

Abstract
Activation of Na+-nutrient cotransport leads to increased tight junction permeability in intestinal absorptive (villus) enterocytes. This regulation requires myosin II regulatory light chain (MLC) phosphorylation mediated by MLC kinase (MLCK). We examined the spatiotemporal segregation of MLCK isoform function and expression along the crypt-villus axis and found that long MLCK, which is expressed as two alternatively spliced isoforms, accounts for 97 ± 4% of MLC kinase activity in interphase intestinal epithelial cells. Expression of the MLCK1 isoform is limited to well differentiated enterocytes, both in vitro and in vivo, and this expression correlates closely with development of Na+-nutrient cotransport-dependent tight junction regulation. Consistent with this role, MLCK1 is localized to the perijunctional actomyosin ring. Furthermore, specific knockdown of MLCK1 using siRNA reduced tight junction permeability in monolayers with active Na+-glucose cotransport, confirming a functional role for MLCK1. These results demonstrate unique physiologically relevant patterns of expression and subcellular localization for long MLCK isoforms and show that MLCK1 is the isoform responsible for tight junction regulation in absorptive enterocytes.
The intestinal epithelial barrier, composed of the cell membranes and tight junctions that seal the paracellular space between adjacent cells, is critical for protecting the interstitium from the harsh luminal environment. However, this barrier is not static; its permeability characteristics change over the course of minutes, in response to luminal Na+ and glucose (1) or bacteria (2), or days, as enterocytes differentiate and migrate from crypt to villus (3, 4). The crypt-villus axis exhibits a large gradient in tight junction permeability; the secretory crypt area contains large pores (50–60Å), whereas the absorbing tip of the villus contains small pores (<5 Å) (4). These small pores at the villus tip also exhibit added functionality, because they are able to increase in number and thus increase the permeability of the epithelial barrier in response to Na+ and glucose (4–6). This acute regulation of paracellular permeability at the villus tip in response to Na+ and glucose is controlled by activation of the Na+-glucose cotransporter SGLT1, triggering a signaling pathway that leads to myosin II regulatory light chain (MLC)1 phosphorylation, contraction of the perijunctional actomyosin ring, and increased tight junction permeability (1, 7). Numerous kinases are known to phosphorylate MLC and thus have the potential to regulate this process, including myosin light chain kinase (MLCK) (8), Rho kinase (9), ZIP kinase (10), and citron kinase (11). Of these kinases, only specific inhibition of MLCK has been shown to block SGLT1-mediated tight junction regulation (1, 12).
The MLCK gene expressed in smooth muscle and nonmuscle cells gives rise to multiple mRNA species, using three different transcriptional promoters as well as alternative splicing. In smooth muscle, the 130-kDa smooth muscle MLCK (short MLCK) and a carboxyl-terminal fragment of MLCK, telokin, are produced as separate mRNA transcripts. In contrast, a 220-kDa MLCK (long MLCK) containing a 922-amino acid amino-terminal extension is expressed in many nonmuscle cells, including embryonic tissues and endothelium (13, 14). Long MLCK is expressed as at least five different splice variants that all contain the amino-terminal extension; these variants are distinguished by the presence or absence of short (<70 amino acids) regions as a result of alternative mRNA splicing (15). Long MLCK isoform 2, or MLCK2, is the predominant isoform expressed and differs from the full-length long MLCK isoform, MLCK1, by the absence of a 207-base pair sequence from nucleotides 1428–1634 (15). The corresponding 69-amino acid region contains Src kinase phosphorylation sites and a potential SH2-binding domain (15). In vitro enzymatic assays have shown that phosphorylation by Src kinase increases the Vmax of MLCK1, whereas the activity of unphosphorylated MLCK1 is similar to MLCK2 (16).
Despite the functional specialization of MLCK1, there have been no reports of differential subcellular or tissue distributions of long MLCK isoforms 1 and 2 that may explain the functional significance, at the cellular level, of the biochemical differences between these splice variants. Moreover, no studies have examined MLCK isoforms in intestinal epithelium, where there is spatial segregation of MLCK-dependent processes such as Na+-nutrient cotransport-dependent barrier regulation. Analysis of MLCK isoforms in differentiating human intestinal epithelium using laser capture microdissection and immunofluorescence microscopy with MLCK isoform-specific antisera as well as in vitro functional analyses have allowed us to document spatiotemporal regulation of MLCK isoform expression. The data show that long MLCK represents the principal myosin light chain kinase of intestinal enterocytes and is responsible for Na+-nutrient cotransport-dependent tight junction regulation. Within the intestinal epithelium, only MLCK1 and MLCK2 isoforms are expressed, and MLCK1 expression is limited to villus enterocytes, where it is concentrated within the perijunctional actomyosin ring. MLCK1 expression correlates with the development of the ability to increase tight junction permeability in response to Na+-glucose cotransport, and selective knockdown of MLCK1 decreases tight junction permeability. These data demonstrate a unique function and localization for a single splice variant of MLCK within the intestinal epithelium, implying that the short sequences unique to individual MLCK splice variants confer these distinct functional and spatial properties.
MATERIALS AND METHODS
Cell Culture
Caco-2 BBe cells (17) expressing the intestinal Na+-glucose cotransporter SGLT1 (18) were plated on Transwell semipermeable supports (Corning-Costar, Acton, MA) as described previously (1). For RT-PCR, monolayers were scraped into TRIzol (Invitrogen). RNA was extracted with chloroform, precipitated with isopropanol, and resuspended in diethyl pyrocarbonate-treated water.
Electrophysiology
Electrophysiological measurements were made with agar bridges and Ag-AgCl calomel electrodes as previously described (1). Briefly, monolayers were transferred from culture medium to Hank’s balanced salt solution with 15 mm HEPES, pH 7.4. The media also contained either 25 mm glucose to activate Na+-glucose cotransport or 5 mm glucose, 20 mm mannose, and 2 mm phloridzin to inhibit Na+-glucose cotransport. For inhibitor experiments, either 250 μm PIK (12), 10 μm Y27632 (Calbiochem, San Diego, CA), or 50 nm calyculin A (Calbiochem) was also included. Potential differences were measured before and after application of a 50-μA current, and the transepithelial resistance (TER) was determined using Ohm’s law. TER values are reported 2 h after activation or inhibition of Na+-glucose cotransport, at which point TER had reached the new steady state value as a function of Na+-glucose cotransport activity (1).
Kinase Immunodepletion Assay
Lysates (in 20 mm MOPS, pH 7.4, 2 mm MgCl2, 0.2 mM CaCl2, 1 mM dithiothreitol, 0.5% Igepal CA630, and 0.5% Triton X-100) of confluent Caco-2 cells were immunoprecipitated with K36 monoclonal anti-MLCK antibody (Sigma-Aldrich) and protein G Plus/protein A-agarose beads (Calbiochem). Immunoblot analyses showed that this depleted the lysate of the 215-kDa long MLCK. The immunodepleted lysate and control lysates (immunoprecipitated using an irrelevant antibody) were used for in vitro kinase assays as previously described (12). Briefly, the lysates were incubated with 0.2 μm calmodulin, 5 μm recombinant human intestinal epithelial MLC, and [γ-32P]ATP for 5 min at 30 °C, the reaction was terminated by the addition of SDS-PAGE sample buffer, and MLC phosphorylation was determined by SDS-PAGE and autoradiography.
RT-PCR Sequencing of Long MLCK
cDNA was generated from 1 μg of Caco-2 cell RNA using Thermoscript reverse transcriptase and random hexamer primers in a 25-μl reaction (Invitrogen). 1 μl of this reaction was used for PCR amplification of long MLCK using primers 1–10 to allow for analysis of splice variants (see Fig. 2). Primer pairs 1 + 2 (CAACAAACAACAGAGAAGACGG + AGTCTTCTGAAGGACCG-GG), 3 + 4 (TCTGAGAAGAACGGCATG + ACTTCAGGGGGTGGAT-TC), 5 + 6 (CCTCCTTCTCCAGTGTCC + TGGGAGTCCCCTTCTT-GG), and 7 + 8 (GAGTTGGTGCTGATGGTGG + ATGAGGAGCCATA-CCAGG) used a 57 °C annealing temperature; primer pair 9 + 10 (CAGCAGGCAGGCCCAGGTCAAC + CTTAGAAACTGCTTTTCTCTGGC) used a 66 °C annealing temperature. The full-length long MLCK was amplified using primers 1 and 10 and a 60 °C annealing temperature. All of the reactions were cycled 36 times in a Bio-Rad iCycler thermal cycler (Bio-Rad). The PCR products were separated on a 1% agarose gel and visualized using ethidium bromide. Bands of appropriate size were excised and purified by gel extraction (Qiagen). The bands were sequenced by the University of Chicago DNA Sequencing Facility using primers 1–10. Long MLCK from normal human intestine was amplified from a human intestinal epithelial cDNA library generated by cloning poly(dT) primed cDNA into the HybriZAP bacteriophage vector followed by amplification and in vivo mass excision to generate a two-hybrid library in pAD-GAL4 (19). Amplification used primers sets 1 + 2, 3 + 4, 5 + 6, 7 + 8, and 9 + 10 as described above. Primers 4–6 were used for sequencing to generate a 1-kb section of long MLCK sequence, which was used for BLAST searches.
Fig. 2. Long MLCK is found in intestinal epithelium and is primarily responsible for Na+-glucose cotransport-induced tight junction regulation.
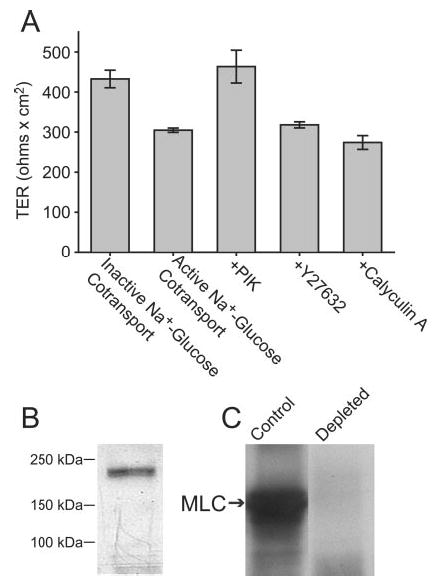
A, Caco-2 monolayers were incubated in Hank’s balanced salt solution with 25 mm glucose (active Na+-glucose cotransport) or 5 mm glucose, 20 mm mannitol, and 2 mm phloridzin (inactive Na+-glucose cotransport), as in Fig. 1, or with 25 mm glucose and the MLCK inhibitor PIK (250 μm), the Rho kinase inhibitor Y27632 (10 μm), or the phosphatase 1 and 2A inhibitor calyculin A (10 nm). TER after 2 h, at which time TER had stabilized, is shown. Activation of Na+-glucose cotransport reduced TER by 30 ± 4%. The addition of PIK elevated TER to that of monolayers with inactive Na+-glucose cotransport. Y27632 and calyculin A had no effect on TER, indicating that MLCK is primarily responsible for Na+-glucose cotransport-dependent tight junction regulation. B, analysis of Caco-2 cell lysates by SDS-PAGE and immunoblot with broadly reactive anti-MLCK monoclonal antibody detects a single band of ~215 kDa. C, immunodepletion of Caco-2 lysates using the same anti-MLCK monoclonal antibody removed 97 ± 4% of MLC kinase activity.
Laser Capture Microdissection
Surgically excised portions of normal human jejunum were embedded in optimal cutting temperature media and snap frozen within minutes of resection. The protocol for use of human tissues was approved by the Institutional Review Board of The University of Chicago. Frozen sections of normal human jejunum (10 μm) were cut on membrane-based laser microdissection slides (Leica Microsystems, Wetzlar, Germany) and jejunal epithelial cells from villus tip, mid-villus, and crypt were collected using an ASLMD laser microdissection system (Leica). mRNA was isolated from the samples using a Quick-Prep micro mRNA purification kit (Amersham Biosciences).
RT-PCR Quantitation of mRNA Levels
Quantitation of MLCK1 and MLCK2 mRNA levels from Caco-2 cells was performed using primers 3 and 4 as described above. cDNA obtained from laser microdissection was used for the amplification of MLCK1, MLCK2, BRK, and keratin 8. Preliminary assays showed that 40 cycles of amplification was within the linear range of amplification for each of the primer sets under the conditions used. MLCK2 was amplified with primers 3 and 4 using an annealing temperature of 66 °C. MLCK1 was amplified with primers ACCCCCGTGAGGAGACAG and ACTTCAGGGGGTGGATTC and an annealing temperature of 58 °C. BRK was amplified with primers ATCCAGGCCATGAGAAGC and TGGATGTAATTCTGCGACTCC and an annealing temperature of 62.5 °C. Keratin 8 was amplified with primers TTGCCTCCTTCATAGACAAGG and TGTTGTCCATGTTGCTTCG and an annealing temperature of 62.5 °C. All of the reactions were separated on a 1% agarose gel and photographed with a FluorChem 8800 (AlphaInnotech, San Leandro, CA), and band intensity was measured with ImageQuant software (Amersham Biosciences).
MLCK1 Antibody Production
An 18-mer peptide (KARTRDSGTYSCTASNAQ) corresponding to residues within the 69-amino acid sequence that is unique to human MLCK1 was synthesized by the University of Chicago Cancer Research Center Peptide Facility. BLAST searches confirmed that this sequence was not expressed in other known or predicted sequences in the human genome. To avoid generating phosphospecific antiserum, described phosphorylation sites at Tyr464 and Tyr471 (16) were excluded from the peptide. The peptide was conjugated to keyhole limpet hemocyanin (Calbiochem) using glutaraldehyde. Rabbits were primed with the peptide-keyhole limpet hemocyanin conjugate emulsified in complete Freund’s adjuvant and boosted with the same peptide-keyhole limpet hemocyanin conjugate emulsified in incomplete Freund’s adjuvant at 3-week intervals (Zymed Laboratories Inc., South San Francisco, CA). A peptide-specific enzyme-linked immunosorbent assay was performed on test bleed serum to determine the specific antibody titers. The anti-peptide antiserum was purified through an affinity gel (Affi-Gel 10; Bio-Rad) coupled with the same peptide, and the antibodies bound specifically to the peptides were eluted with 3m KSCN and dialyzed exhaustively against phosphate-buffered saline.
Immunoblotting
Human endothelial cell MLCK1 and MLCK2 were prepared using the Bac-to-Bac baculovirus expression system (Invitrogen) as described previously (16). For large scale expression and purification, Sf9 cells were infected with baculovirus (multiplicity of infection = 1), and the cells producing human endothelial MLCK1 and MLCK2 were harvested. The recombinant long MLCK1 or MLCK2 proteins, 0.5 μg each, or Caco-2 extracts, 10 μg, were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes. Duplicate blots were probed with affinity-purified anti-MLCK1-specific rabbit antibody (10 μg/ml) or K36 pan-MLCK monoclonal antibody (10 μg/ml). These were detected with appropriate horseradish peroxidase-conjugated secondary antisera (Cell Signaling, Beverly, MA), and the blot was visualized by enhanced chemiluminescence as described previously (1).
Immunofluorescent Microscopy
Immunostaining was performed as previously described (20). Briefly, 5-μm frozen sections were collected on coated slides, fixed in 1% paraformaldehyde, and washed thrice with phosphate-buffered saline. Nonspecific binding was blocked with 5% normal goat serum in phosphate-buffered saline. After incubation with affinity-purified anti-MLCK1-specific rabbit antibody (10 μg/ml), affinity-purified anti-phosphorylated MLC specific rabbit antibody (10 μg/ml) (20), K36 pan-MLCK monoclonal antibody (10 μg/ml), or mouse monoclonal anti-ZO-1 antibody (2.5 μg/ml, Zymed Laboratories Inc.), sections were washed and incubated with appropriate Alexa 488- or 594-conjugated secondary antisera, Alexa 488-conjugated phalloidin, and/or Hoechst 33342 (Molecular Probes, Eugene, OR). Sections were imaged using a Leica DMLB epifluorescence microscope equipped with an 88000 filter set (Chroma Technology, Brattleboro, VT), and a Cool-snap HQ camera (Roper Scientific, Tucson, AZ) controlled by MetaMorph 6 (Universal Imaging Corporation, Downingtown, PA).
RNAi
The 207-base pair sequence from nucleotides 1428–1634 unique to MLCK1 was used to design the MLCK1 siRNA SMARTpool (Dharmacon, Lafayette, CO). For transfection, 10 μl of 50 μm SMART-pool or 25 μl of 20 μm nonspecific siRNA mix was added to 500 μl of Optimem (Invitrogen) and allowed to incubate for 5 min at 25 °C. The siRNA solution was then added to 500 μl of Optimem containing 30 μl of siLentFect reagent (Bio-Rad) and allowed to incubate for 30 min at 25 °C to create the transfection mix. At the same time, ~107 Caco-2 BBe cells were trypsinized with 1.5 ml of trypsin for 20 min. After trypsinization was complete, the cells were resuspended in 5 ml of Dulbecco’s modified Eagle’s medium with 4.5 g glucose/liter (Mediatech, Herndon, VA). The cells were recovered by gentle centrifugation (500 × g for 5 min), washed once more in Dulbecco’s modified Eagle’s medium followed by OptiMEM (Invitrogen), separated into two equal aliquots, and resuspended in 800 μl of OptiMEM. This was added to 1 ml of previously prepared transfection mix. The cell suspension/siRNA mix was then plated at high density on Transwell semipermeable supports (Corning-Costar) at 375,000 cells/cm2 and cultured for 4 days to allow for tight junction assembly and polarization before use in electrophysiology experiments.
RESULTS
Na+-Glucose Cotransport-dependent Barrier Regulation Is Differentiation-dependent
Na+-nutrient cotransport-dependent regulation of tight junction permeability is a characteristic of small intestinal epithelium that is restricted to terminally differentiated absorptive villus enterocytes (4). We have developed an in vitro model of this regulation in an intestinal Na+-glucose cotransporter-expressing clone of the adenocarcinoma-derived Caco-2 cell line. Like other Caco-2-derived cell lines, these cells acquire features of differentiated villus enterocytes during culture after confluence in a manner that recapitulates crypt to villus enterocyte differentiation (21, 22). We noted that Na+-glucose cotransport-dependent tight junction regulation did not occur in relatively undifferentiated monolayers, despite the presence of intact tight junctions. We thus assessed the development of Na+-glucose cotransport-dependent tight junction regulation during Caco-2 differentiation (Fig. 1). We measured TER, a sensitive marker that is inversely related to tight junction permeability. In undifferentiated monolayers, <4 days post-confluence, TER was not significantly different with active or inactive Na+-glucose cotransport (p = 0.79). In contrast, Na+-glucose cotransport-dependent tight junction regulation was observed in more differentiated monolayers ≥6 days post-confluence; the average TER with active Na+-glucose cotransport was ~40% less than with inhibited Na+-glucose cotransport (p = 0.002). Thus, the ability of intestinal epithelial monolayers to increase tight junction permeability in response to Na+-glucose cotransport is differentiation-dependent, suggesting that factor(s) responsible for this tight junction regulation are expressed or activated during enterocyte differentiation.
Fig. 1. Tight junction regulation in response to Na+-glucose cotransport is dependent on intestinal epithelial differentiation.
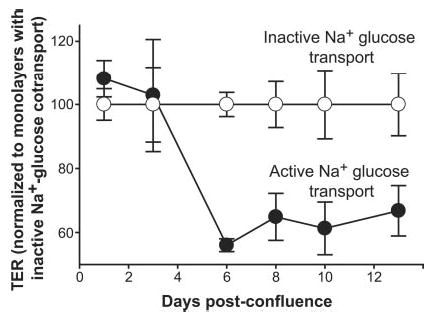
At the indicated times after reaching confluence, Caco-2 monolayers were incubated in Hank’s balanced salt solution containing 25 mm glucose (black circles) or 5 mm glucose, 20 mm mannitol, and 2 mm phloridzin, a Na+-glucose cotransport inhibitor (white circles). TER was measured after 2 h of incubation, at which time steady-state TER was established. The data are normalized to TER in monolayers with inactive Na+-glucose cotransport (i.e. in Hank’s balanced salt solution with phloridzin). No significant responses were seen in monolayers less than 4 days post-confluence (p = 0.79). At 6 or more days post-confluence, a robust tight junction regulatory response was present (p = 0.002).
MLCK Is Necessary for Na+-Glucose Cotransport-dependent Tight Junction Regulation
Na+-glucose cotransport-dependent tight junction regulation requires MLC phosphorylation, which is enhanced after activation of Na+-glucose cotransport (1). Several kinases can phosphorylate MLC, including MLCK (8), Rho kinase (9), citron kinase (11), and ZIP kinase (10). Of these, only MLCK and Rho kinase have been implicated in tight junction regulation (1, 12, 23–25). MLC phosphorylation is also regulated by MLC phosphatase, which can be controlled by Rho kinase (26). To determine the contribution of these regulators of MLC phosphorylation to Na+-glucose contransport-dependent tight junction regulation, we measured the effect of MLCK, Rho kinase, and phosphatase inhibitors on TER in monolayers with active Na+-glucose contransport (Fig. 2A). In control monolayers with active Na+-glucose cotransport, TER was 30 ± 4% less than that of monolayers with inactive Na+-glucose cotransport. PIK, a membrane permeant peptide inhibitor of MLCK (1, 12), raised the TER of monolayers with active Na+-glucose cotransport to that of monolayers with inactive Na+-glucose cotransport, confirming the critical role of MLCK in this regulatory pathway. In contrast, neither the Rho kinase inhibitor Y27632 nor the phosphatase inhibitor calyculin A significantly altered the TER of monolayers with active Na+-glucose cotransport, suggesting that neither Rho kinase nor myosin phosphatase are critically involved in Na+-glucose cotransport-dependent tight junction regulation.
A 215-kDa MLCK Is the Principal Caco-2 Cell MLC Kinase
As we have shown previously (27), immunoblot of Caco-2 cell lysates with a monoclonal antibody that reacts broadly with myosin light chain kinases detected only a single ~215-kDa protein consistent with long MLCK (Fig. 2B). To determine whether this was the principal MLC kinase expressed in Caco-2 cells, this protein was removed from Caco-2 lysates by immunodepletion. This removed 97 ± 4% of MLC kinase activity from the lysates (Fig. 2C). Thus, the 215-kDa MLCK is the principal enzyme responsible for MLC phosphorylation in Caco-2 intestinal epithelial cells.
Splice Variants 1 and 2 of Long MLCK Are Expressed in the Intestinal Epithelium
Two primary isoforms of MLCK are transcribed from the smooth muscle MLCK gene using separate promoters: a 130-kDa short MLCK and a 210-kDa long MLCK (14). Based on the size of the MLCK detected in Fig. 2B, we hypothesized that long MLCK is the 215-kDa MLCK found in Caco-2 intestinal epithelial cells. To determine whether the 215-kDa Caco-2 MLCK is long MLCK, we cloned and sequenced long MLCK from Caco-2 cell mRNA. A full-length 5.8-kb transcript was detected, consistent with long MLCK (Fig. 3A). Previous analysis of endothelial long MLCK has identified at least five splice variants of long MLCK as a result of 207-, 204-, and 153-base pair in-frame deletions (15). These variations in length are too small to be detectable by agarose gel electrophoresis of the full-length 5.8-kb transcript; thus we amplified and sequenced smaller portions of long MLCK cDNA individually (Fig. 3B and C). Two splice variants corresponding to long MLCK isoforms MLCK1 and MLCK2 were identified by RT-PCR, but mRNA transcripts corresponding to other splice variants were not present. The sequence data obtained (GenBank™ accession numbers AY424269 and AY424270) were nearly identical to the published sequence of endothelial long MLCK (GenBank™ accession number NM053025). A third band that migrated just ahead of the MLCK1 PCR product in agarose gels was also present in PCR products of Caco-2 cells. Despite multiple attempts, we could not obtain sequence data for this band, whose intensity on ethidium bromide-stained gels correlated with that of MLCK1. This band was only present in PCRs using Caco-2-derived cDNA and was not detected in the products of PCRs using cDNA derived from native human intestinal epithelium. This band was therefore excluded from further analyses. Thus, intestinal epithelial cells produce mRNA transcripts for MLCK1 and MLCK2 but not other MLCK isoforms.
Fig. 3. mRNA transcripts of long MLCK isoforms 1 and 2 are present in Caco-2 cells.
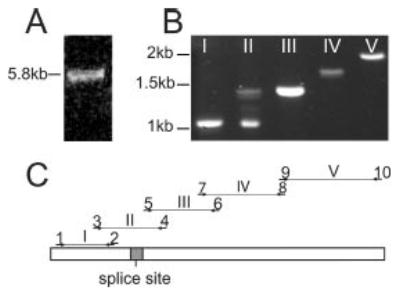
A, a single 5.8-kb full-length PCR product corresponding to long MLCK is obtained from Caco-2 cDNA. B, PCR using primers to assess alternative splicing of long MLCK shows only a single product in lanes I, III, IV, and V, confirming that alternative splicing of these regions of long MLCK does not occur in intestinal epithelia. In contrast, three products are detected in lane II, showing that alternative splicing occurs in this region. Sequence analysis of these products confirmed that the upper 1.4-kb band is MLCK1 and the lower 1.1-kb band is MLCK2, whereas a faint, unknown band is located directly below MLCK1. C, diagram of the long MLCK gene and the primer pairs used for amplification and sequencing of Caco-2 cDNA. The shaded region marked splice site designates the location of the 207-base pair region that is present in MLCK1 and absent from MLCK2.
MLCK1 mRNA Transcription Is Increased during Differentiation and Correlates with the Ability to Regulate Tight Junction Permeability in Response to Na+-Glucose Cotransport
Given that Na+-glucose cotransport-dependent tight junction regulation correlates with differentiation state (Fig. 1) and requires MLCK activity (Fig. 2), we considered the hypothesis that MLCK isoform expression might correlate with differentiation state. Thus, we measured the relative abundance of MLCK1 and MLCK2 mRNA during Caco-2 monolayer differentiation. Semiquantitiative RT-PCR analysis of mRNA from monolayers harvested during differentiation, i.e. at intervals before and after confluence, showed that MLCK2 mRNA represented the majority of long MLCK transcripts in subconfluent and newly post-confluent monolayers but that MLCK1 mRNA content began to increase progressively at 3 days post-confluence (Fig. 4). By 6 days post-confluence MLCK1 mRNA was more abundant than MLCK2 mRNA. Thus, an inversion of long MLCK isoform predominance occurs during Caco-2 cell differentiation, with MLCK2 predominating in undifferentiated cells and MLCK1 predominating in differentiated cells.
Fig. 4. Relative MLCK1 mRNA levels increase during Caco-2 differentiation.
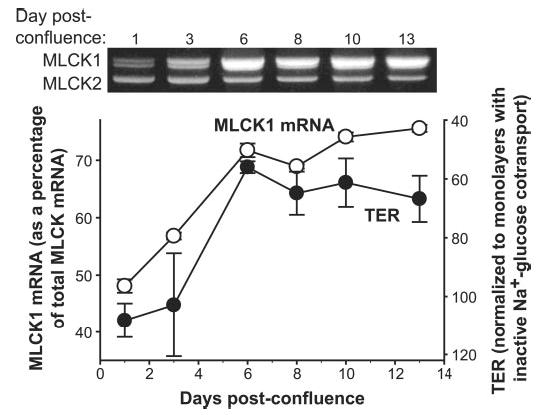
Monolayers of Caco-2 cells were harvested at indicated points after reaching confluence. MLCK isoforms 1 and 2 were detected using primers 3 and 4 flanking the MLCK1 splice site. Thus, a single PCR allowed detection of both isoforms (see Fig. 3). As Caco-2 cells differentiated the amount of MLCK1 mRNA (white circles) as a percentage of total MLCK mRNA increased. The TER response of these monolayers to activation of Na+-glucose cotransport (as in Fig. 1) is shown for comparison (black circles). Development of the characteristic response of TER to Na+-glucose cotransport correlates strongly with increases in relative MLCK1 mRNA content (R2 = 0.92).
Our data show that both MLCK1 expression and tight junction regulation following Na+-glucose cotransport are related to Caco-2 differentiation; thus we asked whether these processes occur in parallel. Strikingly, when identical monolayers were analyzed it became apparent that MLCK1 mRNA expression and the ability to regulate tight junctions in response to Na+-glucose cotransport were closely related (Fig. 4). This strong correlation (R2 = 0.92) suggests that MLCK1 expression may be related to physiological Na+-glucose cotransport-dependent tight junction regulation.
MLCK1 mRNA and MLC Phosphorylation Are Primarily Found in Well Differentiated Villus Enterocytes in Vivo
To determine whether human intestinal enterocytes express the same long MLCK isoforms in vivo that are seen in Caco-2 cells in vitro, we applied our MLCK primer sets shown in Fig. 3C to a human intestinal epithelial cDNA library (19). Again, only isoforms 1 and 2 were present. Sequencing of portions of long MLCK mRNA transcripts, including the MLCK2 splice site, demonstrated that the sequence was nearly identical to the corresponding regions of the Caco-2 sequence.
To determine whether the long MLCK isoform switch observed during Caco-2 enterocyte differentiation in vitro also occurred in native human small intestinal enterocytes in vivo, we used laser capture microdissection to isolate epithelial cells from the villus tip, mid-villus, or crypt of normal human jejunum (Fig. 5A). Semi-quantitative RT-PCR confirmed that the mRNA content of keratin 8, which is known to be expressed uniformly along the crypt-villus axis, was constant in all samples. The quality of the mRNA preparations was also established by analysis of BRK tyrosine kinase transcripts. BRK is preferentially expressed in villus epithelium (28), and, in our preparations, BRK mRNA content increased markedly from crypt to mid-villus to villus tip fractions. Normalized to keratin 8 mRNA, BRK mRNA increased 4.5 ± 0.9- and 6.8 ± 0.5-fold in mid-villus and villus tip, respectively, relative to the crypt (p < 0.02; Fig. 5B). Thus, in addition to verifying previous observations (28), these data validate the quality of these mRNA preparations. MLCK2 mRNA was relatively abundant throughout the crypt-villus axis in human jejunum and did not change from crypt to villus relative to keratin 8 mRNA. This abundance of MLCK2 mRNA relative to MLCK1 mRNA rendered the single primer set used to evaluate Caco-2 mRNA unsuitable for analysis of MLCK1 mRNA content. To circumvent this problem, a primer pair specific for MLCK1 was used to assess MLCK1 mRNA content. MLCK1 mRNA increased 1.6 ± 0.7-and 4.2 ± 0.4-fold in mid-villus and villus tip, respectively, relative to the crypt (p < 0.03) (Fig. 5B). Thus, MLCK1 mRNA is present in only small amounts in human jejunal crypt and mid-villus enterocytes but increases markedly at the villus tip.
Fig. 5. Phosphorylated MLC and MLCK1 mRNA are concentrated at the villus tip.
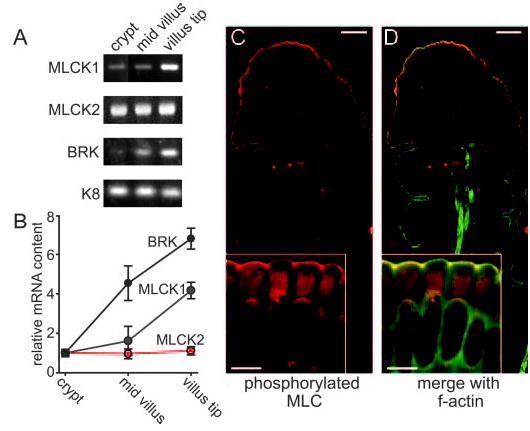
A, long MLCK1 RNA expression is greatest at the villus tip, whereas MLCK2 expression is constant along the crypt-villus axis as detected by RT-PCR. As controls, mRNA content of BRK and keratin 8 (K8) are also shown. BRK is known to be expressed preferentially at the villus tip, and keratin 8 is known to be expressed uniformly along the crypt-villus axis (28). B, densitometric analysis demonstrates MLCK1 enhancement at the villus tip. MLCK1 (blue), MLCK2 (red), and BRK (black) mRNA content was normalized to keratin 8 content. These ratios were then normalized to crypt mRNA content. Both BRK and MLCK1 show a gradient of increasing expression from crypt to villus, although MLCK1 mRNA is more restricted to the villus tip. C, phosphorylated MLC (red) is predominantly seen in villus enterocytes. The inset shows that phosphorylated MLC is specifically enhanced at areas of intercellular junctions. The scale bar is 20 μm (5 μm in the inset). D, when merged with F-actin (green), phosphorylated MLC colocalizes with the perijunctional actomyosin ring, producing a yellow color. The inset shows that phosphorylated MLC is specifically enhanced at areas of intercellular junctions within the perijunctional actomyosin ring. The scale bar is 20 μm (5 μm in the inset).
Given that MLCK1 mRNA is found primarily in the well differentiated cells of the villus tip, we examined the crypt-villus distribution of MLC phosphorylation that is the result of MLCK activity. Immunofluorescent labeling with antisera specific for MLC phosphorylated at serine-19 (20) showed that MLC phosphorylation is enhanced at the villus tip (Fig. 5C and D). Examination at higher magnification showed that phosphorylated MLC is concentrated within the perijunctional actomyosin ring and, as we have previously shown (20), enhanced at tight junctions. These data suggest that, similar to MLCK1 transcript content, MLCK activity is increased in terminally differentiated villus enterocytes.
MLCK1 Protein Is Localized to the Villus Tip
The data presented above show that MLCK1 mRNA content is enhanced within the well differentiated absorptive enterocytes of the villus tip. Given that MLCK2 mRNA content is unchanged from crypt to villus, the observation that MLC phosphorylation is enhanced at the same region as MLCK1 mRNA suggests that this phosphorylation may be mediated by MLCK1. However, this conclusion would first require that MLCK1 protein expression parallels mRNA content. Thus, we developed a polyclonal anti-peptide antibody to a unique 18-mer peptide derived from within the MLCK1 69-amino acid region that is absent in MLCK2. The affinity-purified rabbit anti-MLCK1 was characterized by SDS-PAGE immunoblot of recombinant MLCK1 and MLCK2 proteins and Caco-2 lysates (Fig. 6A). Comparable loading of recombinant MLCK1 and MLCK2 proteins was confirmed by immunoblots with the monoclonal antibody that recognizes both isoforms. These blots show that the affinity-purified anti-MLCK1 antibody reacts with recombinant MLCK1 but not MLCK2. Immunoblots of Caco-2 cell lysates confirmed that the affinity-purified anti-MLCK1 antibody reacted with a single protein of ~215 kDa that was also detected by the monoclonal antibody.
Fig. 6. MLCK1 is localized to the perijunctional actomyosin ring at the villus tip in human jejunum.
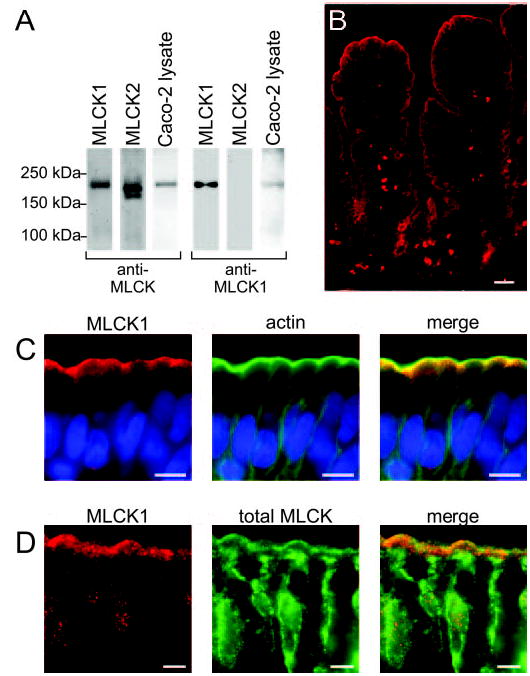
A, recombinant MLCK1, MLCK2, or Caco-2 lysates were blotted for total MLCK (anti-MLCK) or with the MLCK1-specific antisera (anti-MLCK1). The total MLCK antibody detects both recombinant MLCK1 and recombinant MLCK2, as well as a corresponding band in the Caco-2 lysate. The affinity-purified anti-MLCK1 antiserum detects recombinant MLCK1 but not recombinant MLCK2. A 215-kDa band is detected in Caco-2 lysates. B, MLCK1 detected by the anti-MLCK1 antiserum (red) is present primarily within enterocytes at the villus tip (scale bar, 20 μm). C, MLCK1 (red) is found in a narrow band just subapical to the brush border and displays enhancement in the area of cell-cell junctions (scale bar, 5 μm). A Hoechst stain for DNA is shown for reference (blue). When merged with f-actin (green), it is apparent that MLCK1 localizes to the peri-junctional actomyosin ring. D, total MLCK (green) can be found in two intracellular pools. The major pool is concentrated in the cytoplasm near the nucleus, whereas a more limited pool of MLCK is seen as a faint line near the brush border. This second pool colocalizes with MLCK1 (red; scale bar, 5 μm).
The affinity-purified anti-MLCK1 antibody was used to label normal human jejunum. As with mRNA, MLCK1 protein expression was predominantly seen within villus tip enterocytes (Fig. 6B). This staining was specific, because affinity-purified preimmune serum did not label human small intestine, and preincubation of the affinity-purified anti-MLCK1 antibody with excess of the peptide antigen prevented enterocyte staining (data not shown). MLCK1 within villus enterocytes was restricted to a subapical band corresponding to the perijunctional actomyosin ring (Fig. 6C). There was also enhancement of MLCK1 in the area of intercellular junctions, although this enhancement was not as marked as that observed for phosphorylated MLC (compare with Fig. 5C and D). Thus, both the cellular and subcellular patterns of MLCK1 expression match those of MLC phosphorylation.
To determine whether this perijunctional localization of MLCK1 was typical of the total cellular pool of MLCK, i.e. MLCK1 and MLCK2, human jejunum was double-labeled with anti-MLCK1 antibody and the anti-total MLCK monoclonal antibody that recognizes MLCK1 and MLCK2 equally. Like the anti-MLCK1 antibody, the monoclonal anti-MLCK antibody labeled the perijunctional actomyosin ring, consistent with the presence of MLCK1 at this location (Fig. 6D). However, the majority of monoclonal anti-MLCK staining was localized within the cytoplasm. Given that our initial RT-PCR data showed that MLCK2 mRNA is far more prevalent that that of MLCK1, the small amount of total MLCK found at the perijunctional actomyosin ring suggests that primarily MLCK1 is localized to this area, whereas MLCK2 is excluded from that site and found predominantly in perinuclear cytoplasm. Moreover, the observation that MLC phosphorylation at the tight junction is enhanced in villus enterocytes is consistent with the hypothesis that this enhanced phosphorylation may be catalyzed by MLCK1.
MLCK1 Regulates Tight Junction Permeability
The data above demonstrate that MLCK1 is predominantly expressed within villus enterocytes, where it is localized to the perijunctional actomyosin ring. This is the same population of enterocytes that respond to Na+-glucose cotransport by increasing tight junction permeability via a mechanism that requires MLC phosphorylation. The data also show a striking correlation between acquisition of Na+-glucose cotransport-dependent tight junction regulation and increasing MLCK1 mRNA content during in vitro Caco-2 differentiation. Taken as a whole, these observations suggest the hypothesis that MLCK1 is the MLCK isoform responsible for Na+-glucose cotransport-dependent tight junction regulation. To test this idea, we employed RNAi silencing of the MLCK1 isoform. Caco-2 cells were transfected in suspension with either nonspecific siRNA or a pool of four MLCK1-specific siRNA constructs. Given that siRNA constructs have a half-life of ~4 days, and monolayers do not transfect well, transfected cells were plated as monolayers at high density to accelerate confluence and differentiation. Under these conditions, intact tight junctions were assembled within 4 days. At this time MLCK1 mRNA content of monolayers transfected with control siRNA was 52 ± 3% of the total. MLCK1 mRNA was reduced to 27 ± 2% of total MLCK mRNA, a 47 ± 4% reduction, in monolayers transfected with MLCK1-specific siRNA (Fig. 7A and B). MLCK2 mRNA content was unchanged by the MLCK1-specific siRNA. This specific reduction in MLCK1 mRNA content correlated with reduced MLCK1 protein expression in monolayers transfected with MLCK1-specific siRNA (Fig. 7C). When Na+-glucose cotransport was active, the TER of monolayers transfected with MLCK1 siRNA was 25 ± 7% greater than that of monolayers transfected with nonspecific siRNA (Fig. 7D). Thus, selective knockdown of MLCK1 decreased tight junction permeability in a manner similar to the effect of inhibiting either Na+-glucose cotransport or MLCK in monolayers with active Na+-glucose cotransport. These data suggest that MLCK1 is the MLCK isoform that regulates tight junction permeability.
Fig. 7. Reduction of MLCK1 mRNA via RNAi causes increased TER.
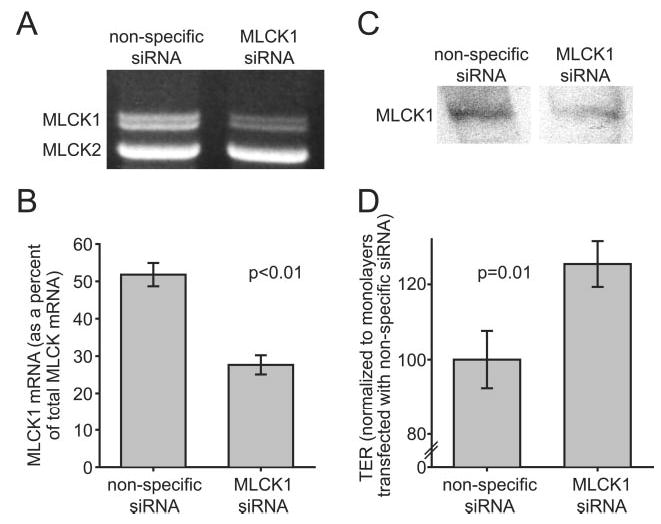
A, Caco-2 monolayers were transfected with either nonspecific siRNA or a pool of 4 siRNAs targeting MLCK1. Semi-quantitative RT-PCR analysis of these transfectants demonstrates a specific decrease in MLCK1 mRNA in monolayers transfected with MLCK1-specific siRNA. B, densitometric analysis of semi-quantitative RT-PCR gels shows that MLCK1, as a percentage of total MLCK, fell from 52 ± 3% in nonspecific siRNA-transfected monolayers to 27 ± 2% in monolayers treated with MLCK1 siRNA. This corresponds to a 47 ± 3% drop in MLCK1 mRNA. C, Caco-2 monolayers were transfected with either nonspecific siRNA or a pool of four siRNAs targeting MLCK1. Lysates of these monolayers were separated by SDS-PAGE and immunoblotted for MLCK1, as in Fig. 6. There was a specific decrease in MLCK1 protein expression in monolayers transfected with MLCK1-specific siRNA. D, the TER of monolayers transfected with MLCK1-specific siRNA is increased by 25 ± 7% over control monolayers transfected with nonspecific siRNA.
DISCUSSION
Maintenance of the intestinal epithelial barrier is a critical function of small intestinal enterocytes. Activation of the Na+-glucose cotransporter, SGLT1, initiates a signaling cascade that results in MLC phosphorylation, actomyosin contraction, and increased tight junction permeability, i.e. reduced barrier function (1, 7). The latter has been proposed to allow paracellular nutrient absorption, thereby increasing the total absorption rate beyond the saturation point of transcellular transporters when luminal nutrient loads are high (5–7). MLCK has been implicated in the control of MLC phosphorylation in this process (1, 12). Although other kinases and MLC phosphatase can also regulate MLC phosphorylation, the data presented in this study suggest that MLCK plays the principal role in Na+-nutrient cotransport-dependent tight junction regulation.
Two major isoforms of smooth muscle MLCK exist: short MLCK, found in smooth muscle, and long MLCK, first described in endothelium (14, 29). In this study we show that long MLCK accounts for the majority of MLC kinase activity in intestinal epithelia. Long MLCK is expressed as multiple splice variants; among these MLCK2 predominates, whereas the full-length transcript, MLCK1, is a minor form (15). The transcripts for these isoforms differ by only 207 base pairs, but insertion of this 69-amino acid domain can increase MLCK1 activity after phosphorylation by Src family kinases (16). This 69-amino acid region also contains a potential SH2-binding domain, raising the possibility that there are protein interactions unique to MLCK1. Previous studies have confirmed that MLCK1 and MLCK2 are coexpressed in a variety of tissues but did not identify differences in expression pattern that could suggest unique roles for each splice variant (15).
Given that multiple MLCK-dependent processes, including Na+-glucose cotransport-dependent barrier regulation and extrusion of apoptotic enterocytes, are precisely localized along the crypt-villus axis, we hypothesized that MLCK isoform expression might parallel epithelial function. We took advantage of the ordered system of enterocyte differentiation to assess MLCK isoform expression, both in the in vitro Caco-2 cell model of enterocyte differentiation (21, 30) and in vivo, as assessed in human intestine. Analysis of mRNA from Caco-2 cells and human intestinal epithelia showed that long MLCK expression was limited to isoforms 1 and 2. Semiquantitative RT-PCR showed that MLCK1 expression increased progressively during enterocyte differentiation, both in vitro and in vivo, and immunofluorescence microscopy confirmed that mRNA increases are matched by increased villus expression of MLCK1.
Our data show that MLCK1 expression is low in undifferentiated, proliferating (crypt) enterocytes but increases markedly during differentiation into absorptive (villus) enterocytes. MLCK1 mRNA expression was regulated similarly in vitro and in vivo: Caco-2 cells demonstrate an increase in MLCK1 levels at 6 days postconfluence, whereas human jejunal enterocytes show increased MLCK1 mRNA at the villus tip, 3–5 days after cells exit the crypt compartment (31). This increased MLCK1 expression strongly correlated with Na+-glucose cotransport-dependent tight junction regulation, which is limited to the villus in native intestine (4) and, as we show here, occurs only in differentiated Caco-2 monolayers ≥6 days post-confluence.
In addition to restriction of MLCK1 expression to well differentiated absorptive enterocytes, MLCK1 demonstrated a unique subcellular localization that suggested functional specialization of this isoform. MLCK1 is restricted to the apical enterocyte cytoplasm in the area of the perijunctional actomyosin ring. Both this crypt to villus distribution and subcellular localization are similar to those described here and previously described (20), respectively, for phosphorylated MLC. Thus, MLCK1 may be responsible for perijunctional MLC phosphorylation in villus enterocytes.
To directly test the role of MLCK1 in tight junction regulation, we selectively knocked down MLCK1 using isoform-specific siRNA. MLCK1 knockdown increased TER, i.e. decreased tight junction permeability, in monolayers with active Na+-glucose cotransport. This result confirms that the localization of MLCK1 to the perijunctional actomyosin ring is functionally significant and explains the strong correlation between development of physiological tight junction regulation and expression of MLCK1 during enterocyte differentiation. Thus, we must ask how a small 69-amino acid insert in a 1914-amino acid protein can result in such specialized localization and function. The answer may lay in definition of the binding partners and enzymatic regulators of motifs within this region. Given that MLCK-mediated barrier dysfunction has been implicated in diverse disease processes (12, 19, 32–34), definition of these regulatory motifs within MLCK1 may both enhance our understanding of physiology and provide novel therapeutic targets for a variety of disorders.
In summary, the data show that the MLCK expressed in intestinal epithelium is long MLCK and that the two isoforms expressed, MLCK1 and MLCK2, exhibit precisely regulated expression along the crypt-villus axis. The expression of MLCK1 is restricted to absorptive villus enterocytes, and increased levels of this isoform correlate strongly with the development of physiologic tight junction regulation. Furthermore, MLCK1 is localized to the perijunctional actomyosin ring at the level of the tight junction. Knockdown of MLCK1 leads to decreased tight junction permeability, demonstrating that MLCK1 controls tight junction regulation in small intestinal enterocytes. Thus, the short sequence that distinguishes MLCK1 from MLCK2 endows this isoform with a unique functional role in intestinal epithelium
Acknowledgments
We appreciate the expert technical assistance of Jason Angle, Dayou Liu, and Sara Palkon. We are also indebted to Dr. S. Milgram for providing the human intestinal epithelial cDNA library, Dr. M. Tretiakova for assistance with the laser capture apparatus, and Drs. A. Tyner, S. C. Meredith, and Y.-X. Fu for helpful discussions.
Footnotes
This work was supported by National Institutes of Health Grant R01DK61931 (to J. R. T.), a Senior Investigator Award from the Crohn’s and Colitis Foundation of America (to J. R. T.), and resources provided by The University of Chicago Digestive Disease Center P30 DK42086 and University of Chicago Cancer Center P30 CA14599.
The abbreviations used are: MLC, myosin II regulatory light chain; MLCK, myosin light chain kinase; TER, transepithelial resistance; PIK, membrane permeant inhibitor of myosin light chain kinase; RT, reverse transcriptase; MOPS, 3-(N-morpholino)propanesulfonic acid.
References
- 1.Turner JR, Rill BK, Carlson SL, Carnes D, Kerner R, Mrsny RJ, Madara JL. Am J Physiol. 1997;273:C1378–C1385. doi: 10.1152/ajpcell.1997.273.4.C1378. [DOI] [PubMed] [Google Scholar]
- 2.Yuhan R, Koutsouris A, Savkovic SD, Hecht G. Gastroenterology. 1997;113:1873–1882. doi: 10.1016/s0016-5085(97)70006-4. [DOI] [PubMed] [Google Scholar]
- 3.Marcial MA, Carlson SL, Madara JL. J Membr Biol. 1984;80:59–70. doi: 10.1007/BF01868690. [DOI] [PubMed] [Google Scholar]
- 4.Fihn BM, Sjoqvist A, Jodal M. Gastroenterology. 2000;119:1029–1036. doi: 10.1053/gast.2000.18148. [DOI] [PubMed] [Google Scholar]
- 5.Madara JL, Pappenheimer JR. J Membr Biol. 1987;100:149–164. doi: 10.1007/BF02209147. [DOI] [PubMed] [Google Scholar]
- 6.Pappenheimer JR, Reiss KZ. J Membr Biol. 1987;100:123–136. doi: 10.1007/BF02209145. [DOI] [PubMed] [Google Scholar]
- 7.Turner JR. Adv Drug Deliv Rev. 2000;41:265–281. doi: 10.1016/s0169-409x(00)00046-6. [DOI] [PubMed] [Google Scholar]
- 8.Adelstein RS. Cell. 1982;30:349–350. doi: 10.1016/0092-8674(82)90232-x. [DOI] [PubMed] [Google Scholar]
- 9.Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K. J Biol Chem. 1996;271:20246–20249. doi: 10.1074/jbc.271.34.20246. [DOI] [PubMed] [Google Scholar]
- 10.Komatsu S, Ikebe M. J Cell Biol. 2004;165:243–254. doi: 10.1083/jcb.200309056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamashiro S, Totsukawa G, Yamakita Y, Sasaki Y, Madaule P, Ishizaki T, Narumiya S, Matsumura F. Mol Biol Cell. 2003;14:1745–1756. doi: 10.1091/mbc.E02-07-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zolotarevsky Y, Hecht G, Koutsouris A, Gonzalez DE, Quan C, Tom J, Mrsny RJ, Turner JR. Gastroenterology. 2002;123:163–172. doi: 10.1053/gast.2002.34235. [DOI] [PubMed] [Google Scholar]
- 13.Birukov KG, Schavocky JP, Shirinsky VP, Chibalina MV, Van Eldik LJ, Watterson DM. J Cell Biochem. 1998;70:402–413. [PubMed] [Google Scholar]
- 14.Kamm KE, Stull JT. J Biol Chem. 2001;276:4527–4530. doi: 10.1074/jbc.R000028200. [DOI] [PubMed] [Google Scholar]
- 15.Lazar V, Garcia JG. Genomics. 1999;57:256–267. doi: 10.1006/geno.1999.5774. [DOI] [PubMed] [Google Scholar]
- 16.Birukov KG, Csortos C, Marzilli L, Dudek S, Ma SF, Bresnick AR, Verin AD, Cotter RJ, Garcia JG. J Biol Chem. 2001;276:8567–8573. doi: 10.1074/jbc.M005270200. [DOI] [PubMed] [Google Scholar]
- 17.Peterson MD, Mooseker MS. J Cell Sci. 1992;102:581–600. doi: 10.1242/jcs.102.3.581. [DOI] [PubMed] [Google Scholar]
- 18.Turner JR, Lencer WI, Carlson S, Madara JL. J Biol Chem. 1996;271:7738–7744. doi: 10.1074/jbc.271.13.7738. [DOI] [PubMed] [Google Scholar]
- 19.Scott RO, Thelin WR, Milgram SL. J Biol Chem. 2002;277:22934–22941. doi: 10.1074/jbc.M202434200. [DOI] [PubMed] [Google Scholar]
- 20.Berglund JJ, Riegler M, Zolotarevsky Y, Wenzl E, Turner JR. Am J Physiol. 2001;281:G1487–G1493. doi: 10.1152/ajpgi.2001.281.6.G1487. [DOI] [PubMed] [Google Scholar]
- 21.Pinto M, Robine-Leon S, Appay MD, Kedinger M, Triadou N, Dussaulx E, Lacroix B, Simon-Assmann P, Haffen K, Fogh J, Zweibaum A. Biol Cell. 1983;47:323–330. [Google Scholar]
- 22.Rousset M. Biochimie (Paris) 1986;68:1035–1040. doi: 10.1016/s0300-9084(86)80177-8. [DOI] [PubMed] [Google Scholar]
- 23.Nusrat A, Giry M, Turner JR, Colgan SP, Parkos CA, Carnes D, Lemichez E, Boquet P, Madara JL. Proc Natl Acad Sci U S A. 1995;92:10629–10633. doi: 10.1073/pnas.92.23.10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hopkins AM, Walsh SV, Verkade P, Boquet P, Nusrat A. J Cell Sci. 2003;116:725–742. doi: 10.1242/jcs.00300. [DOI] [PubMed] [Google Scholar]
- 25.Jou TS, Schneeberger EE, Nelson WJ. J Cell Biol. 1998;142:101–115. doi: 10.1083/jcb.142.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 27.Turner JR, Angle JM, Black ED, Joyal JL, Sacks DB, Madara JL. Am J Physiol. 1999;277:C554–C562. doi: 10.1152/ajpcell.1999.277.3.C554. [DOI] [PubMed] [Google Scholar]
- 28.Llor X, Serfas MS, Bie W, Vasioukhin V, Polonskaia M, Derry J, Abbott CM, Tyner AL. Clin Cancer Res. 1999;5:1767–1777. [PubMed] [Google Scholar]
- 29.Verin AD, Lazar V, Torry RJ, Labarrere CA, Patterson CE, Garcia JG. Am J Respir Cell Mol Biol. 1998;19:758–766. doi: 10.1165/ajrcmb.19.5.3125. [DOI] [PubMed] [Google Scholar]
- 30.Jumarie C, Malo C. J Cell Physiol. 1991;149:24–33. doi: 10.1002/jcp.1041490105. [DOI] [PubMed] [Google Scholar]
- 31.Shorter RG, Moertel CG, Titus JL, Reitemeier RJ. Am J Dig Dis. 1964;9:760–763. doi: 10.1007/BF02231983. [DOI] [PubMed] [Google Scholar]
- 32.Clayburgh DR, Shen L, Turner JR. Lab Invest. 2004;84:282–291. doi: 10.1038/labinvest.3700050. [DOI] [PubMed] [Google Scholar]
- 33.Huang Q, Xu W, Ustinova E, Wu M, Childs E, Hunter F, Yuan S. Shock. 2003;20:363–368. doi: 10.1097/01.shk.0000079425.0000.db. [DOI] [PubMed] [Google Scholar]
- 34.Wainwright MS, Rossi J, Schavocky J, Crawford S, Steinhorn D, Velentza AV, Zasadzki M, Shirinsky V, Jia Y, Haiech J, Van Eldik LJ, Watterson DM. Proc Natl Acad Sci U S A. 2003;100:6233–6238. doi: 10.1073/pnas.1031595100. [DOI] [PMC free article] [PubMed] [Google Scholar]