine promotes vascular barrier and because elevation of cAMP is associated with increased barrier function (2), it is likely that our results represent activation of A2A and/or A2B receptors. The A2B receptor may well be the target, as A2B receptor mRNA is up-regulated by hypoxia, and A2B receptor antagonists neutralize ATP-mediated changes in posthypoxic endothelial barrier function. Indeed, we provide evidence using MRS 1754 and ZM241385 that it is likely that both the A2A and A2B receptors contribute to the changes in vascular barrier during hypoxia. An A2B receptor gene-targeted mouse will probably be required to confirm this conclusion with certainty. Alternatively, inosine could mediate these responses. We think this is unlikely because inosine binds primarily to the A3 receptor (37) (a class of adenosine receptor that mediates decreased cAMP), and it is likely that Cd73 −/− mice retain the ability to generate inosine through the combined action of AMP deaminase and cytoplasmic nucleotidases. Third, we do not know exact molecular mechanisms of adenosine-mediated barrier regulation. Although our previous in vitro work has suggested that adenosine-mediated phosphorylation of tight junction-associated proteins such as vasodilator-stimulated phosphoprotein may be critical in both epithelial and endothelial permeability (38, 39), it is not known if these pathways function at the whole tissue level in vivo. Finally, we do not understand why vascular barrier function is better maintained in the brains of Cd73 −/− mice even under conditions of hypoxia. However, it is perhaps not surprising that the response of brain might be anomalous, given the unique properties of the blood–brain barrier. Perhaps the mechanisms that regulate vascular leakage in the blood–brain barrier are less dependent on adenosine. These issues remain to be resolved.
A consistent finding throughout these experiments was a predominant influence of CD73 in pulmonary vascular leakage. Indeed, lung vascular leakage was highly influenced by exogenous administration of APCP in wild-type animals, and the vascular leakage phenotype was most prominent in the lungs of Cd73 −/− mice. Based on these observations, we addressed lung histology in more detail. Closer examination revealed perivascular interstitial edema with inflammatory infiltrates surrounding the larger conducting vessels of the pulmonary vasculature in Cd73 −/− animals. This phenotype was particularly prevalent in Cd73 −/− mice subjected to hypoxia and was confirmed by assessment of lung wet/dry ratios indicating a >65% increase in lung water content. The mechanism of such vascular leakage remains undetermined.
In summary, these results identify CD73-catalyzed adenosine production as a critical control point for maintenance and regulation of vascular barrier function in multiple tissues during hypoxia. Studies in the lungs of hypoxic Cd73 −/− mice revealed segmental perivascular leakage involving macrovascular barrier disruption. Our ability to reconstitute barrier function with C. atrox 5′-NT suggests that CD73 might function as a therapeutic target for disorders involving vascular leakage syndromes.
Acknowledgments
The authors thank J. Vaughn, A. Laurent, S. Hooker, and R. Cotta for technical assistance; Dr. B. Gordon for helpful discussions; Dr. T. Mak for advice on the design of the targeting vector and methodology for gene targeting; and Dr. K. Jacobson for MRS 1754.
This work was supported by National Institutes of Health grant nos. AI18220 (L.F. Thompson), HL60569 (S.P. Colgan), and DK50189 (S.P. Colgan); and by a fellowship award from the Foundation for Anesthesia Education and Research (J.C. Ibla). L.F. Thompson holds the Putman City Schools Chair in Cancer Research.
The authors have no conflicting financial interests.
C.J. Van De Wiele's present address is Dept. of Surgery, University of Oklahoma College of Medicine, Tulsa, OK 74135.
R. Resta's present address is Albany Regional Cancer Center, Amsterdam, NY 12010.
Abbreviations used in this paper: ANOVA, analysis of variance; APCP, α,β-methylene ADP; E-Ado, etheno-adenosine; E-AMP, etheno-AMP; ES, embryonic stem; NECA, 5′-(N-ethylcarboxamido)-adenosine; 5′-NT, 5′-nucleotidase.
References
- 1.Webb, A.R. 2000. Capillary leak. Pathogenesis and treatment. Minerva Anestesiol. 66:255–263. [PubMed] [Google Scholar]
- 2.Stevens, T., J.G.N. Garcia, D.M. Shasby, J. Bhattacharya, and A.B. Malik. 2000. Mechanisms regulating endothelial cell barrier function. Am. J. Physiol. Lung Cell. Mol. Physiol. 279:L419–L422. [DOI] [PubMed] [Google Scholar]
- 3.Michel, C.C., and F.E. Curry. 1999. Microvascular permeability. Physiol. Rev. 79:703–761. [DOI] [PubMed] [Google Scholar]
- 4.Rippe, B., and B. Haraldsson. 1994. Transport of macromolecules across microvascular walls: the two-pore theory. Physiol. Rev. 74:163–219. [DOI] [PubMed] [Google Scholar]
- 5.Lum, H., and A.B. Malik. 1994. Regulation of vascular endothelial barrier function. Am. J. Physiol. 267:L223–L241. [DOI] [PubMed] [Google Scholar]
- 6.Stevens, T., J. Creighton, and W.J. Thompson. 1999. Control of cAMP in lung endothelial cell phenotypes. Implications for control of barrier function. Am. J. Physiol. 277:L119–L126. [DOI] [PubMed] [Google Scholar]
- 7.Stan, R.V. 2002. Structure and function of endothelial caveolae. Microsc. Res. Tech. 57:350–364. [DOI] [PubMed] [Google Scholar]
- 8.Michel, C.C. 1998. Capillaries, caveolae, calcium and cyclic nucleotides: a new look at microvascular permeability. J. Mol. Cell. Cardiol. 30:2541–2546. [DOI] [PubMed] [Google Scholar]
- 9.Rubin, L.L. 1992. Endothelial cells: adhesion and tight junctions. Curr. Opin. Cell Biol. 4:830–833. [DOI] [PubMed] [Google Scholar]
- 10.Janzer, R.C., and M.C. Raff. 1987. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature. 325:253–257. [DOI] [PubMed] [Google Scholar]
- 11.Dejana, E., R. Spagnuolo, and G. Bazzoni. 2001. Interendothelial junctions and their role in the control of angiogenesis, vascular permeability and leukocyte transmigration. Thromb. Haemost. 86:308–315. [PubMed] [Google Scholar]
- 12.Baxter, G.F. 2002. Role of adenosine in delayed preconditioning of myocardium. Cardiovasc. Res. 55:483–494. [DOI] [PubMed] [Google Scholar]
- 13.Mubagwa, K., and W. Flameng. 2001. Adenosine, adenosine receptors and myocardial protection: an updated overview. Cardiovasc. Res. 52:25–39. [DOI] [PubMed] [Google Scholar]
- 14.Linden, J. 2001. Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu. Rev. Pharmacol. Toxicol. 41:775–787. [DOI] [PubMed] [Google Scholar]
- 15.Sitkovsky, M.V., D. Lukashev, S. Apasov, H. Kojima, M. Koshiba, C. Caldwell, A. Ohta, and M. Thiel. 2004. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu. Rev. Immunol. 22:657–682. [DOI] [PubMed] [Google Scholar]
- 16.Lennon, P.F., C.T. Taylor, G.L. Stahl, and S.P. Colgan. 1998. Neutrophil-derived 5′-adenosine monophosphate promotes endothelial barrier function via CD73-mediated conversion to adenosine and endothelial A2B receptor activation. J. Exp. Med. 188:1433–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eltzschig, H.K., L.F. Thompson, J. Karhausen, R.J. Cotta, J.C. Ibla, S.C. Robson, and S.P. Colgan. 2004. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood. 10.1182/blood.2004-06-2066. [DOI] [PubMed]
- 18.Madara, J.L., T.W. Patapoff, B. Gillece-Castro, S.P. Colgan, C.A. Parkos, C. Delp, and R.J. Mrsny. 1993. 5′-adenosine monophosphate is the neutrophil-derived paracrine factor that elicits chloride secretion from T84 intestinal epithelial cell monolayers. J. Clin. Invest. 91:2320–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Airas, L., J. Hellman, M. Salmi, P. Bono, T. Puurunen, D.J. Smith, and S. Jalkaanen. 1995. CD73 is involved in lymphocyte binding to the endothelium: characterization of lymphocyte–vascular adhesion protein 2 identifies it as CD73. J. Exp. Med. 182:1603–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore, T.M., P.M. Chetham, J.J. Kelly, and T. Stevens. 1998. Signal transduction and regulation of lung endothelial cell permeability. Interaction between calcium and cAMP. Am. J. Physiol. 275:L203–L222. [DOI] [PubMed] [Google Scholar]
- 21.Synnestvedt, K., G.T. Furuta, K.M. Comerford, N. Louis, J. Karhausen, H.K. Eltzschig, K.R. Hansen, L.F. Thompson, and S.P. Colgan. 2002. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 (HIF-1) mediates permeability changes in intestinal epithelia. J. Clin. Invest. 110:993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kobayashi, S., H. Zimmermann, and D.E. Millhorn. 2000. Chronic hypoxia enhances adenosine release in rat PC12 cells by altering adenosine metabolism and membrane transport. J. Neurochem. 74:621–632. [DOI] [PubMed] [Google Scholar]
- 23.Eltzschig, H.K., J.C. Ibla, G.T. Furuta, M.O. Leonard, K.A. Jacobson, K. Enjyoji, S.C. Robson, and S.P. Colgan. 2003. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J. Exp. Med. 198:783–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ledoux, S., I. Runembert, K. Koumanov, J.B. Michel, G. Trugnan, and G. Friedlander. 2003. Hypoxia enhances ecto-5′-nucleotidase activity and cell surface expression in endothelial cells: role of membrane lipids. Circ. Res. 92:848–855. [DOI] [PubMed] [Google Scholar]
- 25.Burger, R.M., and J.M. Lowenstein. 1975. 5′-Nucleotidase from smooth muscle of small intestine and from brain. Inhibition by nucleotides. Biochemistry. 14:2362–2366. [DOI] [PubMed] [Google Scholar]
- 26.Yamashita, Y., S.W. Hooker, H. Jiang, A.B. Laurent, R. Resta, K. Khare, A. Coe, P.W. Kincade, and L.F. Thompson. 1998. CD73 expression and fyn-dependent signaling on murine lymphocytes. Eur. J. Immunol. 28:2981–2990. [DOI] [PubMed] [Google Scholar]
- 27.Thompson, L.F., G.R. Boss, H.L. Spiegelberg, I.V. Jansen, R.D. O'Connor, T.A. Waldmann, R.N. Hamburger, and J.E. Seegmiller. 1979. Ecto-5′-nucleotidase activity in T and B lymphocytes from normal subjects and patients with congenital X-linked agammaglobulinemia. J. Immunol. 123:2475–2478. [PubMed] [Google Scholar]
- 28.Khavandgar, S., H. Homayoun, A. Torkaman-Boutorabi, and M.R. Zarrindast. 2002. The effects of adenosine receptor agonists and antagonists on morphine state-dependent memory of passive avoidance. Neurobiol. Learn. Mem. 78:390–405. [DOI] [PubMed] [Google Scholar]
- 29.Gutensohn, W., R. Resta, Y. Misumi, Y. Ikehara, and L.F. Thompson. 1995. Ecto-5′-nucleotidase activity is not required for T cell activation through CD73. Cell. Immunol. 161:213–217. [DOI] [PubMed] [Google Scholar]
- 30.Thompson, L.F., J.M. Ruedi, M.G. Low, and L.T. Clement. 1987. Distribution of ecto-5′-nucleotidase on subsets of human T and B lymphocytes as detected by indirect immunofluorescence using goat antibodies. J. Immunol. 139:4042–4048. [PubMed] [Google Scholar]
- 31.Resta, R., Y. Yamashita, and L.F. Thompson. 1998. Ecto-enzyme and signaling functions of lymphocyte CD73. Immunol. Rev. 161:95–109. [DOI] [PubMed] [Google Scholar]
- 32.Stelzner, T.J., R.F. O'Brien, K. Sato, and J.V. Weil. 1988. Hypoxia-induced increases in pulmonary transvascular protein escape in rats. Modulation by glucocorticoids. J. Clin. Invest. 82:1840–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burnstock, G. 2002. Potential therapeutic targets in the rapidly expanding field of purinergic signalling. Clin. Med. 2:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang, Q.D., J. Pernow, P.O. Sjoquist, and L. Ryden. 2002. Pharmacological possibilities for protection against myocardial reperfusion injury. Cardiovasc. Res. 55:25–37. [DOI] [PubMed] [Google Scholar]
- 35.Narravula, S., P.F. Lennon, B.U. Mueller, and S.P. Colgan. 2000. Regulation of endothelial CD73 by adenosine: paracrine pathway for enhanced endothelial barrier function. J. Immunol. 165:5262–5268. [DOI] [PubMed] [Google Scholar]
- 36.Pinsky, D.J., M.J. Broekman, J.J. Peschon, K.L. Stocking, T. Fujita, R. Ramasamy, E. Connolly Jr., J. Huang, S. Kiss, Y. Zhang, et al. 2002. Elucidation of the thromboregulatory role of CD39/ectoapyrase in the ischemic brain. J. Clin. Invest. 109:1031–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hasko, G., M.V. Sitkovsky, and C. Szabo. 2004. Immunomodulatory and neuroprotective effects of inosine. Trends Pharmacol. Sci. 25:152–157. [DOI] [PubMed] [Google Scholar]
- 38.Comerford, K.M., D.W. Lawrence, K. Synnestvedt, B.P. Levi, and S.P. Colgan. 2002. Role of vasodilator-stimulated phosphoprotein in PKA-induced changes in endothelial junctional permeability. FASEB J. 16:583–585. [DOI] [PubMed] [Google Scholar]
- 39.Lawrence, D.W., K.M. Comerford, and S.P. Colgan. 2002. Role of VASP in reestablishment of epithelial tight junction assembly after Ca2+ switch. Am. J. Physiol. Cell Physiol. 282:C1235–C1245. [DOI] [PubMed] [Google Scholar]