

Abstract
Initiation of Na+-glucose cotransport in intestinal absorptive epithelia causes NHE3 to be translocated to the apical plasma membrane, leading to cytoplasmic alkalinization. We reported recently that this NHE3 translocation requires ezrin phosphorylation. However, the kinase that phosphorylates ezrin in this process has not been identified. Because Akt has also been implicated in NHE3 translocation, we investigated the hypothesis that Akt phosphorylates ezrin. After initiation of Na+-glucose cotransport, Akt is activated with kinetics that parallel those of ezrin phosphorylation. Inhibition of p38 MAP kinase, which blocks ezrin phosphorylation, also prevents Akt activation. Purified Akt directly phosphorylates recombinant ezrin at threonine 567 in vitro in an ATP-dependent manner. This in vitro phosphorylation can be prevented by Akt inhibitors. In intact cells, inhibition of either phosphoinositide 3-kinase, an upstream regulator of Akt, or inhibition of Akt itself using inhibitors validated in vitro prevents ezrin phosphorylation after initiation of Na+-glucose cotransport. Specific small interfering RNA knockdown of Akt2 prevented ezrin phosphorylation in intact cells. Pharmacological Akt inhibition or Akt2 knockdown also prevented NHE3 translocation and activation after initiation of Na+-glucose cotransport, confirming the functional role of Akt2. These studies therefore identify Akt2 as a critical kinase that regulates ezrin phosphorylation and activation. This Akt2-dependent ezrin phosphorylation leads to NHE3 translocation and activation.
Ezrin is a member of the ezrin-radixin-moesin (ERM)1 protein family (1, 2). These proteins share at least 70% sequence homology and a common structure (2, 3). Ezrin activity is regulated by intramolecular interactions between N- and C-terminal ERM association domains. In the inactive state, these intramolecular interactions mask protein 4.1-ERM and actin-binding domains (4–7), resulting in inactive ezrin monomers and oligomers. Ezrin phosphorylation at threonine 567 releases these intramolecular interactions, allowing active ezrin monomers to link target molecules to the actin cytoskeleton (8–10). Thus, phosphorylation at threonine 567 is a critical regulator of ezrin function.
Given the widespread involvement of ezrin family members in diverse cellular functions, it is not surprising that several kinases have been implicated in threonine 567 ezrin phosphorylation. Protein kinase C (PKC) α, PKCθ, and rho kinase have all been implicated in ezrin phosphorylation (9, 11–13). For example, inhibition of rho kinase can prevent intracellular ezrin threonine 567 phosphorylation (14, 15), whereas expression of activated rhoA can stimulate ezrin phosphorylation at threonine 567 (15). Consistent with a direct effect of rho kinase, recombinant radixin is phosphorylated in vitro by rho kinase at the homologous threonine residue (9). Despite these data, rho can regulate ezrin phosphorylation via other mechanisms (16–18). Moreover, in Drosophila melanogaster, it is clear that the ERM protein moesin can regulate rho (19), suggesting that rho and ERM proteins may regulate one another in a feedback fashion. Thus, although studied intensively, the role of rho in ezrin activation remains incompletely understood. More importantly, there are many examples in which PKCα, PKCθ, and rho kinase are clearly not involved in ezrin phosphorylation (18). In these cases, the specific kinase responsible for ezrin phosphorylation remains unknown.
NHE3, the apical Na+-H+ exchanger expressed in renal and intestinal epithelia, plays a crucial role in organismal Na+-absorption, acid-base homeostasis, and volume regulation. For example, NHE3 knockout mice suffer from mild diarrhea and are acidotic and hypotensive (20). NHE3 activity can be regulated by the actin cytoskeleton (21–23). Although this may be caused in part by regulation of NHE3 at the plasma membrane, it is also clear that translocation of NHE3 to the plasma membrane contributes significantly to NHE3 up-regulation (24, 25). Although the mechanisms that regulate NHE3 translocation are incompletely understood, recent studies suggest that Akt may be involved in this process. For example, transfection of fibroblasts with constitutively active Akt increases surface NHE3 activity (26). Although that study did not measure surface NHE3 in Akt-transfected cells (26), NHE3 translocation and Akt activation both occur as a consequence of lysophosphatidic acid treatment (24).
We have shown that NHE3 activity is increased after the initiation of SGLT1-mediated Na+-glucose cotransport (27). This may reflect a global signal to up-regulate enterocyte nutrient and ion transport upon the detection of nutrients, i.e. glucose, in the intestinal lumen. This increased NHE3 activity requires ezrin phosphorylation at threonine 567 and is at least partly the result of NHE3 exocytosis (25). Although both ezrin phosphorylation and NHE3 activation require p38 MAP kinase activation (25, 27), we considered it unlikely that p38 MAP kinase could phosphorylate ezrin directly because the residues surrounding threonine 567 lack the proline residues typical of p38 MAP kinase substrates. The goal of these studies was to identify the kinase that phosphorylates ezrin, thereby triggering NHE3 exocytosis, after the initiation of Na+-glucose cotransport. We show here that Akt2-mediated ezrin phosphorylation is necessary for NHE3 translocation and activation after initiation of Na+-glucose cotransport. This is the first demonstration of Akt-mediated ezrin phosphorylation and suggests a complex functional association between these critical signaling molecules.
MATERIALS AND METHODS
Cell Culture and Stimulation of SGLT1-mediated Na+-Glucose Cotransport
SGLT1-expressing Caco-2 cells were maintained as described previously (25, 28). For experimental use, cells were grown as confluent monolayers (25). For studies using dominant negative ezrin, Caco-2 cells stably transfected with VSVG tagged N-terminal (1–309) ezrin were used (25). For these studies, Caco-2 cells stably transfected with VSVG-tagged wild-type ezrin were used as controls, as described previously (25).
Before initiation of SGLT1-mediated Na+-glucose cotransport, monolayers were pre-incubated for 20 min at 37 °C in nominally HCO3−-free HBSS with 25 mm mannose and 0.5 mm phloridzin, to inhibit Na+-glucose cotransport. When used, LY294002, PD169316, H-89, and NL-71-101 (Calbiochem) were included during this pre-incubation as well as all subsequent media before cell lysis. Na+-glucose cotransport was initiated by transfer to nominally HCO3−-free Hanks’ balanced salt solution containing 25 mm glucose. At the times indicated in the figures, monolayers were scraped directly into 4 °C SDS-PAGE sample buffer and immediately boiled for 5 min before immunoblot analysis.
In Silico Analysis of Kinases
Analysis of potential kinases that may phosphorylate ezrin at threonine 567 was performed using Scansite 2.0 (29). A 46-residue sequence, ARDENKRTHNDIIHNENMRQGRDKYK-TLRQIRQGNTKQRIDEFEAL, composed of residues 541–586 of human ezrin, was input and scanned against all kinase groups at low stringency.
Immunoblot Analysis
Cell lysates were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes, as described previously (25). Ezrin and Akt phosphorylation were detected using polyclonal antisera against ezrin phosphorylated at threonine 567 and Akt phosphorylated at threonine 308 or serine 473 (Cell Signaling Technology, Beverly, MA). Monoclonal 3C12 anti-total ezrin (Sigma) and polyclonal anti-total Akt (Cell Signaling Technology) were used to probe total levels of these proteins. Isoform-specific anti-Akt1, anti-Akt2, and anti-Akt3 antibodies were from Cell Signaling Technology. Antibody binding was detected with goat anti-mouse or anti-rabbit IgG peroxidase-conjugated antibodies (Cell Signaling Technology) and chemiluminescence (Pierce Biotechnology, Rockford, IL) on Eastman Kodak BioMax MR film. Signal intensity, corrected for background, was analyzed by densitometry using NIH ImageJ 1.29. For quantitative analysis of protein phosphorylation, paired aliquots of each sample were immunoblotted for phosphorylated or total protein and the ratio of these values was considered the phosphorylation state of that sample in arbitrary units. Experiments were performed in triplicate or greater. These values were grouped for statistical analysis.
In Vitro Kinase Assay
Caco-2 monolayers were harvested in lysis buffer (20 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerol phosphate, 1 mm Na3VO4, and 1 μg/ml leupeptin) after initiation of Na+-glucose cotransport. Lysates were pre-cleared by centrifugation at 15,000 × g for 5 min at 4 °C, and immunoprecipitated by incubation at 4 °C with Sepharose-conjugated monoclonal anti-Akt 1G1 antibody, which binds preferentially to Akt phosphorylated at serine 473 (Cell Signaling Technology) for 2.5 h. Beads were then washed twice with lysis buffer, twice with kinase buffer (25 mm Tris, pH 7.5, 5 mm β-glycerol phosphate, 2 mm dithiothreitol, 0.1 mm Na3VO4, and 10 mm MgCl2), and resuspended in kinase buffer supplemented with 70 μm ATP and 1 μg of recombinant-full-length GST-ezrin with or without pharmacological inhibitors, as indicated. After 30 min at 30 °C reactions were terminated by addition of 0.5 volumes of 3× SDS sample buffer. Samples were analyzed by SDS-PAGE and immunoblotted for threonine 567 phosphorylated and total ezrin.
siRNA Knockdown of Akt2 Expression
Caco-2 monolayers were transfected with either SignalSilence Akt2 siRNA duplexes or nonspecific control GC content-matched siRNA duplexes (Cell Signaling Technology). These siRNA duplexes are reported to inhibit expression of Akt2, but not Akt1 or Akt3. Our preliminary optimization studies showed that optimal protein knockdown occurred 4 days after transfection of 200 nm siRNA using Lipofectamine 2000 (Invitrogen). At these times, immunoblot analyses showed that expression of Akt1 remained low but detectable, Akt3 remained undetectable, and expression of NHE3, ezrin, and actin were unaffected by siRNA treatment. Similar immunoblots were routinely used to verify specific Akt2 knockdown (i.e. knockdown of Akt2 without knockdown of Akt1, ezrin, NHE3, or actin) in all experiments.
Immunofluorescent Detection of Surface NHE3
Surface NHE3 expression was performed as described previously (25). In brief, Caco-2 monolayers were fixed in 1% paraformaldehyde in PBS for 15 min, washed, blocked, and incubated with affinity-purified rabbit polyclonal antisera at 4 °C. After washing, bound anti-NHE3 antibodies were detected using Alexa-594 conjugated goat anti-rabbit antisera (Molecular Probes, Eugene, OR). Total NHE3 detection was performed identically except for the inclusion of 0.05% saponin in wash and antibody incubation buffers. Stained monolayers were mounted in Slowfade (Molecular Probes), and images were collected using a 100× Plan Apo objective and a DMLB epifluorescence microscope (Leica Microsystems, Bannockburn, IL) equipped with an 88000 filter set (Chroma Technology Corp, Brattleboro, VT) and Coolsnap HQ camera (Roper Scientific, Duluth, GA) controlled by Metamorph 6 (Universal Imaging, Downingtown, PA). Post-acquisition analyses of mean pixel intensity was performed using Metamorph. In all image acquisition and analysis, identically timed exposures within a single experiment were used for quantitative comparisons, as described previously (25).
Measurement of pHi
Confluent monolayers were incubated in 25 mm mannose containing 3.5 μm of acetoxymethyl ester of 2′-7′-bis(2-carboxyethyl)-5(6)carboxyfluorescein (Molecular Probes, Eugene, OR) for 15 min. 2′-7′-bis(2-Carboxyethyl)-5(6)carboxyfluorescein-loaded cells were analyzed using a Model RC-M fluorometer (Photon Technology International, Monmouth Junction, NJ). Fluorescence was measured at excitation wavelengths of 439 and 507 nm with emission at 535 nm, as described previously (25). Data were analyzed using Felix32 software (Photon Technology International). pHi responses were compared at 2 min after initiation of Na+-glucose cotransport, as described previously (25).
RESULTS
In Silico Analysis of Ezrin Threonine 567
To identify potential ezrin kinases in a non-biased manner, a 46-residue sequence composed of residues 541–586 of human ezrin was analyzed using Scansite 2.0 (29). Scansite 2.0 analyzes more than 60 protein phosphorylation sequence motifs based on data obtained from peptide library and phage display phosphorylation studies (29). Our low stringency analysis of this ezrin domain identified only three kinases, protein kinase A (PKA), PKCζ, and Akt, as potential ezrin threonine 567 kinases. Because PKA is a well known stimulus that inhibits NHE3 (30), we did not consider it further. We also did not consider PKCζ further because it has not been previously implicated in either NHE3 regulation or ezrin phosphorylation, and the sequence recognition score reported by Scansite suggested that PKCζ was the least likely of these three potential ezrin kinases. It is noteworthy that although Akt has never been implicated in ezrin phosphorylation, it has been associated with NHE3 translocation. This prompted further investigation.
Ezrin and Akt Activation Follow Initiation of Na+-Glucose Cotransport
To evaluate the potential role of Akt in ezrin phosphorylation, we first sought to determine whether Akt activation coincided with the ezrin phosphorylation that follows initiation of Na+-glucose cotransport. Akt activation was assessed as phosphorylation at the threonine 308 and serine 473 regulatory sites, which are known to correlate with Akt kinase activity (31). Increased phosphorylation at both sites, indicating Akt activation, increased after initiation of Na+-glucose cotransport (p < 0.01; Fig. 1). This Akt phosphorylation occurred with kinetics that correlated closely with ezrin phosphorylation at threonine 567 (r = 0.99; Fig. 1). Although these data do not prove a causative association, they are consistent with a potential role for Akt in this process.
Fig. 1. Initiation of Na+-glucose cotransport results in increased ezrin phosphorylation at threonine 567 and a corresponding rise in Akt phosphorylation at both threonine 308 and serine 473.
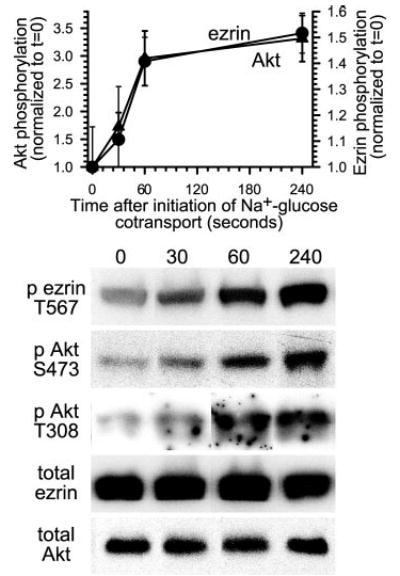
Monolayers were lysed at indicated times after initiation of Na+-glucose cotransport, and lysates were immunoblotted for threonine 567 phosphorylated ezrin, total ezrin, threonine 308 phosphorylated Akt, serine 473 phosphorylated Akt, and total Akt. Densitometric analysis of triplicate samples from this experiment are shown and represent the mean and S.D. of the ratios of threonine 567 phosphorylated ezrin to total ezrin (circles) and serine 473 phosphorylated Akt to total Akt (triangles). These ratios increased by 52 ± 3% and 231 ± 29%, respectively, at 240 s after initiation of Na+-glucose cotransport (p < 0.01). Increases in threonine 308 Akt phosphorylation were similar to serine 473 phosphorylation. Representative immunoblots are shown. Results are typical of at least three independent experiments, each with triplicate samples.
We have shown previously that p38 MAP kinase activation is required for ezrin phosphorylation at threonine 567 (25). Thus, to further explore the relationship between ezrin and Akt phosphorylation, we examined whether p38 MAP kinase activity was required for Akt phosphorylation, as it is for ezrin phosphorylation (25). Similar to its effect on ezrin phosphorylation, p38 MAP kinase inhibition prevented phosphorylation of Akt (Fig. 2). Thus, phosphorylation of both Akt and ezrin seems to occur downstream of signaling events that include p38 MAP kinase activation.
Fig. 2. Inhibition of p38 MAP kinase prevents both ezrin and Akt activation.
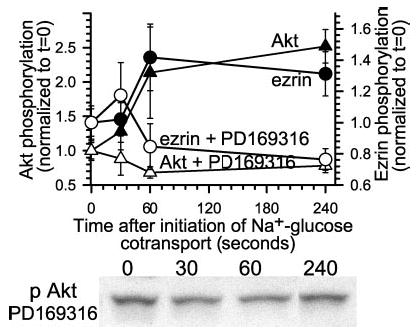
Monolayers were pretreated with the p38 MAP kinase inhibitor PD169316 (10 μm) and lysed at indicated times after initiation of Na+-glucose cotransport. Lysates were immunoblotted for threonine 567 phosphorylated ezrin, total ezrin, serine 473 phosphorylated Akt, and total Akt. Rather than a 152 ± 15% increase in serine 473 Akt phosphorylation 240 s after initiation of Na+-glucose cotransport (black triangles), serine 473 Akt phosphorylation decreased 22 ± 3% in PD169316-treated monolayers (white triangles). Likewise, as we have reported previously (25), p38 MAP kinase inhibition blocked increases in ezrin phosphorylation after initiation of Na+-glucose cotransport (black and white circles). Representative immunoblots of Akt phosphorylated at serine 473 are shown. Results are typical of more than three independent experiments, each with triplicate samples.
Na+-Glucose Cotransport-dependent Akt Activation Does Not Require Ezrin Function
The data above are consistent with the hypothesis that Akt directly phosphorylates ezrin at threonine 567. However, they are also consistent with the converse (i.e. that ezrin activation leads to downstream activation of Akt). The latter has been suggested based on defective Akt activation in kidney-derived epithelia expressing mutant and dominant-negative ezrin constructs (32, 33). We have shown that expression of a dominant-negative ezrin construct consisting of the amino-terminal 309 amino acids (Nter) completely inhibits NHE3 activation after initiation of Na+-glucose cotransport (25). Thus, we used cells expressing the Nter dominant-negative construct to ask whether ezrin function is required for Akt activation to occur. Akt phosphorylation occurred after initiation of Na+-glucose cotransport in control cells transfected with full-length wild-type ezrin (Fig. 3). Cells stably transfected with Nter ezrin demonstrated similar Akt phosphorylation after Na+-glucose cotransport (Fig. 3). Thus, the Akt phosphorylation that follows Na+-glucose cotransport does not require upstream ezrin activation.
Fig. 3. Expression of dominant-negative ezrin does not prevent Akt activation.
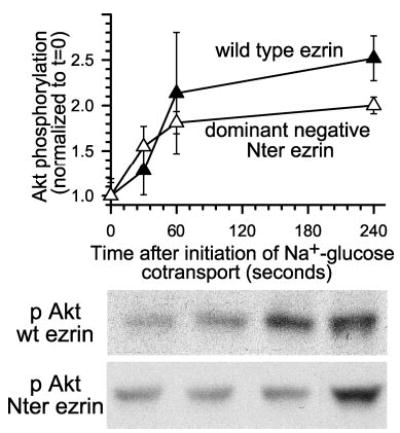
Monolayers stably transfected with wild-type (wt, black triangles) or truncated amino-terminal dominant negative (Nter, white triangles) ezrin constructs (25), were lysed and immunoblotted as above. At 240 s after initiation of Na+-glucose cotransport, Akt phosphorylation increased by 154 ± 7% or 100% ± 5% in wild-type and dominant-negative ezrin-transfected monolayers, respectively. Representative immunoblots of Akt phosphorylated at serine 473 in dominant-negative ezrin transfectants are shown. Results are typical of at least three independent experiments, each with triplicate samples.
Akt Directly Phosphorylates Ezrin at Threonine 567
Although they support the hypothesis that Akt directly phosphorylates ezrin, the data above do not exclude activation of an intermediate kinase, downstream of Akt, that is directly responsible for ezrin phosphorylation. To assess whether Akt can directly phosphorylate ezrin, in vitro kinase assays were performed using immunopurified active Akt and recombinant ezrin. Active Akt was isolated from cells lysed after initiation of Na+-glucose cotransport using an immobilized monoclonal antibody that recognizes activated Akt. The activated Akt beads were then combined with recombinant GST-ezrin produced in bacteria. Akt catalyzed ezrin phosphorylation at threonine 567 (Fig. 4). This ezrin phosphorylation was strictly dependent on the presence of ATP. To pharmacologically confirm that the immunopurified Akt was responsible for the ezrin phosphorylation observed, two compounds known to inhibit Akt, H-89 NL-71-101 (34, 35), were included in the kinase reaction. NL-71-101 is a more specific inhibitor of Akt that was derived from H-89 (34, 35). NL-71-101 (34, 35) reduced in vitro ezrin phosphorylation at threonine 567 by 14 ± 2% at 10 μm and 64 ± 5% at 50 μm (Fig. 4; p < 0.05). However, NL-71-101 also inhibits PKA with an IC50 only 2.4-fold greater than that required for Akt inhibition (35). To determine whether the PKA-inhibitory activity of NL-71-101 could be related to its effect on ezrin phosphorylation, NL-71-101 was compared with its parent compound, H-89, which inhibits Akt and PKA 1.5- and 257-fold more potently than NL-71-101, respectively (35). 10 μm H-89 inhibited ezrin phosphorylation by 32 ± 3% (Fig. 4; p < 0.05), consistent with partial inhibition of Akt, but not the complete PKA inhibition that would occur at that dose. Thus, the inhibition of ezrin phosphorylation by NL-71-101 and H-89 is caused by inhibition of Akt and not PKA. Therefore, in a reductionist assay using immunopurified Akt and ezrin, Akt directly phosphorylates ezrin at threonine 567.
Fig. 4. Akt directly phosphorylates ezrin threonine 567 in vitro.
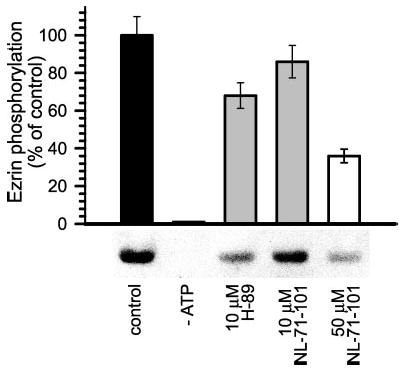
Immunopurified Akt was incubated with full-length GST-ezrin with or without Akt inhibitors for the in vitro kinase assay. Reaction products were analyzed by SDS-PAGE and immunoblot for threonine 567 phosphorylated and total ezrin. Akt phosphorylated ezrin at threonine 567 in the presence, but not absence, of exogenous ATP (p < 0.01). This phosphorylation was blocked by NL-71-101 (p < 0.05) in a dose-dependent manner. 10 μm H-89 inhibited ezrin phosphorylation (p < 0.05) to an extent similar to that of 10 μm NL-71-101, demonstrating that the inhibitory effect of these drugs on ezrin phosphorylation is not caused by PKA inhibition. Results are typical of five independent experiments, each with duplicate samples.
Akt Inhibition Prevents Ezrin Phosphorylation after Initiation of Na+-Glucose Cotransport
Based on the observations that Akt and ezrin are both phosphorylated after initiation of Na+-glucose cotransport and that Akt can directly phosphorylate ezrin in an in vitro kinase assay, we sought to determine whether Akt activity was required for ezrin phosphorylation to occur in intact monolayers. To address this question, three complementary approaches were used. First, the PI3 kinase-Akt pathway was blocked using the PI3 kinase inhibitor LY294002 (36). PI3 kinase activity is frequently required for Akt activation (37–40). Consistent with this requirement, LY294002 suppressed Akt phosphorylation in Caco-2 cells to undetectable levels (i.e. below basal levels in cells without active Na+-glucose cotransport (data not shown)) and completely prevented ezrin phosphorylation (Fig. 5; p < 0.02). Akt kinase activity was also inhibited directly with 50 μm NL-71-101, which completely prevented increases in threonine 567 ezrin phosphorylation after initiation of Na+-glucose cotransport (Fig. 5; p < 0.01). These data suggest that, in intact epithelial monolayers, ezrin phosphorylation after initiation of Na+-glucose cotransport requires Akt activity. Rho kinase is known to phosphorylate ezrin (9) and has also been implicated in NHE3 inhibition (22, 41). Thus, we considered the possibility that rho kinase regulates the coordinate activation of ezrin and NHE3 after initiation of Na+-glucose cotransport. Inhibition of rho kinase using Y-27632 did not prevent increases in ezrin phosphorylation after Na+-glucose cotransport and also failed to prevent NHE3 activation and the resulting increase in cytoplasmic pH after initiation of Na+-glucose cotransport (data not shown). Therefore, ezrin phosphorylation and NHE3 activation after initiation of Na+-glucose cotransport are independent of rho kinase.
Fig. 5. Pharmacological inhibition of Akt in intact cells prevents ezrin phosphorylation.

Monolayers were lysed at each specific time point after initiation of Na+-glucose cotransport and immunoblotted for threonine 567 phosphorylated and total ezrin. Inhibition of Akt activity with 50 μm NL-71-101 (inverted triangles; p < 0.01) or 50 μm LY294002 (squares; p < 0.02) caused ezrin phosphorylation to fall by 26 ± 5% and 35 ± 4%, respectively, relative to control monolayers (circles), in which ezrin phosphorylation increased by 115 ± 23%. Representative immunoblots of ezrin phosphorylated at threonine 567 are shown. Results are typical of at least four independent experiments, each with triplicate samples.
To define the specific Akt isoform involved in ezrin regulation, Akt isoform expression was assessed in Caco-2 cells. SDS-PAGE immunoblots suggested that Akt2 is the predominant isoform expressed in these cells (Fig. 6A), consistent with the pattern of expression in rabbit ileal villus enterocyte brush border membranes (42). Akt1 was detected only faintly using isoform-specific antibodies, and Akt3 was not detected at all. To determine whether Akt2 is also the isoform responsible for ezrin phosphorylation, isoform-specific siRNA targeted to Akt2 was used to specifically knock down Akt2 protein levels by 55 ± 9% relative to monolayers treated with nonspecific siRNA (Fig. 6B; p < 0.05). Expression of related proteins, including Akt1, ezrin, NHE3, and actin, was not affected by either siRNA treatment. Akt2 knockdown prevented increases in threonine 567 ezrin phosphorylation after initiation of Na+-glucose cotransport (Fig. 6C; p < 0.05). Therefore, because both pharmacological Akt inhibition and specific knockdown of Akt2 prevent ezrin phosphorylation after initiation of Na+-glucose cotransport, this ezrin phosphorylation requires Akt2 activity.
Fig. 6. Ezrin activation after initiation of Na+-glucose cotransport requires Akt2 expression.
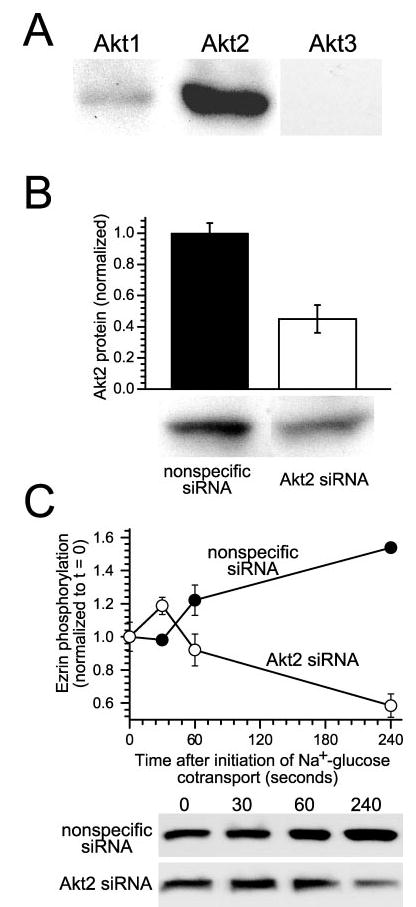
A, Caco-2 intestinal epithelial cells primarily express Akt2. To determine which Akt isoforms are expressed in intestinal epithelia, confluent monolayers were lysed and immunoblotted using isoform-specific antisera. Both Akt1 and 2 are present, but Akt2 is the predominant isoform expressed in intestinal epithelial cells. Akt1 was detected only weakly. No signal was detected in immunoblots using anti-Akt3 antibodies. Results are typical of three independent experiments, each with duplicate samples. B, Akt2 protein expression can be specifically knocked down using siRNA. Treatment of monolayers with Akt2-specific siRNA resulted in knockdown of protein levels by 55 ± 9% relative to control monolayers transfected with nonspecific siRNA (p < 0.05). Expression of Akt1, ezrin, NHE3, and actin was not changed by either specific or nonspecific siRNA (data not shown). Results are typical of five independent experiments, each with triplicate samples. C, knockdown of Akt2 expression in intact cells blocks ezrin phosphorylation after initiation of Na+-glucose cotransport. Monolayers transfected with either nonspecific siRNA (black circles) or Akt2-specific siRNA (white circles) were lysed at indicated timepoints after initiation of Na+-glucose cotransport. After 240 s, ezrin phosphorylation fell by 42 ± 5% in monolayers with knocked down Akt2 expression (white circles), whereas ezrin phosphorylation increased by 154 ± 4% in monolayers transfected with nonspecific siRNA (black circles; p < 0.05). Results are typical of three independent experiments, each with triplicate samples.
NHE3 Translocation and Activation after Na+-Glucose Cotransport Requires Akt2
Initiation of Na+-glucose cotransport triggers NHE3 translocation to the plasma membrane, thereby activating Na+-H+ exchange and causing cytoplasmic alkalinization via a mechanism that requires ezrin phosphorylation (25). If Akt2-dependent ezrin phosphorylation is critical to NHE3 translocation and cytoplasmic alkalinization after initiation of Na+-glucose cotransport, Akt inhibition or Akt2 knockdown should prevent both. In support of this hypothesis, the Akt inhibitor NL-71-101 completely prevented NHE3 translocation to the plasma membrane after initiation of Na+-glucose cotransport (Fig. 7A; p < 0.01). Similar to the effect of 50 μm NL-71-101 on ezrin phosphorylation, surface NHE3 levels fell by 26 ± 1% after initiation of Na+-glucose cotransport in NL-71-101-treated monolayers. This inhibition of NHE3 translocation was matched by 97 ± 4% and 77 ± 6% inhibition of NHE3-dependent cytoplasmic alkalinization by LY294002 and 50 μm NL-71-101, respectively (Fig. 7B; p < 0.01). In a pattern similar to partial inhibition of ezrin phosphorylation, 10 μm NL-71-101 and 10 μm H-89 only partially inhibited NHE3-dependent cytoplasmic alkalinization, by 51 ± 8% and 39 ± 5%, respectively (Fig. 7B; p < 0.05), confirming that the effect of NL-71-101 is not caused by PKA inhibition.
Fig. 7. Akt activity is required for NHE3 translocation and cytoplasmic alkalinization after Na+-glucose cotransport.
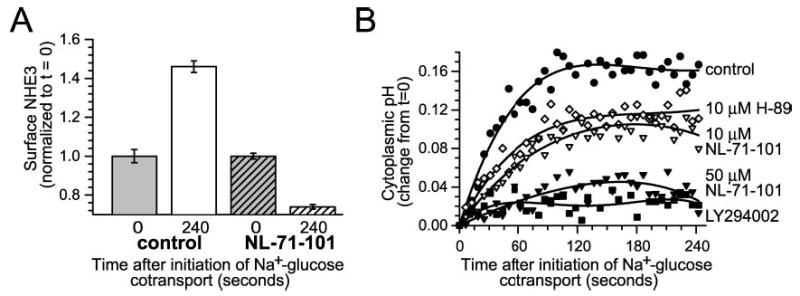
A, Akt activity is necessary for ezrin translocation after initiation of Na+-glucose cotransport. Monolayers were fixed 240 s after initiation of Na+-glucose cotransport in the absence or presence of 50 μm NL-71-101. Surface NHE3 was assessed by surface immunofluorescence of nonpermeabilized cells, as described previously (25). 50 μm NL-71-101 completely prevented increases in surface NHE3 expression after initiation of Na+-glucose cotransport (p < 0.01). Results are typical of three independent experiments, each with triplicate samples in which each sample was evaluated over at least 10 separate fields. B, Akt activity is required for NHE3-dependent cytoplasmic alkalinization. Pharmacological inhibition of Akt with 50 μm NL-71-101 (black inverted triangles) or of PI3 kinase with LY294002 (black squares) almost completely prevented NHE3-dependent cytoplasmic alkalinization after initiation of Na+-glucose cotransport (p < 0.01). In contrast, 10 μm H-89 (white diamonds) or 10 μm NL-71-101 (white inverted triangles) only partially inhibited cytoplasmic alkalinization, paralleling their incomplete inhibition of ezrin phosphorylation in vitro (see Fig. 4). Results are typical of five independent experiments.
Although the results comparing NL-71-101 and H-89 support a role for Akt in this ezrin-dependent NHE3 activation, they suffer from limitations of many studies that depend on pharmacological agents. Moreover, these drugs are not able to distinguish between Akt isoforms. Thus, we reduced Akt2 protein expression by using siRNA knockdown using isoform-specific siRNA as in Fig. 6B. siRNA-mediated Akt2 knockdown prevented NHE3 translocation to the plasma membrane by 78 ± 3% (Fig. 8A; p < 0.01). Cytoplasmic alkalinization after initiation of Na+-glucose cotransport was reduced by 41 ± 3% after Akt2 siRNA treatment (Fig. 8B; p < 0.05). Thus, the NHE3 translocation and activation that occurs secondary to ezrin phosphorylation requires Akt2 to be both present and active.
Fig. 8. Akt2 expression is required for NHE3 translocation and cytoplasmic alkalinization after Na+-glucose cotransport.
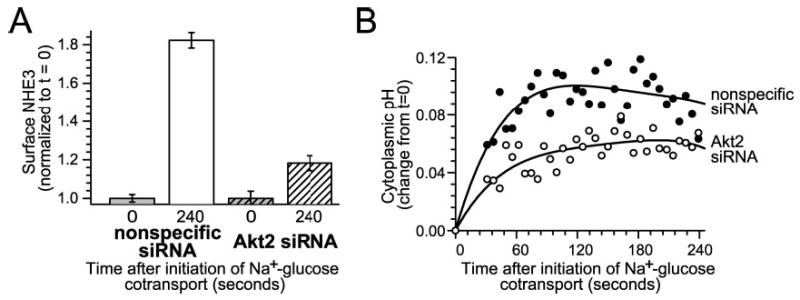
A, Akt2 knockdown prevents NHE3 translocation to the plasma membrane after initiation of Na+-glucose cotransport. Monolayers transfected with either nonspecific siRNA or Akt2-specific siRNA were fixed 240 s after initiation of Na+-glucose cotransport. Surface NHE3 was assessed as above. Akt2 knockdown prevented 78% ± 3% of the increase in surface NHE3 expression detected in monolayers transfected with nonspecific siRNA (p < 0.01). Results are typical of three independent experiments, each with triplicate samples in which each sample was evaluated over at least 10 separate fields. B, NHE3-dependent cytoplasmic alkalinization is inhibited by Akt2 knockdown. Akt2 knockdown (white circles) inhibited 41 ± 3% of NHE3-dependent cytoplasmic alkalinization after initiation of Na+-glucose cotransport (p < 0.05), relative to monolayers transfected with nonspecific siRNA (black circles), roughly paralleling the 55 ± 9% knockdown of Akt2 expression in these monolayers. Results are typical of six independent experiments.
DISCUSSION
We have characterized the physiological NHE3 activation and translocation to the surface that follows initiation of Na+-glucose cotransport as a process that depends on ezrin phosphorylation (25). This ezrin phosphorylation requires upstream p38 MAP kinase activation but is unlikely to be mediated directly by p38 MAP kinase, because ezrin threonine 567 lacks the proline residues that are typically present within p38 MAP kinase targets. Rho kinase, PKA, and some PKC isoforms have been reported to phosphorylate ezrin or other ERM-related proteins (9, 11, 12, 43). However, rho kinase (27) or PKA inhibition does not prevent NHE3 activation after initiation of Na+-glucose cotransport, and both PKA and PKC activation are well known to reduce NHE3 activity and surface expression (30, 44). Thus, we sought an alternative kinase that might be directly responsible for ezrin phosphorylation in this physiologically relevant model of NHE3 activation. We performed in silico analysis, which suggested Akt might phosphorylate ezrin at threonine 567. Akt has not been previously reported as a regulator of ezrin phosphorylation. However, Akt has been associated with translocation of NHE3 to the surface by undefined mechanism(s) (26). We therefore tested the hypothesis that the role of Akt might be to phosphorylate ezrin at threonine 567.
We found that Akt is activated after initiation of Na+-glucose cotransport with kinetics that parallel those of ezrin phosphorylation. We therefore asked if p38 MAP kinase inhibition could prevent Akt activation, because we have previously shown that, in this model, ezrin phosphorylation requires upstream p38 MAP kinase activation. The data show that, like ezrin phosphorylation, Akt phosphorylation requires p38 MAP kinase activity. This is consistent with previous studies in smooth muscle and mesenchymal cells suggesting a relationship between p38 MAP kinase and downstream Akt activation (45), including a recent report that p38 MAP kinase and its downstream effector MAPKAP-2 can mediate Akt activation (46). A separate study, also in muscle cells, demonstrated that Akt serine 473 is a substrate for MAPKAP-2 phosphorylation (31). Our data suggest that this signaling pathway linking p38 MAP kinase to Akt may also be active in epithelia.
To test whether Akt could directly phosphorylate ezrin, we performed in vitro kinase assays using immunopurified active Akt and recombinant ezrin. These studies show that Akt can phosphorylate ezrin at threonine 567. This result was predicted by the in silico analysis, but contrasts with previous data suggesting that, in regulation of cell survival, cells expressing dominant-negative ezrin fail to activate Akt (32, 33). Thus, we examined Akt activation in cells expressing dominant-negative ezrin. This dominant-negative Nter ezrin expression is sufficient to block NHE3 translocation and activation (25). However, the data show that Akt is activated normally after initiation of Na+-glucose cotransport in monolayers of cells expressing dominant-negative ezrin. Thus, we conclude that Akt activation after initiation of Na+-glucose cotransport does not require ezrin activation.
Based on these data, we concluded that Akt is able to phosphorylate ezrin at threonine 567. However, we also wanted to know whether Akt is responsible for the ezrin phosphorylation that occurs in living cells after initiation of Na+-glucose cotransport. Thus, we used three complementary approaches to determine whether Akt is responsible for ezrin phosphorylation and downstream NHE3 translocation and activation. First, we inhibited the upstream Akt regulator PI3 kinase. This reduced Akt phosphorylation to undetectable levels and completely blocked increases in ezrin phosphorylation. Likewise, pharmacological inhibition of Akt blocked increases in ezrin phosphorylation. Finally, after showing that Akt2 is the predominant isoform in Caco-2 intestinal epithelial cells, we used siRNA to knock down Akt2 expression. This also prevented increases in ezrin phosphorylation after initiation of Na+-glucose cotransport. In addition, either pharmacological inhibition or siRNA knockdown of Akt prevented both NHE3-dependent cytoplasmic alkalinization and NHE3 surface translocation. Thus, Akt2 is required for ezrin phosphorylation as well as downstream NHE3 translocation and activation after initiation of Na+-glucose cotransport.
Although the role of ezrin in NHE3 regulation has generally been considered to be mediated through NHE3 regulatory factors (21, 23, 30, 47, 48), cell surface NHE3 expression is not altered by expression of mutant NHERF-1 (47), and brush border NHE3 content is not reduced in NHERF-1 knockout mice (49). Thus, another mechanism of ezrin-dependent NHE3 regulation (e.g. a direct interaction mediated by a putative ezrin-binding site at the C-terminal of NHE3 (21)) may be involved. Although Akt has been previously associated with translocation of NHE3 (24, 26, 42), the mechanisms of this regulation are incompletely defined. For example, EGF-mediated stimulation of PI3-kinase results in increased Akt activity within early endosome and brush border plasma membrane fractions that correlates with increased total and phosphorylated Akt2 at those sites. This PI3-kinase-dependent increase in Akt activity correlated with translocation of NHE3 to the brush border (50). Separate studies have shown that a significant fraction of brush border NHE3 is found in detergent-insoluble lipid rafts (50) that also contain Akt2 (42) and ezrin (51, 52). Until now, however, a functional relationship between ezrin and Akt2 has not been proposed. The data presented here suggest that one critical role of Akt2 in NHE3 regulation is to phosphorylate ezrin. Although other Akt2 targets may still be involved in this process, it is notable that dominant-negative ezrin prevents NHE3 translocation without altering Akt2 activation. Thus, when considered as a whole, these data and those from previous studies suggest that Akt2 works through ezrin to effect translocation of NHE3 from an intracellular pool to lipid raft domains within the brush border.
This Akt- and ezrin-dependent NHE3 translocation may also be relevant to studies of other transporters and receptors and explain otherwise unrelated data from disparate systems. For example, GLUT4 is translocated from intracellular pools to the adipocyte plasma membrane by Akt- and p38 MAP kinase-dependent mechanisms after insulin stimulation (53–55). In these cells, PI3 kinase mediates Akt activation (56, 57), but PI3 kinase-independent events are also required (58). These include activation of the small GTPase TC10, whose activity within lipid rafts regulates cortical actin (59) and is essential for docking and fusion of GLUT4-containing vesicles with the plasma membrane (60, 61). Thus, aspects of signal transduction pathways that mediate both NHE3 and GLUT4 trafficking and activation include p38 MAP kinase, PI3 kinase, Akt2, and actin. Although no role for ezrin in GLUT4 translocation has been reported, there are also no reports that it has been considered. Taken together, these data suggest the existence of a signalosome including p38 MAP kinase, PI3-kinase, Akt2, and ezrin that may regulate traffic between the plasma membrane and intracellular vesicles.
In summary, we show here that Akt2 phosphorylates ezrin after initiation of Na+-glucose cotransport. Akt2 activation requires p38 MAP kinase activity but is independent of ezrin function. Akt2-dependent ezrin phosphorylation is required for subsequent NHE3 translocation and activation. Thus, these studies identify ezrin activation as a critical role of Akt2 in NHE3 regulation. In addition to showing that Akt phosphorylates ezrin in this pathway, these data may also lead to understanding of other examples of rho-kinase independent ezrin activation as well as Akt-dependent trafficking events.
Acknowledgments
We thank Dr. Monique Arpin for generously sharing the dominant-negative and wild-type ezrin constructs used to create the Caco-2 transfected cell lines and Drs. Christopher Weber, Daniel Clayburgh, and Judith Turner for their critical reviews of this manuscript.
Footnotes
This work was supported by the National Institutes of Health (Grants R01-DK61931 to J. R. T. and R01-DK038510 to E. B. C.), The Crohn’s Colitis Foundation of America (to J. R. T.), The University of Chicago Digestive Disease Center (P30-DK42086), and the University of Chicago Cancer Center (P30-CA14599).
The abbreviations used are: ERM, ezrin-radixin-moesin; Nter, dominant negative ezrin construct consisting of the amino terminal 309 amino acids; PKA, protein kinase A; PKC, protein kinase C; MAP, mitogen-activated protein; siRNA, small interfering RNA; PI3, phosphoinositide 3.
References
- 1.Gautreau A, Louvard D, Arpin M. Curr Opin Cell Biol. 2002;14:104–109. doi: 10.1016/s0955-0674(01)00300-3. [DOI] [PubMed] [Google Scholar]
- 2.Bretscher A, Edwards K, Fehon RG. Nat Rev Mol Cell Biol. 2002;3:586–599. doi: 10.1038/nrm882. [DOI] [PubMed] [Google Scholar]
- 3.Turunen O, Wahlstrom T, Vaheri A. J Cell Biol. 1994;126:1445–1453. doi: 10.1083/jcb.126.6.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pearson MA, Reczek D, Bretscher A, Karplus PA. Cell. 2000;101:259–270. doi: 10.1016/s0092-8674(00)80836-3. [DOI] [PubMed] [Google Scholar]
- 5.Gary R, Bretscher A. Mol Biol Cell. 1995;6:1061–1075. doi: 10.1091/mbc.6.8.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bretscher A, Gary R, Berryman M. Biochemistry. 1995;34:16830–16837. doi: 10.1021/bi00051a034. [DOI] [PubMed] [Google Scholar]
- 7.Finnerty CM, Chambers D, Ingraffea J, Faber HR, Karplus PA, Bretscher A. J Cell Sci. 2004;117:1547–1552. doi: 10.1242/jcs.01038. [DOI] [PubMed] [Google Scholar]
- 8.Fievet BT, Gautreau A, Roy C, Del Maestro L, Mangeat P, Louvard D, Arpin M. J Cell Biol. 2004;164:653–659. doi: 10.1083/jcb.200307032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsui T, Maeda M, Doi Y, Yonemura S, Amano M, Kaibuchi K, Tsukita S. J Cell Biol. 1998;140:647–657. doi: 10.1083/jcb.140.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gautreau A, Louvard D, Arpin M. J Cell Biol. 2000;150:193–203. doi: 10.1083/jcb.150.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ng T, Parsons M, Hughes WE, Monypenny J, Zicha D, Gautreau A, Arpin M, Gschmeissner S, Verveer PJ, Bastiaens PI, Parker PJ. EMBO J. 2001;20:2723–2741. doi: 10.1093/emboj/20.11.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pietromonaco SF, Simons PC, Altman A, Elias L. J Biol Chem. 1998;273:7594–7603. doi: 10.1074/jbc.273.13.7594. [DOI] [PubMed] [Google Scholar]
- 13.Matsui T, Yonemura S, Tsukita S. Curr Biol. 1999;9:1259–1262. doi: 10.1016/s0960-9822(99)80508-9. [DOI] [PubMed] [Google Scholar]
- 14.Tran Quang C, Gautreau A, Arpin M, Treisman R. EMBO J. 2000;19:4565–4576. doi: 10.1093/emboj/19.17.4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shaw RJ, Henry M, Solomon F, Jacks T. Mol Biol Cell. 1998;9:403–419. doi: 10.1091/mbc.9.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawano Y, Fukata Y, Oshiro N, Amano M, Nakamura T, Ito M, Matsumura F, Inagaki M, Kaibuchi K. J Cell Biol. 1999;147:1023–1038. doi: 10.1083/jcb.147.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fukata Y, Kimura K, Oshiro N, Saya H, Matsuura Y, Kaibuchi K. J Cell Biol. 1998;141:409–418. doi: 10.1083/jcb.141.2.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yonemura S, Matsui T, Tsukita S. J Cell Sci. 2002;115:2569–2580. doi: 10.1242/jcs.115.12.2569. [DOI] [PubMed] [Google Scholar]
- 19.Speck O, Hughes SC, Noren NK, Kulikauskas RM, Fehon RG. Nature. 2003;421:83–87. doi: 10.1038/nature01295. [DOI] [PubMed] [Google Scholar]
- 20.Schultheis PJ, Clarke LL, Meneton P, Miller ML, Soleimani M, Gawenis LR, Riddle TM, Duffy JJ, Doetschman T, Wang T, Giebisch G, Aronson PS, Lorenz JN, Shull GE. Nat Genet. 1998;19:282–285. doi: 10.1038/969. [DOI] [PubMed] [Google Scholar]
- 21.Kurashima K, D’Souza S, Szaszi K, Ramjeesingh R, Orlowski J, Grinstein S. J Biol Chem. 1999;274:29843–29849. doi: 10.1074/jbc.274.42.29843. [DOI] [PubMed] [Google Scholar]
- 22.Szaszi K, Kurashima K, Kapus A, Paulsen A, Kaibuchi K, Grinstein S, Orlowski J. J Biol Chem. 2000;275:28599–28606. doi: 10.1074/jbc.M001193200. [DOI] [PubMed] [Google Scholar]
- 23.Cha B, Kenworthy A, Murtazina R, Donowitz M. J Cell Sci. 2004;117:3353–3365. doi: 10.1242/jcs.01180. [DOI] [PubMed] [Google Scholar]
- 24.Lee-Kwon W, Kawano K, Choi JW, Kim JH, Donowitz M. J Biol Chem. 2003;278:16494–16501. doi: 10.1074/jbc.M300580200. [DOI] [PubMed] [Google Scholar]
- 25.Zhao H, Shiue H, Palkon S, Wang Y, Cullinan P, Burkhardt JK, Musch MW, Chang EB, Turner JR. Proc Natl Acad Sci U S A. 2004;101:9485–9490. doi: 10.1073/pnas.0308400101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee-Kwon W, Johns DC, Cha B, Cavet M, Park J, Tsichlis P, Donowitz M. J Biol Chem. 2001;276:31296–31304. doi: 10.1074/jbc.M103900200. [DOI] [PubMed] [Google Scholar]
- 27.Turner JR, Black ED. Am J Physiol. 2001;281:C1533–C1541. doi: 10.1152/ajpcell.2001.281.5.C1533. [DOI] [PubMed] [Google Scholar]
- 28.Turner JR, Lencer WI, Carlson S, Madara JL. J Biol Chem. 1996;271:7738–7744. doi: 10.1074/jbc.271.13.7738. [DOI] [PubMed] [Google Scholar]
- 29.Obenauer JC, Cantley LC, Yaffe MB. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lamprecht G, Weinman EJ, Yun CH. J Biol Chem. 1998;273:29972–29978. doi: 10.1074/jbc.273.45.29972. [DOI] [PubMed] [Google Scholar]
- 31.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 32.Gautreau A, Poullet P, Louvard D, Arpin M. Proc Natl Acad Sci U S A. 1999;96:7300–7305. doi: 10.1073/pnas.96.13.7300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu KL, Khan S, Lakhe-Reddy S, Jarad G, Mukherjee A, Obejero-Paz CA, Konieczkowski M, Sedor JR, Schelling JR. J Biol Chem. 2004;279:26280–26286. doi: 10.1074/jbc.M400814200. [DOI] [PubMed] [Google Scholar]
- 34.Murakami M, Kataoka K, Fukuhara S, Nakagawa O, Kurihara H. Eur J Biochem. 2004;271:3330–3339. doi: 10.1111/j.1432-1033.2004.04267.x. [DOI] [PubMed] [Google Scholar]
- 35.Reuveni H, Livnah N, Geiger T, Klein S, Ohne O, Cohen I, Benhar M, Gellerman G, Levitzki A. Biochemistry. 2002;41:10304–10314. doi: 10.1021/bi0202530. [DOI] [PubMed] [Google Scholar]
- 36.Vlahos C, Matter W, Hui K, Brown R. J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- 37.Zhang HM, Rao JN, Guo X, Liu L, Zou T, Turner DJ, Wang JY. J Biol Chem. 2004;279:22539–22547. doi: 10.1074/jbc.M314337200. [DOI] [PubMed] [Google Scholar]
- 38.Fujita T, Azuma Y, Fukuyama R, Hattori Y, Yoshida C, Koida M, Ogita K, Komori T. J Cell Biol. 2004;166:85–95. doi: 10.1083/jcb.200401138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu J, Liao K. J Biol Chem. 2004;279:35914–35922. doi: 10.1074/jbc.M402297200. [DOI] [PubMed] [Google Scholar]
- 40.Zhuang S, Dang Y, Schnellmann RG. Am J Physiol. 2004;287:F365–F372. doi: 10.1152/ajprenal.00035.2004. [DOI] [PubMed] [Google Scholar]
- 41.Hayashi H, Szaszi K, Coady-Osberg N, Furuya W, Bretscher AP, Orlowski J, Grinstein S. J Gen Physiol. 2004;123:491–504. doi: 10.1085/jgp.200308979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li X, Leu S, Cheong A, Zhang H, Baibakov B, Shih C, Birnbaum MJ, Donowitz M. Gastroenterology. 2004;126:122–135. doi: 10.1053/j.gastro.2003.10.061. [DOI] [PubMed] [Google Scholar]
- 43.Alfthan K, Heiska L, Gronholm M, Renkema GH, Carpen O. J Biol Chem. 2004;279:18559–18566. doi: 10.1074/jbc.M313916200. [DOI] [PubMed] [Google Scholar]
- 44.Lee-Kwon W, Kim JH, Choi JW, Kawano K, Cha B, Dartt DA, Zoukhri D, Donowitz M. Am J Physiol. 2003;285:C1527–C1536. doi: 10.1152/ajpcell.00017.2003. [DOI] [PubMed] [Google Scholar]
- 45.Horowitz JC, Lee DY, Waghray M, Keshamouni VG, Thomas PE, Zhang H, Cui Z, Thannickal VJ. J Biol Chem. 2004;279:1359–1367. doi: 10.1074/jbc.M306248200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taniyama Y, Ushio-Fukai M, Hitomi H, Rocic P, Kingsley MJ, Pfahnl C, Weber DS, Alexander RW, Griendling KK. Am J Physiol. 2004;287:C494–C499. doi: 10.1152/ajpcell.00439.2003. [DOI] [PubMed] [Google Scholar]
- 47.Weinman EJ, Steplock D, Wade JB, Shenolikar S. Am J Physiol. 2001;281:F374–F380. doi: 10.1152/ajprenal.2001.281.2.F374. [DOI] [PubMed] [Google Scholar]
- 48.Yun CH, Lamprecht G, Forster DV, Sidor A. J Biol Chem. 1998;273:25856–25863. doi: 10.1074/jbc.273.40.25856. [DOI] [PubMed] [Google Scholar]
- 49.Weinman EJ, Steplock D, Shenolikar S. FEBS Lett. 2003;536:141–144. doi: 10.1016/s0014-5793(03)00043-7. [DOI] [PubMed] [Google Scholar]
- 50.Li X, Galli T, Leu S, Wade JB, Weinman EJ, Leung G, Cheong A, Louvard D, Donowitz M. J Physiol. 2001;537:537–552. doi: 10.1111/j.1469-7793.2001.00537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Itoh K, Sakakibara M, Yamasaki S, Takeuchi A, Arase H, Miyazaki M, Nakajima N, Okada M, Saito T. J Immunol. 2002;168:541–544. doi: 10.4049/jimmunol.168.2.541. [DOI] [PubMed] [Google Scholar]
- 52.Brdickova N, Brdicka T, Andera L, Spicka J, Angelisova P, Milgram SL, Horejsi V. FEBS Lett. 2001;507:133–136. doi: 10.1016/s0014-5793(01)02955-6. [DOI] [PubMed] [Google Scholar]
- 53.Sweeney G, Somwar R, Ramlal T, Volchuk A, Ueyama A, Klip A. J Biol Chem. 1999;274:10071–10078. doi: 10.1074/jbc.274.15.10071. [DOI] [PubMed] [Google Scholar]
- 54.Somwar R, Niu W, Kim DY, Sweeney G, Randhawa VK, Huang C, Ramlal T, Klip A. J Biol Chem. 2001;276:46079–46087. doi: 10.1074/jbc.M109093200. [DOI] [PubMed] [Google Scholar]
- 55.Cong LN, Chen H, Li Y, Zhou L, McGibbon MA, Taylor SI, Quon MJ. Mol Endocrinol. 1997;11:1881–1890. doi: 10.1210/mend.11.13.0027. [DOI] [PubMed] [Google Scholar]
- 56.Sakaue H, Ogawa W, Takata M, Kuroda S, Kotani K, Matsumoto M, Sakaue M, Nishio S, Ueno H, Kasuga M. Mol Endocrinol. 1997;11:1552–1562. doi: 10.1210/mend.11.10.9986. [DOI] [PubMed] [Google Scholar]
- 57.Katome T, Obata T, Matsushima R, Masuyama N, Cantley LC, Gotoh Y, Kishi K, Shiota H, Ebina Y. J Biol Chem. 2003;278:28312–28323. doi: 10.1074/jbc.M302094200. [DOI] [PubMed] [Google Scholar]
- 58.Chiang SH, Baumann CA, Kanzaki M, Thurmond DC, Watson RT, Neudauer CL, Macara IG, Pessin JE, Saltiel AR. Nature. 2001;410:944–948. doi: 10.1038/35073608. [DOI] [PubMed] [Google Scholar]
- 59.Chunqiu Hou J, Pessin JE. Mol Biol Cell. 2003;14:3578–3591. doi: 10.1091/mbc.E03-01-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Watson RT, Shigematsu S, Chiang SH, Mora S, Kanzaki M, Macara IG, Saltiel AR, Pessin JE. J Cell Biol. 2001;154:829–840. doi: 10.1083/jcb.200102078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Inoue M, Chang L, Hwang J, Chiang SH, Saltiel AR. Nature. 2003;422:629–633. doi: 10.1038/nature01533. [DOI] [PubMed] [Google Scholar]