

Sir—Celecoxib is a widely used nonsteroidal anti-inflammatory agent that acts through selective inhibition of COX-2. Here we demonstrate that celecoxib also significantly increases nuclear localization of the glucocorticoid receptor (GR), increases GR binding to its DNA responsive element and enhances GR-mediated gene transcription in association with inhibition of p38 MAP kinase. These results expand the intracellular targets of COX-2 inhibitors while widening their potential clinical application to multiple disorders associated with impaired GR function including major depression.
Glucocorticoids play an essential role in shaping and eventually restraining physiological responses to environmental stressors, including early, innate, immune responses (inflammation); activation of the sympathetic nervous system and stimulation of neuroendocrine pathways, all of which are capable of producing a host of adverse health outcomes if allowed to continue unabated after stressor cessation.1 A number of diseases including certain auto-immune, infectious and inflammatory disorders as well as a subgroup of neuropsychiatric disorders such as major depression have been associated with decreased responsiveness to glucocorticoids and impaired functioning of the GR.2,3 One potential mediator of disrupted GR function in these diseases is proinflammatory cytokines.3 Each of these disorders has been characterized by increased circulating and/or tissue expression of proinflammatory cytokines. Moreover, proinflammatory cytokines through their signaling pathways have been found to negatively regulate GR function. For example, IL-1 alpha activation of p38 MAPK pathways has been shown to decrease GR DNA binding and GR-mediated gene transcription.4 Similar decreases in GR activity have been found following activation of JNK and NF-kB inflammatory signaling pathways.5,6
In this study, rat neuronal PC12 cells stably transfected with MMTV-luciferase construct were used to investigate the direct impact of COX inhibition on GR function. Treatment of cells with the selective COX-2 inhibitor, celecoxib, caused significant dose-dependent induction of GR-mediated luciferase activity at both 5 and 24 h of treatment (Figure 1a). Similar results were found with the nonselective COX inhibitor, ibuprofen (100–1000 μM). Interestingly, valerylsalicylic acid, a COX-1 selective inhibitor, had no effects on GR-mediated luciferase activity, indicating that the effects of COX inhibition on GR function are COX-2 specific. To determine the GR specificity of the effects of celecoxib on luciferase activity, we cotreated PC12 cells with celecoxib and the GR antagonist, RU486 (40 μM), alone and in combination, for 5 h (data not shown). RU486 significantly reduced the effects of celecoxib on GR-mediated luciferase activity (32% decrease, n = 3 per group, P < 0.05 using Newman–Keuls Multiple Comparison Test), although, as has been previously reported, RU486 alone induced a mild increase in GR function.7 The GR is primarily located in cytosol under basal conditions and its nuclear accumulation is essential for GR DNA binding and/or interaction with other relevant transcription factors. We therefore probed GR protein in nuclear extracts of cells treated with celecoxib with or without cotreatment with the synthetic glucocorticoid, dexamethasone (Dex). As seen in Figure 1b, celecoxib caused a noticeable increase of GR nuclear localization both alone and in combination with Dex. Of note, GR nuclear localization was accompanied by evidence of increased binding of the GR to its DNA glucocorticoid responsive element (GRE) as determined by electrophoretic mobility shift assay (EMSA) (Figure 1c). A significant increase in GR-GRE binding was also observed as a result of celecoxib (Cbx) treatment using the TansAM GR transcription factor assay (Active Motif, Carlsbad, CA, USA) (mean absorbance: control 177.7 (SD 4.0); Cbx 50 μM 5 h 227.8 (SD 15.4); Cbx 50 μM 24 h 252.0 (SD 15.4); Dex 10 nM 2 h 348.7 (SD 20.1), F(3,8) = 69.0, P < 0.001). As certain COX inhibitors have also been reported to inhibit p38 MAPK,8 to explore the molecular mechanism of COX-2 inhibitor-mediated GR function enhancement, we investigated the direct effects of celecoxib on p38 MAPK. Results in Figure 1d show that celecoxib alone (50 μM) caused significant reduction of phosphorylated p38 MAPK. To further investigate p38 involvem ent, a selective p38 activator, anisomycin, was employed. Anisomycin (50 ng/ml) showed strong time-dependent induction of p38 phosphorylation (Figure 1d). Moreover, results showed that cotreatment with anisomycin significantly reversed celecoxib’s effect on GR function (Figure 1a). The results of this study are the first to show that a widely used selective COX-2 inhibitor, celecoxib, induces GR nuclear localization and enhances GR-mediated gene transcription. Celecoxib’s positive effect on GR function appears to be through inhibition of p38 MAPK. The data from this study suggest that inhibition of COX pathways may represent a therapeutic target for normalization of GR function in disorders characterized by reduced responsiveness to glucocorticoids. Interestingly, aside from its relevance to inflammatory disorders, reduced responsiveness to glucocorticoids is commonly seen in neuropsychiatric disorders including, most notably, major depression. These data suggest that COX-2 inhibitors may represent novel therapeutic agents for the treatment of depression. Indeed, like celecoxib, antidepressant agents have been shown to translocate the GR and increase GR-mediated gene transcription.9 Given the negative impact of inflammatory signaling pathways including p38 and COX-2 on long-term potentiation (LTP)10 and neurogenesis,11 inhibition of COX-2 may represent a unique approach to multiple pathophysiologic targets, including, but not limited to the GR, in affective disorders.
Figure 1.
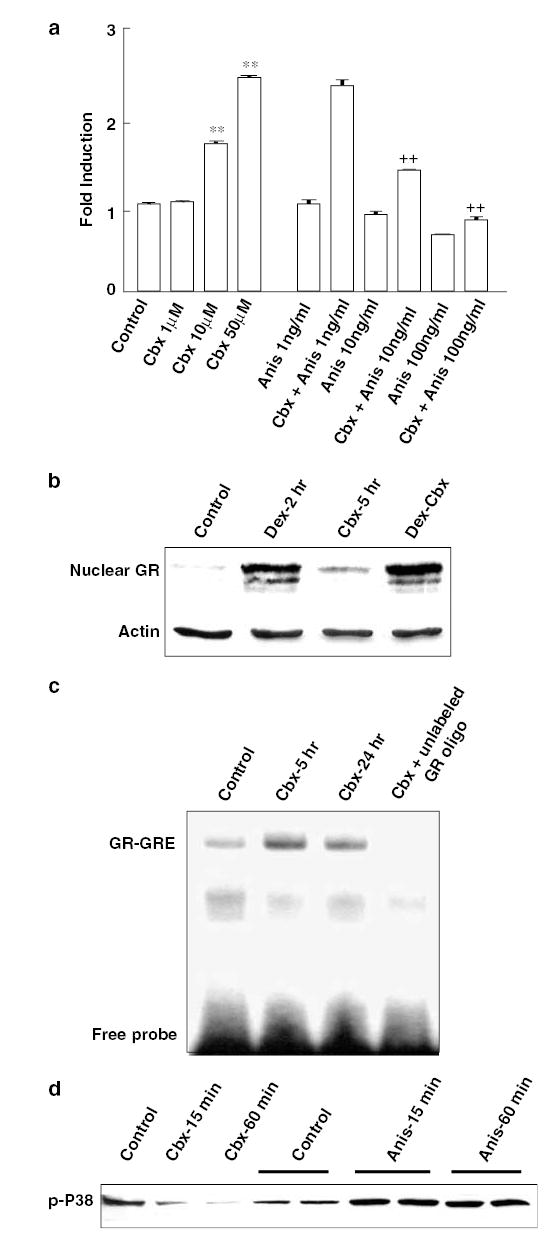
(a) PC12 cells stably transfected with an MMTV-luciferase construct were grown in 12-well culture plates to 80% confluency and treated with celecoxib (Cbx) and anisomycin (Anis) alone or in combination at the concentrations indicated. Cells treated with anisomycin plus celecoxib were incubated for 1 h with anisomycin prior to addition of celecoxib. Cells were harvested after 5 h incubation, and luciferase activity was assessed. Each data point represents the mean ± SE from at least three separate experiments. **Indicates P < 0.001 compared to control, ** Indicates P < 0.001 compared to Cbx alone. (b) To assess GR nuclear translocation, PC12 cells were treated with celecoxib (50 μM) for 5 h in the presence or absence of Dex (10 nM, 2 h). Nuclear extracts (25 μg) from each treatment group were analyzed by Western blot using a GR antibody from ABR-Affinity BioReagents (Golden, CO, USA). Nuclear extracts were obtained by resuspending nuclear pellets in high-salt buffer (20 mM HEPES, pH 7.9, 420 mM NaCl, 10 mM KCl, 0.1 mM Na3VO4, 1 mM EDTA, 1 mM EGTA, 20% glycerol, 0.5 mM PMSF and 1 mM DTT) and vigorously rocking for 30 min at 4°C. Nuclear extracts were then collected after centrifugation for 30 min at 48 000 rpm (4°C). GR—glucocorticoid receptor. Actin was used as a loading control. (c) The effect of celecoxib (50 μM) on GR-GRE binding was examined in 10 μg nuclear extracts (prepared as above) from PC12 cells using EMSA and 32P-labeled synthetic double-strand GRE oligonucleotides (5′-AAG ATT CAG GTC ATG ACC TGA GGA GA). Data are from a representative experiment of three separate experiments. (d) To examine the effects of treatments on p38 MAPK, total cell extracts were isolated after cells were treated with celecoxib (50 μM) and/or anisomycin (50 ng/ml) as indicated. Protein (50 μg) from each treatment group was analyzed by Western blot using a monoclonal antibody against phosphorylated p38 MAPK.
Acknowledgments
This work was supported in part by grants from the National Institute of Mental Health (MH-067041) and the Emory University Research Committee Award.
References
- 1.Raison CL, et al. Am J Psychiatry. 2003;160:1554–1565. doi: 10.1176/appi.ajp.160.9.1554. [DOI] [PubMed] [Google Scholar]
- 2.Licinio J, et al. Mol Psychiatry. 1999;4:317–327. doi: 10.1038/sj.mp.4000586. [DOI] [PubMed] [Google Scholar]
- 3.Pariante CM, et al. Biol Psychiatry. 2001;49:391–404. doi: 10.1016/s0006-3223(00)01088-x. [DOI] [PubMed] [Google Scholar]
- 4.Wang X, et al. Mol Psychiatry. 2004;9:65–75. doi: 10.1038/sj.mp.4001339. [DOI] [PubMed] [Google Scholar]
- 5.Rogatsky I, et al. Proc Natl Acad Sci USA. 1998;95:2050–2055. doi: 10.1073/pnas.95.5.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McKay LI, et al. Mol Endocrinol. 1998;12:45–56. doi: 10.1210/mend.12.1.0044. [DOI] [PubMed] [Google Scholar]
- 7.Pariante CM, et al. J Endocrinol. 2001;169:309–320. doi: 10.1677/joe.0.1690309. [DOI] [PubMed] [Google Scholar]
- 8.Paccani SR, et al. J Biol Chem. 2002;277:1509–1513. doi: 10.1074/jbc.M110676200. [DOI] [PubMed] [Google Scholar]
- 9.Pariante CM, et al. Mol Pharmacol. 1997;52:571–581. doi: 10.1124/mol.52.4.571. [DOI] [PubMed] [Google Scholar]
- 10.Murray HJ, et al. Neuropharmacology. 2003;44:374–380. doi: 10.1016/s0028-3908(02)00375-1. [DOI] [PubMed] [Google Scholar]
- 11.Monje ML, et al. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]