

Abstract
An unprecedented [4Fe-4S] iron-sulfur cluster was found in RumA, the enzyme that methylates U1939 in Escherichia coli 23 S ribosomal RNA (Agarwalla, S., Kealey, J. T., Santi, D. V., and Stroud, R. M. (2002) J. Biol. Chem. 277, 8835–8840; Lee, T. T., Agarwalla, S., and Stroud, R. M. (2004) Structure 12, 397–407). Methyltransferase reactions do not involve a redox step. To understand the structural and functional roles of the cluster in RumA, we have characterized redox reactions of the iron-sulfur cluster. As isolated aerobically, RumA exhibits a visible absorbance maximum at 390 nm and is EPR silent. It cannot be reduced by anaerobic additions of dithionite. Photoreduction by deazariboflavin/EDTA gives EPR spectra, the quantity (56% of S = 1/2 species) and details (gav ~ 1.96–1.93) of which indicate a [4Fe-4S]1+ cluster in the reduced RumA. Oxidation of RumA by ferricyanide leads to loss of the 390-nm band and appearance of lower intensity bands at 444 and 520 nm. EPR spectra of ferricyanide-oxidized RumA show a fraction (<8%) of the FeS cluster trapped in the [3Fe-4S]1+ form (gav ~ 2.011) together with unusual radical-like spectrum (g′ values 2.015, 2.00, and 1.95). RumA also reacts with nitric oxide to give EPR spectra characteristic of the protein-bound iron dinitrosyl species. Oxidation of the cluster leads to its decomposition and that could be a mechanism for regulating the activity of RumA under conditions of oxidative stress in the cell. Sequence data base searches revealed that RumA homologs are widespread in various kingdoms of life and contain a conserved and unique iron-sulfur cluster binding motif, CX5CGGC.
RumA, an S-adenosyl-l-methionine-dependent methyltransferase specifically catalyzes the methylation of U1939 of 23 S ribosomal RNA to yield 5-methyluridine (m5U)1 or ribothymidine (1). This enzyme was found to contain a [4Fe-4S] cluster, a prosthetic group usually not associated with methyltransferase enzymes. The x-ray structure of RumA revealed that the cluster is located in the RNA binding domain of the three-domain protein (2). It is held by four Cys-iron bonds provided by Cys81, Cys87, Cys90, and Cys162 and is buried in a largely hydrophobic region, but one of its sulfur atoms is quite exposed to solvent. The closest distance between the cluster and the sulfur of the catalytic nucleophile, Cys389, is ~19 Å. RumA is a member of a growing list of nucleic acid modifying enzymes where the function of the iron-sulfur (FeS) cluster remains enigmatic.
The reaction mechanism of m5U methyltransferases is similar to that of thymidylate synthase, and has been studied mainly employing the tRNA m5U54 methyltransferase, TrmA (3–5). It involves Michael addition of the catalytic Cys to carbon 6 (C-6) of pyrimidine, which activates C-5 for a nucleophilic attack on the methyl group of S-adenosyl-l-methionine. Following the methyl transfer, the enzyme is resolved by abstraction of a proton from C-5 and β-elimination. TrmA, as well as DNA and RNA 5-methylcytidine methyltransferases, which employ a similar catalytic mechanism (6), do not posses an FeS cluster or any other prosthetic group. Lack of a redox step in the catalytic mechanism, and absence of the cluster in enzymes that employ a similar mechanism preclude a direct role for [4Fe-4S] cluster in the RumA-catalyzed reaction.
Two features distinguish the FeS cluster in RumA from the clusters in a different class of S-adenosyl-l-methionine-dependent enzymes that proceed by free radical mechanisms. The RumA cluster is ligated by 4 cysteines and the iron-binding sequence motif is CX5CX2CXnC. In contrast, the radical S-adenosyl-l-methionine enzymes, of which MiaB (7) and biotin synthase (8) are examples, have a [4Fe-4S] cluster built of a CX3CX2C sequence that leaves one iron of the cluster free to participate in catalysis. MiaB is a member of a family of enzymes catalyzing addition of sulfur to tRNA bases (9). The FeS sequence motifs in the DNA repair enzymes, endonuclease III (10) and MutY (11), are CX6CX2CX5C, somewhat similar to the RumA sequence. Although the FeS clusters in these enzymes are usually regarded as structural, recently it was shown that the redox potential of MutY is altered by DNA binding and that a change in charge of the [4Fe-4S]3+/2+ cluster might alter its affinity for DNA (12).
The redox potentials of [4Fe-4S] clusters vary widely (13–16). Of particular interest in many contemporary studies of FeS proteins is understanding the factors that influence the potentials, and thus functions, of these clusters (17, 18). Proteins with four Cys-iron bonds to the [4Fe-4S] cluster fall into two general redox groups, ones with an accessible 2+/1+ couple, exemplified by some ferredoxins (19) and ones, generally called HiPiPs, in which the 2+ form can be oxidized to a stable 3+ form. The potential of the 2+/1+ couple is variable among known [4Fe-4S] clusters. Clusters with very low reduction potentials (~ −600 mV) (20) may be photoreduced by deazariboflavin in the presence of EDTA, a hydrogen atom donor (21–23). The 2+ HiPiPs are readily oxidized by ferricyanide (24). The FeS cluster in family 4 uracil-DNA glycosylases was recently shown to have redox properties of HiPiPs (25).
In contrast, some [4Fe-4S] proteins rearrange when they are oxidized. Aconitase, with only three Cys-iron bonds in the intact [4Fe-4S] cluster, is the best studied example of these (13). It loses iron to form a cuboidal [3Fe-4S]1+ cluster. Simple one-electron oxidative loss of one iron is precluded in 4Fe-4S proteins with four Cys-iron bonds and the clusters may completely decompose after treatment with excess oxidant. Oxidative decomposition of 4-Cys-4Fe-4S clusters has been observed in some cases, for instance, in Azotobacter vinlandii ferredoxin I (FdI) and in endonuclease III (26, 27). The initial steps in 4-Cys-4Fe-4S cluster decomposition of A. vinlandii FdI have been examined by oxidizing the crystalline protein. X-ray analysis showed that unique iron and inorganic sulfur atoms were separating from the cluster after reaction with three oxidizing equivalents (28). The electron paramagnetic resonance (EPR) spectra resulting from oxidation of the A. vinlandii FdI 4Fe-4S cluster have characteristics of a sulfur-based radical and have been attributed to a cysteinyl disulfide radical (29, 30). In A. vinlandii FdI, formation of this radical involves a Cys residue (Cys24) that is not initially a cluster iron ligand (31).
The spectroscopic signatures of FeS clusters are well documented for numerous proteins and model compounds of determined structures (13, 32–34). Paramagnetic species resulting from redox reactions of [4Fe-4S] clusters give characteristic EPR spectra, summarized by average g′ values, gav, and anisotropy, Δg (16). Representative values of gav and (Δg) of [4Fe-4S]1+ are ~1.95 (~0.1), of [4Fe-4S]3+ are ~2.06 (~0.1), and of [3Fe-4S]1+ are ~2.01 (~0.02). The EPR silent clusters derived from 4-iron clusters are those with even numbers of unpaired electrons. The cysteinyl disulfide radical studied in oxidized A. vinlandii FdI has EPR spectra in the gav ~ 2.01 range, but these differ in temperature dependence from spectra of the paramagnetic FeS cluster forms. The [3Fe-4S]1+ clusters have EPR signals of maximum intensity between 10 and 12 K, and can scarcely be observed at 40 K, because of thermal population of low-lying excited states. In contrast, sulfur-based radical signals typically have maximum intensity around 30 –60 K (29, 35).
To determine possible functional roles of the FeS cluster in RumA, we examined the redox reactions of the cluster by both optical and EPR spectroscopy. We have also performed sequence analysis on the genomic data bases and have identified the putative FeS cluster binding sequences and consensus motifs in RumA homologs in genomes of various organisms.
EXPERIMENTAL PROCEDURES
Materials
5-Deazariboflavin was a gift from Gordon Tollin (Tucson, AZ). The nitric oxide (NO) donor, PAPA NONOate ((Z)-1-[N-(3-amino-propyl)-N-(n-propyl)amino]diazen-1-ium-1,2-diolate), was from Cayman Chemical (Ann Arbor, MI). It dissociates with a half-life of 1.3 h at pH 7.4 (Cayman Chemical) to form two nitric oxide molecules. Solid calcium oxide (98%) with a minor manganous impurity for calibration of g′ values was from Aldrich (catalog number 24,856-8). It was sealed in an evacuated small quartz capillary.
Isolation and Purification of Native RumA
The expression and aerobic purification of RumA were reported earlier (1). Protein concentrations were determined using an extinction coefficient, ε280 = 64600 m−1 cm−1, obtained by quantitative amino acid analysis. Iron content in RumA was estimated by inductively coupled plasma resonance spectroscopy (Garatt-Callahan Co., Millbrae, CA) that yielded 4.6 mol of iron/mol of protein. Inorganic sulfur was estimated by the method of Beinert (36) and a value of 3.6 mol of sulfur/mol of protein was obtained. The protein samples (0.1–0.4 mm) for both EPR and optical spectroscopy were in buffer (pH 7.0) composed of 50 mm potassium phosphate, 0.1 m potassium chloride, and 10% (v/v) glycerol (buffer A).
UV-visible Spectroscopy
15 μm RumA solution was prepared in buffer A that was purged with argon. K3Fe(CN)6 solution was freshly prepared in the same buffer. Protein sample was successively titrated with the oxidant, and spectra were recorded on a Shimadzu UV-2401PC spectrophotometer. Spectra of K3Fe(CN)6 solutions were subtracted from that of the titrated protein.
Preparation of EPR Samples
For photoreduction, oxidized 5-deaza-riboflavin (56 μm) was dissolved in buffer A plus 0.5 mm EDTA and diluted 10-fold into RumA solutions. Anaerobic samples for photoreduction were prepared by slow equilibration (1 h, 4 °C) of RumA and deazaflavin in an evacuated chamber (Wilmad, Buena NJ; tip-off manifold number 552-5) attached to an EPR tube. After several pump/equilibration cycles, the anaerobic sample was transferred to the EPR tube. The EPR tube was placed in a room temperature water bath and photoreduction was initiated by the white light of a 300 W projector bulb.
For oxidation of RumA, potassium ferricyanide solutions were freshly prepared in buffer A at 10 –100 times the protein concentration. The ferricyanide solution was placed in the EPR tube first and RumA was added to it. The sample was frozen in liquid nitrogen at the first sign of bleaching of the yellow/brown color of RumA. Oxidations were examined under both aerobic and anaerobic conditions. The highest yield of EPR signal of the oxidized protein was obtained when 0.1 mm RumA and 130 units of copper/zinc superoxide dismutase (bovine kidney superoxide dismutase, Sigma S-2139)) were added anaerobically to a 10-fold excess of potassium ferricyanide.
Solutions of PAPA NONOate ((Z)-1-[N-(3-aminopropyl)-N-(n-propyl)amino]diazen-1-ium-1,2-diolate) (0.1 m) were prepared in 8 mm potassium hydroxide, diluted 10-fold into 0.1 m phosphate (pH 7.4) 1–2 h before a further 10-fold dilution into RumA, resulting in ~2 mm NO in the EPR samples. These reactions were carried out in aerobic buffers.
EPR Spectroscopy
X-band EPR spectra were recorded on a Varian E 109 spectrometer equipped with an Oxford Instruments ESR9/10 pumped, liquid helium-flow cryostat. Figure legends contain relevant instrumental parameters. The sample buffer was used to record baselines under conditions identical to those with which the sample spectra were obtained. These baselines were subtracted from RumA spectra shown in the figures. X-band frequencies were measured at the bridge. Values of the g factor at the sample were calibrated using a sealed quartz tube (0.5 mm inner diameter) of CaO containing a manganous impurity, frozen in a 4-mm X-band quartz EPR tube of sample buffer. Spin concentrations in RumA samples were determined by double integration of the EPR signal over a range of 2 kgauss and comparison with double integrals of signals of 0.1 mm cupric acetate in sample buffer. The microwave power was 0.1 mW for accurate spin determination, and the temperature was 40 K.
The reported g′ values (Table I) of RumA EPR spectra were assigned by computer simulation. Simulations were performed using the XSophe program (37) (available from Bruker BioSpin). Simple simulations were used, considering only a spin of 1/2 with g factor and line width anisotropy. Simulation line shapes were gaussian. For the simulations, usually the magnetic field range was partitioned into 1.0 mT segments and the spatial grid, the Sydney OPera House grid (38), into θ increments of ~3°.
Table I.
Parameters for simulation of RumA EPR spectra
| Figurea | gx | gy | gz | ga | Width (x, y, z; G) |
|---|---|---|---|---|---|
| 2, inset, left | 2.01 | 1.92 | 1.85 | 1.96 | 20, 20, 30 |
| 2, inset, right | 2.005 | 1.95 | 1.92 | 1.96 | 50, 50, 50 |
| 4A | 2.024 | 2.015 | 1.995 | 2.011 | 8, 30, 15 |
| 4C | 2.015 | 2.00 | 1.95 | 1.99 | 10, 12, 20 |
The first column refers to the figure in which the simulation appears. For all simulations, the frequency was 9.26 GHz.
Sequence Analysis
Blast searches (39) were performed on completed eubacterial, Archaea, and eukaryotic genomes available in the NCBI data set on April 20, 2004. In cases where the completed genome from more than one bacterial species or strain of the same genus were available, only one member of each genus was arbitrarily selected for analyses. Eschericha coli RumA, RumB, and TrmA sequences were used as probes.
RESULTS
Optical Survey of Redox Properties of RumA
The optical spectrum of native RumA contains an absorption band with a maximum at 390 nm, a shoulder around 325 nm, and a minimum at 350 nm, besides the usual 280-nm band because of the aromatic residues (Fig. 1). Successive titration of 15 μm RumA with ferricyanide resulted in the loss of the 325- and 390-nm bands. By 180 μm ferricyanide, intensities of these bands were significantly reduced and new bands at 444 nm and at about 520 nm (very weak) emerged. Further addition of the oxidant or longer incubation caused precipitation of the protein. Anaerobic addition of sodium dithionite or Ti(III) citrate did not alter the absorption spectrum of the native RumA.
Fig. 1. UV-visible spectra of 15 μm RumA, and in the presence of varying concentrations of ferricyanide.
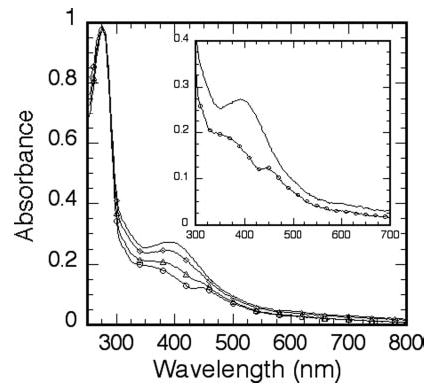
Ferricyanide concentrations shown are 0 μm (no symbol), 60 μm (⋄), 120 μm (Δ), and 180 μm (○). Inset, expansion of the spectra in the absence and presence of 180 μm ferricyanide.
EPR Study of Reduction of RumA FeS Cluster
Native RumA has >99% of the FeS cluster in a state that has essentially no EPR spectrum (see below, discussion of Fig. 3). Initial attempts to reduce RumA by anaerobic addition of dithionite gave no observed change in the EPR spectra, so a stronger reductant, 5-deazariboflavin semiquinone radical (21–23), was used. Brief irradiation (30 s) of RumA (0.1 mm), deazaflavin (5.6 μm), and EDTA (50 μm), followed by freezing, leads to an EPR spectrum of one electron-reduced RumA (Fig. 2). Based on comparison with the double integrals of the spectrum of a Cu2+ standard and of the RumA spectrum, the signal in Fig. 2 is estimated to represent 56% of the protein concentration. The shape of the reduced RumA EPR spectrum does not change between 10 and 30 K and is most intense at 10 –12 K. Below ~10 K, the signal broadens, and much above 30 K it is undetectable. At 12 K, the signal intensity does not begin to saturate until >4 mW. In the temperature range examined, there is no evidence of selective saturation of any portion of the reduced RumA EPR spectrum. The observed EPR signal is reasonably simulated as the sum of two equally weighted components (see Table I) of gx, gy, and gz values 2.005, 1.95, 1.92 and 2.01, 1.92, 1.85. This simulation is shown as an inset in Fig. 2. A continuum of g′ values between these extremes would also provide a good fit.
Fig. 3. EPR spectra (9.26 GHz) of oxidized RumA at two temperatures.
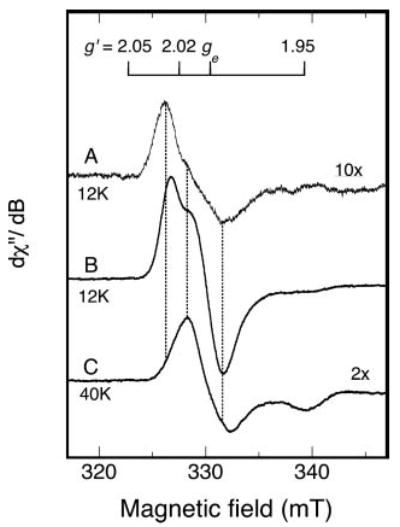
A, spectrum (12 K) of aerobically isolated RumA (0.4 mm); B, spectrum (12 K) of ferricyanide-oxidized RumA (0.35 mm RumA, 0.37 mm potassium ferricyanide); C, higher temperature spectrum (40 K) of the same oxidized RumA sample as in B. Instrument parameters were the same as those listed in the legend to Fig. 2 except the time constant was 0.25 s. Spectrum B was recorded at a receiver gain of 2000 and the amplitudes of spectra A and C, recorded with different gains (6,300 and 12,500 respectively), were adjusted to a gain of 2,000 and then multiplied by the factors indicated on the figure for visual comparison. The vertical dotted lines show alignment of maxima and minima, and are positioned at g′ values of 2.028, 2.016, and 1.995. The free electron g value, ge, is a constant.
Fig. 2. Experimental EPR spectrum (9.26 GHz) of photoreduced RumA.
Deoxygenated RumA (0.1 mm), 5-deazariboflavin (0.0056 mm), and EDTA (0.05 mm) were irradiated briefly at room temperature and then frozen for EPR. The inset shows how two separate line shapes, both with parameters (Table I) characteristic of a [4Fe4S]1+ center, might combine to give the observed line shape. The experimental EPR spectrum was recorded at 12 K and the instrument parameters were: microwave power, 10 mW; modulation amplitude, 1.6 mT; sweep rate, 12.5 mT/min; time constant, 0.5 s.
EPR Study of Oxidative Rearrangement of RumA FeS Cluster
The usual procedure adopted for EPR studies of RumA upon treatment with ferricyanide was to use two or fewer equivalents of the reagent and to freeze the sample when cloudiness first appeared (after about 1–2 min) because the addition of this reagent causes precipitation of the protein. RumA is slightly more stable to nitric oxide additions and samples treated with this reagent were frozen when cloudiness first appeared. For example, 0.1 mm RumA mixed with 2 mm NO, aerobically and at 4 °C, is stable for about 20 min.
RumA, isolated aerobically and before oxidation, exhibits variable amounts of a weak EPR signal (Fig. 3A). The low-field maximum occurs at a g′ value of 2.0278. In wider field sweeps, variable levels of g = 4.3 signal, characteristic of free ferric iron, are observed in different sample preparations (not shown). Addition of one-tenth oxidizing equivalent of ferricyanide per protein slightly enhances the intensity of the spectrum shown in Fig. 3A, whereas addition, at once, of 1 eq gives a much larger EPR spectrum of somewhat altered shape (Fig. 3B). Spectra of the same sample are compared at 12 and 40 K in Fig. 3, B and C, respectively. At 12 K, the maximum at g = 2.024 is the most intense peak and is slightly shifted in the g′ value from the low-field maximum of 2.028 seen in Fig. 3A. At 40 K the maximum is at g = 2.016 (see vertical lines in Fig. 3). The EPR signal in Fig. 3C falls to half the extrapolated linear intensity versus (microwave power)1/2 (P1/2) at ~13 mW of microwave power. There is little change above 40 K in the shape of the EPR signal from that of Fig. 3C. All of the spectral features are broadened at temperatures 8 K and below. Double integration and comparison with a copper standard indicates that the fraction of EPR detectable spin 1/2 species at 40 K (Fig. 3C) is 4 –8% of the initial protein concentration. The higher yield is obtained when the oxidation is carried out anaerobically. Because thermal population of excited states and relaxation both influence the intensity of the signal in Fig. 3A, assignment of the fraction of the protein concentration giving rise to the EPR-detectable species in Fig. 3A as <1% is simply based on comparisons of the double integrals of spectra in Fig. 3, A–C, together with the simulation discussed below.
Fig. 4 provides simulations of the spectra in Fig. 3, A and C, based on g′ values given in Table I. The summation shows that 12 K EPR spectra of oxidized RumA samples may be as simple as the sum of 2 components. However, a broader magnetic field scan of the sample giving the experimental spectrum in Fig. 3C shows some absorbance above baseline at ~g′ = 2.12 (Fig. 5A). A line shape model and more rhombic set of g′ values than the ones used in calculating Fig. 4C were not found to give an adequate simulation of the g′ = 2.13 feature. However, it is possible that there is some absorbance associated with the 40 K EPR of oxidized RumA that is so broad, or canceled by underlying spectra, that a double integral of the experimental spectrum (Fig. 5A) underestimates the spin concentration. If so, the number of spins as given in the spectra in Fig. 3C, or Fig. 5A, could be significantly greater than 8%.
Fig. 4. Simulated EPR spectra approximating the experimental spectra in Fig. 3.
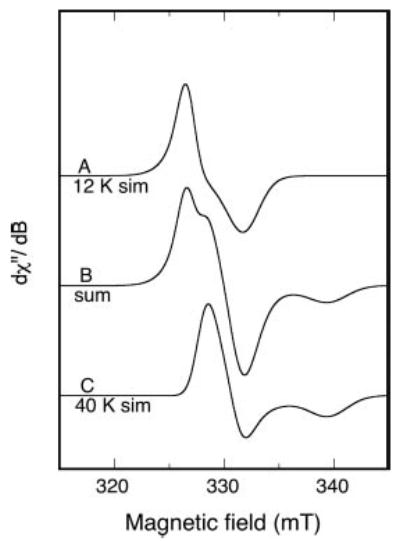
Parameters used in the simulation are given in Table I. The variation in simulation of g′ values giving good fits is about ±0.005. A sum of 53% simulated spectrum A and 47% simulated spectrum C (doubly integrated intensity) is shown as B.
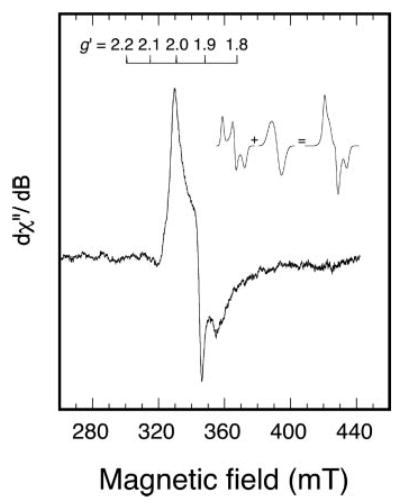
Fig. 5. An EPR spectrum of oxidized RumA compared with the dinitrosyl RumA derivative.
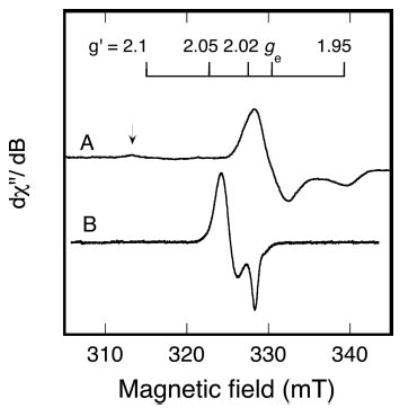
A, the oxidized RumA spectrum (40 K) is the same as that shown in Fig. 3C, but here a low-field region with possible additional absorption is also shown. The arrow indicates minor absorption at ~g′ value = 2.13. B, nitric oxide (from 1 mm nitric oxide donor) was added to deoxygenated RumA (0.17 mm) and the EPR spectrum was recorded (15 K). The fraction of the FeS cluster in the iron-dinitrosyl state, based on double integration, was 33%. Instrument parameters for A are given in the legend to Fig. 3C. For B, the parameters were: microwave frequency, 9.26 GHz; microwave power, 0.1 mW; modulation amplitude, 0.5 mT; sweep rate, 1.3 mT/min; time constant, 0.5 s.
Other Reactions of RumA
The reaction of RumA with nitric oxide has also been examined. The EPR spectrum shown in Fig. 5B is obtained with a molar ratio of nitric oxide to protein of 12:1. Based on integration, the Fig. 5B signal intensity corresponds to 33% of the concentration of the protein converted to an S = 1/2 iron dinitrosyl species by the time the reaction is quenched by freezing.
Preliminary measurements of the potential of the cluster are consistent with the EPR observations on oxidation and reduction of RumA. Square wave voltametry measurements done under anaerobic conditions, on gold electrode, yielded two peaks at −430 and +42 mV (versus standard hydrogen electrode). The peak at the lower potential could be because of the cluster reduction but could also be consistent with reduction of a S(Cys)-gold bond. However, precipitation of protein during these measurements precluded reliable measurements.2
Sequence Analyses and the Cluster Binding Motif
Blast searches (39) were performed on completed eubacterial genomes from 72 genera, using E. coli RumA, and homologous RumB (23 S rRNA 747 m5U methyltransferase) (40) and TrmA (tRNA 54 m5U methyltransferase) sequences as probes. Sequences with bit scores higher than 105 were considered orthologs of RumA, when the data base was probed with the E. coli RumA sequence. These sequences had an expected value lower than e-22. All, except one, of these sequences yielded significantly lower bit scores when probed with RumB and TrmA sequences. RumA orthologs were thus identified very widely throughout the eubacterial kingdom. Most of these high scoring RumA homologs contain the FeS binding motif with the consensus CXX(FYA)XXCGGC (Fig. 6). Organisms from Bacillales and Clostridia taxa had two high scoring homologs of RumA and both of these contain the consensus iron-sulfur binding motif. In contrast, several members of Lactobacillales taxon also have two high scoring homologs of RumA but only one of these two sequences has the FeS binding motif. RumA homologs from the Actinobacteria taxon have further diverged from the E. coli sequence. In the RumA homologs of several of the these organisms, the FeS sequence motif has deviated from that in the E. coli and has the sequence: C-Xn (n = 7–8)-C-C. In all other eubacteria where RumA could be identified, very few of them had deviations from the consensus FeS motif. Members of the taxon, Mollicutes, did not contain high scoring homologs that can be designated as RumA.
Fig. 6. Iron-sulfur cluster binding motif in RumA homologs from representative organisms from different kingdoms of life.
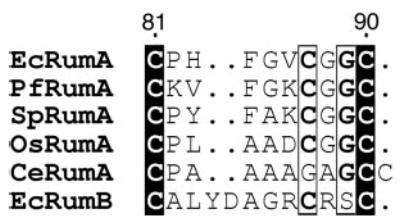
The cluster binding sequence from another ribosomal RNA 5-methyluridine methyltransferase, RumB, is also shown. Absolutely conserved residues are in solid blocks with white letters, and residues within hollow blocks vary at only one sequence in this subset. The residue positions indicated on top are for E. coli RumA. This figure was generated by ESPript (53). The two-letter species abbreviations used are: Ec, E. coli; Pf, Pyrococcus furiosus; Sp, Schizosaccharomyces pombe; Os, Oryza sativa; and Ce, Corynebacterium efficiens.
Archaea and eukaryotic genomic data bases were also probed for the presence of RumA homologs. Among archaebacteria, Pyrococcus species had two each and Nanoarchaeum equitans had one high scoring homolog of RumA. All of these contained the consensus FeS binding motif. Among the completed sequence data bases from fungi, most organisms including Saccharomyces pombe, Neurospora crassa, Eremothecium gossypii, and Aspergillus nidulans genomes had RumA homologs containing the FeS binding motif. Several plant species like Arabidopsis thaliana and Oryza sativa (rice) also had RumA homologs with the FeS binding motif. Any high scoring homolog of RumA was absent in the genomes from the animal kingdom.
RumB, another ribosomal RNA m5U methyltransferase in E. coli, also possesses an FeS binding sequence motif with a consensus CXX(FY)XXXXC(RTH)SC. This motif resides at the extreme N terminus of the protein sequence with the first cysteine located at position three in the E. coli RumB. Blast analyses revealed that the RumB gene is present only among the organisms from the γ subdivision of the proteobacteria family and is not as prevalent as RumA.
DISCUSSION
The structurally relevant conclusion of these EPR studies of the FeS cluster in RumA is that the cluster is in the [4Fe-4S]2+ state in the aerobically isolated protein, and also under conditions that would prevail in its normal cellular environment. The majority of the cluster can be reduced to [4Fe-4S]1+, but the reduction requires strongly reducing conditions. FeS clusters requiring photochemical reduction by deazaflavin semiquinones may have potentials for the 2+/1+ couple as low as −600 mV (20), far below any cellular reducing power. EPR spectra of [4Fe-4S]1+ clusters may vary with their solvent environment (41), reflecting different delocalization of electrons within the cluster. The spectrum observed for photoreduced RumA (Fig. 2) has a distribution of g′ values, but the average g′ value (gav = 1.96, Table I) identifies the species as the 4Fe cluster of 1+ charge.
Fragmentation of the FeS clusters under oxidizing conditions is common and may be related to function or may reflect on how the cluster is assembled. Ferricyanide oxidation of a number of [4Fe-4S] proteins leads to EPR signals with either an average g′ value (gav) of ~2.01 and narrow ′g range, as exemplified by A. vinlandii FdI (26), endonuclease III (27), and aconitase (13), or to a broader signal covering the g′ value range from ~2.12 to 2.02, found for HiPiPs (24, 42). The EPR spectra of oxidized RumA (Fig. 3) are clearly in the g′ = 2.01 category, although a portion of the signal appears to be that of a radical. The narrow g′ = 2.01 cluster signals arise from [3Fe-4S]1+, a fact that has been established by comparison of several types of spectroscopy and x-ray structures for various preparations of aconitase (32, 33) and the 3Fe center of 7-iron ferredoxins (35). For [4Fe-4S] clusters with only 3 iron-Cys bonds, as in aconitase, the fourth, Cys-free iron is the one lost. For those clusters with all irons attached to a cysteine, the fragmentation pattern is less obvious.
Temperature-dependent EPR signals, similar to those exhibited by RumA (Fig. 3), are observed in other proteins with [3Fe-4S]1+ clusters. The EPR spectra of the oxidized [3Fe-4S]1+ cluster in A. vinlandii FdI, recorded at 11 and ≥30 K (29), somewhat resemble those of RumA at 12 and 40 K, respectively. The spectra collected at the lower temperature emphasize the signal from [3Fe-4S]1+, whereas the higher temperature spectra reflect magnetic properties of a radical species. Substoichiometric oxidation (0.1 oxidizing equivalent) of RumA leads first to the [3Fe-4S]1+ EPR spectrum. Thus, the EPR spectrum emphasized at 40 K (Fig. 3C), which appears upon addition of one or more oxidizing equivalents, could result from further oxidation of [3Fe-4S]1+ or it could be the product of a different fragmentation. The large fraction of RumA iron unaccounted for (EPR silent) under oxidizing conditions could result from dissociation of the [4Fe-4S]2+ cluster to give a [2Fe-2S]2+ form, also EPR silent. The absorption spectrum of the oxidized RumA (Fig. 1), which is similar to that of, as isolated, MiaB, supports this notion. The spectrum of MiaB was ascribed to the [2Fe-2S]2+ cluster (7). The [4Fe-4S]2+ to [2Fe-2S]2+ conversion is part of the oxygen-sensing mechanism of the transcription factor FNR (fumarate nitrate reduction), for instance (43, 44).
Because the time courses of RumA oxidation and resulting precipitation are similar, the stoichiometry for radical formation cannot be determined by an optical titration as illustrated in Fig. 1. In comparison to the 3-electron oxidized A. vinlandii Fd EPR signal, attributed to Cys-S·S-Cys (29), the oxidized RumA radical EPR signal (Fig. 3C) lacks the expected well defined peak at g = ~2.09. Assuming this low field region may be ignored, the simulation in Fig. 4C accounts reasonably for the EPR signal in Fig. 3C. This signal is unusual in having two close principal g′ values (2.015, 2.00) at lower field than the unique g′ value at higher field (1.95). Although a similar order of g′ values is exhibited by some R-S·S-R radicals, for instance, the cystine radical (45), no radical in this category has a gz value as low as 1.95. A precedent for an EPR signal with g′ values almost identical to those of the RumA radical has been reported recently in studies of SdhC, an expressed subunit of the respiratory complex II of an archebacterium (46). Like the RumA signal, the EPR of SdhC has maximum intensity at 30 –40 K. The EPR signal from the isolated subunit SdhC is substoichiometric, and therefore, its origin was difficult to identify. However, SdhC contains a [2Fe-2S] cluster. One may speculate that both oxidized RumA and the isolated SdhC contain some novel oxidized form of a 2Fe cluster. Alternatively, they might arise from a disulfide radical in close proximity to iron.
A second example of the fragmentation of the RumA cluster is the reaction with nitric oxide (Fig. 5B). When [4Fe-4S-4Cys] proteins react with limited amounts of nitric oxide, one or more S3-Fe-Cys- motifs in the native cluster are replaced by a spin 1/2 iron-dinitrosyl motif (S2Fe(NO)2) (47). The EPR signal resulting from nitric oxide addition to RumA is identical to the dinitrosyl spectra reported for many FeS proteins, including HiPiP (47), endonuclease III (48), and aconitase (49). The nitric oxide complexes of FeS proteins are of interest because they suggest physiological mechanisms by which the proteins might be regulated. For instance, regulation of transcription by the E. coli fumarate/nitrate reduction transcription factor is influenced by either oxygen or NO (50). In the case of endonuclease III, it has been possible to reverse the NO modification by addition of iron and sulfur (48). RumA reacts readily (radical concentration equal 33% that of the protein in 5 min) with additions of NO. The possibility that this reaction can be driven to completion and reversed, and therefore may be physiologically relevant, remains to be explored.
The two ribosomal RNA m5U methyltransferases in E. coli, RumA and RumB, both contain FeS clusters whereas, the tRNA m5U methyltransferase, TrmA does not. The cluster binding motif is highly conserved among RumA homologs even in phylogenetically distinct species. The motifs in RumA and RumB are different (Fig. 6), and that might influence their respective reduction potentials. Some organisms contain two homologs of RumA that are very similar among themselves. It is unclear if the two homologous enzymes within the same organism modify the same or different target uridines. A very widespread presence of the rumA gene in bacteria, fungi, and plant kingdoms suggests an ancient evolutionary origin for RumA.
The physiological role of the methylation of uridine 1939 in the ribosome is presently unknown. In the crystal structure of the 70 S ribosome in complex with tRNAs, the rRNA loop harboring U1939 is seen to protrude into the major groove of the acceptor stem of A-site tRNA at the CCA tail (51). The E. coli 23 S rRNA sequence between 1934 and 1949 is highly complementary (14 of 16 nucleotides) to the sequence in the tRNA-like domain of tmRNA, strongly indicating that it has the potential to base pair with the tmRNA. A cryo-EM structure of the ribosome-tmRNA complex places the tmRNA in a location proximal to U1939 (52). These observations suggest that the region around U1939 has a role in tRNA and tmRNA binding. It can be speculated that methylation of U1939 may subtly affect the interactions between tRNA or tmRNA and the ribosome.
Ease of oxidation of the cluster in RumA by ferricyanide and NO indicates that the cluster can be readily oxidized under physiological conditions of oxidative stress in the cell. This could be a way to abolish the activity of RumA in the cell, because oxidation of RumA leads to cluster decomposition and subsequent precipitation of the protein. Absence of RumA-mediated methylation in the ribosome might have certain advantages to ribosomal functions under conditions of oxidative stress. Additionally, far fewer turnovers of an enzyme involved in modification of the ribosome are required than of other enzymes and electron transfer FeS proteins, so the RumA FeS cluster may be a sacrificial one to provide iron and sulfur for other clusters, after the ribosome is methylated.
The crystal structure of RumA in complex with the substrate RNA shows that a four-nucleotide stack of RNA is in close proximity to the FeS cluster.3 One of the inorganic sulfurs of the cluster forms a water-mediated hydrogen bond to an RNA base at one end of this stack. Barton and colleagues (12) have demonstrated efficient charge migration to long distance through stacked nucleic acid bases. They postulated that release of an electron from the [4Fe-4S]2+ cluster might be involved in long range signaling through DNA-mediated charge transfer (12). It remains to be explored whether the observed interaction of the cluster in RumA with the RNA base stack has any functional significance or a role in charge transfer.
Acknowledgments
We thank Fayi Wu for determining spin concentrations for EPR spectra. We also thank to Gordon Tollin for the gift of 5-deazariboflavin.
Footnotes
This work was supported by National Institutes of Health Grants GM65268 (to B. J. G.) and GM51232 (to R. M. S.).
The abbreviations used are: m5U, 5-methyluridine; EPR, electron paramagnetic resonance; NO, nitric oxide; FeS, iron-sulfur; HiPiP, high potential iron-sulfur protein; g′ value, proportional to (EPR frequency/resonance magnetic field); mT, milliTesla (10 mT = 1 gauss); K3Fe(CN)6, ferricyanide; FdI, ferredoxin I.
Prof. Marco Sola, personal communication.
T. T. Lee, S. Agarwalla, and R. Stroud, personal communication.
References
- 1.Agarwalla S, Kealey JT, Santi DV, Stroud RM. J Biol Chem. 2002;277:8835–8840. doi: 10.1074/jbc.M111825200. [DOI] [PubMed] [Google Scholar]
- 2.Lee TT, Agarwalla S, Stroud RM. Structure (Lond) 2004;12:397–407. doi: 10.1016/j.str.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Ivanetich KM, Santi DV. Prog Nucleic Acids Res Mol Biol. 1992;42:127–156. doi: 10.1016/s0079-6603(08)60575-9. [DOI] [PubMed] [Google Scholar]
- 4.Gu X, Santi DV. Biochemistry. 1992;31:10295–10302. doi: 10.1021/bi00157a017. [DOI] [PubMed] [Google Scholar]
- 5.Kealey JT, Gu X, Santi DV. Biochimie (Paris) 1994;76:1133–1142. doi: 10.1016/0300-9084(94)90042-6. [DOI] [PubMed] [Google Scholar]
- 6.Wu JC, Santi DV. Prog Clin Biol Res. 1985;198:119 –129. [PubMed] [Google Scholar]
- 7.Pierrel F, Hernandez HL, Johnson MK, Fontecave M, Atta M. J Biol Chem. 2003;278:29515–29524. doi: 10.1074/jbc.M301518200. [DOI] [PubMed] [Google Scholar]
- 8.Berkovitch F, Nicolet Y, Wan JT, Jarrett JT, Drennan CL. Science. 2004;303:76 –79. doi: 10.1126/science.1088493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jager G, Leipuviene R, Pollard MG, Qian Q, Bjork GR. J Bacteriol. 2004;186:750 –757. doi: 10.1128/JB.186.3.750-757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuo CF, McRee DE, Fisher CL, O’Handley SF, Cunningham RP, Tainer JA. Science. 1992;258:434 –440. doi: 10.1126/science.1411536. [DOI] [PubMed] [Google Scholar]
- 11.Lu AL, Wright PM. Biochemistry. 2003;42:3742–3750. doi: 10.1021/bi0269198. [DOI] [PubMed] [Google Scholar]
- 12.Boon EM, Livingston AL, Chmiel NH, David SS, Barton JK. Proc Natl Acad Sci U S A. 2003;100:12543–12547. doi: 10.1073/pnas.2035257100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beinert H, Kennedy MC, Stout CD. Chem Rev. 1996;96:2335–2374. doi: 10.1021/cr950040z. [DOI] [PubMed] [Google Scholar]
- 14.Beinert H, Holm RH, Munck E. Science. 1997;277:653–659. doi: 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- 15.Beinert H. J Biol Inorg Chem. 2000;5:2–15. doi: 10.1007/s007750050002. [DOI] [PubMed] [Google Scholar]
- 16.Bentrop, D., Capozzi, F., and Luchinat, C. (2001) in Handbook on Metalloproteins (Bertini, I., Sigel, A., and Sigel, H., eds) pp. 357–460, Marcel Dekker, Inc., New York
- 17.Glaser T, Bertini I, Moura JJ, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 2001;123:4859 –4860. doi: 10.1021/ja0155940. [DOI] [PubMed] [Google Scholar]
- 18.Glaser T, Rose K, Shadle SE, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 2001;123:442–454. doi: 10.1021/ja002183v. [DOI] [PubMed] [Google Scholar]
- 19.Chen K, Jung YS, Bonagura CA, Tilley GJ, Prasad GS, Sridhar V, Armstrong FA, Stout CD, Burgess BK. J Biol Chem. 2002;277:5603–5610. doi: 10.1074/jbc.M108916200. [DOI] [PubMed] [Google Scholar]
- 20.Blankenhorn G. Eur J Biochem. 1976;67:67–80. doi: 10.1111/j.1432-1033.1976.tb10634.x. [DOI] [PubMed] [Google Scholar]
- 21.Massey V, Hemmerich P. Biochemistry. 1978;17:9 –16. doi: 10.1021/bi00594a002. [DOI] [PubMed] [Google Scholar]
- 22.Knappe WR, Hemmerich P, Duchstein HJ, Fenner H, Massey V. Biochemistry. 1978;17:16 –17. [PubMed] [Google Scholar]
- 23.Walker MC, Pueyo JJ, Navarro JA, Gomez-Moreno C, Tollin G. Arch Biochem Biophys. 1991;287:351–358. doi: 10.1016/0003-9861(91)90489-6. [DOI] [PubMed] [Google Scholar]
- 24.Dunham WR, Hagen WR, Fee JA, Sands RH, Dunbar JB, Humblet C. Biochim Biophys Acta. 1991;1079:253–262. doi: 10.1016/0167-4838(91)90066-9. [DOI] [PubMed] [Google Scholar]
- 25.Hinks JA, Evans MC, De Miguel Y, Sartori AA, Jiricny J, Pearl LH. J Biol Chem. 2002;277:16936 –16940. doi: 10.1074/jbc.M200668200. [DOI] [PubMed] [Google Scholar]
- 26.Morgan TV, Stephens PJ, Devlin F, Burgess BK, Stout CD. FEBS Lett. 1985;183:206 –210. doi: 10.1016/0014-5793(85)80777-8. [DOI] [PubMed] [Google Scholar]
- 27.Cunningham RP, Asahara H, Bank JF, Scholes CP, Salerno JC, Surerus K, Munck E, McCracken J, Peisach J, Emptage MH. Biochemistry. 1989;28:4450 –4455. doi: 10.1021/bi00436a049. [DOI] [PubMed] [Google Scholar]
- 28.Sridhar V, Prasad GS, Burgess BK, Stout CD. J Biol Inorg Chem. 1998;3:140 –149. [Google Scholar]
- 29.Morgan TV, Stephens PJ, Devlin F, Stout CD, Melis KA, Burgess BK. Proc Natl Acad Sci U S A. 1984;81:1931–1935. doi: 10.1073/pnas.81.7.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu Z, Jollie D, Burgess BK, Stephens PJ, Munck E. Biochemistry. 1994;33:14475–14485. doi: 10.1021/bi00252a014. [DOI] [PubMed] [Google Scholar]
- 31.Shen B, Jollie DR, Diller TC, Stout CD, Stephens PJ, Burgess BK. Proc Natl Acad Sci U S A. 1995;92:10064 –10068. doi: 10.1073/pnas.92.22.10064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robbins AH, Stout CD. Proc Natl Acad Sci U S A. 1989;86:3639 –3643. doi: 10.1073/pnas.86.10.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Surerus KK, Kennedy MC, Beinert H, Munck E. Proc Natl Acad Sci U S A. 1989;86:9846 –9850. doi: 10.1073/pnas.86.24.9846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou J, Hu ZG, Munck E, Holm RH. J Am Chem Soc. 1996;118:1966 –1980. [Google Scholar]
- 35.Jung YS, Bonagura CA, Tilley GJ, Gao-Sheridan HS, Armstrong FA, Stout CD, Burgess BK. J Biol Chem. 2000;275:36974–36983. doi: 10.1074/jbc.M004947200. [DOI] [PubMed] [Google Scholar]
- 36.Beinert H. Anal Biochem. 1983;131:373–378. doi: 10.1016/0003-2697(83)90186-0. [DOI] [PubMed] [Google Scholar]
- 37.Wang D, Hansen GR. J Magn Res A. 1995;117:1–8. [Google Scholar]
- 38.Griffin M, Mays A, Noble C, Wang D, Eldershaw C, Gates KE, Burrage K, Hanson GR. Mol Phys Rep. 1999;26:60 –84. [Google Scholar]
- 39.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Nucleic Acids Res. 1997;25:3389 –3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Madsen CT, Mengel-Jorgensen J, Kirpekar F, Douthwaite S. Nucleic Acids Res. 2003;31:4738 –4746. doi: 10.1093/nar/gkg657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu A, Graslund A. J Biol Chem. 2000;275:12367–12373. doi: 10.1074/jbc.275.17.12367. [DOI] [PubMed] [Google Scholar]
- 42.Bertini I, Gaudemer A, Luchinat C, Piccioli M. Biochemistry. 1993;32:12887–12893. doi: 10.1021/bi00210a042. [DOI] [PubMed] [Google Scholar]
- 43.Jordan PA, Thomson AJ, Ralph ET, Guest JR, Green J. FEBS Lett. 1997;416:349 –352. doi: 10.1016/s0014-5793(97)01219-2. [DOI] [PubMed] [Google Scholar]
- 44.Popescu CV, Bates DM, Beinert H, Munck E, Kiley PJ. Proc Natl Acad Sci U S A. 1998;95:13431–13435. doi: 10.1073/pnas.95.23.13431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hadley JH, Jr, Gordy W. Proc Natl Acad Sci U S A. 1974;71:4409 –4413. doi: 10.1073/pnas.71.11.4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iwasaki T, Kounosu A, Aoshima M, Ohmori D, Imai T, Urushiyama A, Cosper NJ, Scott RA. J Biol Chem. 2002;277:39642–39648. doi: 10.1074/jbc.M207312200. [DOI] [PubMed] [Google Scholar]
- 47.Foster HW, Cowan JA. J Am Chem Soc. 1999;121:4093–4100. [Google Scholar]
- 48.Rogers PA, Eide L, Klungland A, Ding H. DNA Repair (Amst) 2003;2:809 –817. doi: 10.1016/s1568-7864(03)00065-x. [DOI] [PubMed] [Google Scholar]
- 49.Kennedy MC, Antholine WE, Beinert H. J Biol Chem. 1997;272:20340 –20347. doi: 10.1074/jbc.272.33.20340. [DOI] [PubMed] [Google Scholar]
- 50.Cruz-Ramos H, Crack J, Wu G, Hughes MN, Scott C, Thomson AJ, Green J, Poole RK. EMBO J. 2002;21:3235–3244. doi: 10.1093/emboj/cdf339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JH, Noller HF. Science. 2001;292:883–896. doi: 10.1126/science.1060089. [DOI] [PubMed] [Google Scholar]
- 52.Valle M, Gillet R, Kaur S, Henne A, Ramakrishnan V, Frank J. Science. 2003;300:127–130. doi: 10.1126/science.1081798. [DOI] [PubMed] [Google Scholar]
- 53.Gouet P, Courcelle E, Stuart DI, Metoz F. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]