

Abstract
Nerve cells may survive or die after axonal or dendritic transection. After neurite transection near (<50μm) the cell body of Fura-2 loaded B104 neuroblastoma (rat brain derived) cells, the somal calcium concentration (SCC) undergoes a three-phase transient change: a rapid (0 to 0.15 min posttransection (PT)) rise phase followed by an early (0.15 to 1.5 min PT) rapid decay phase, succeeded by a late (1.5 to 60 min PT) slower decay phase that restores SCC to preinjury levels. The SCC in a critical interval (1.5 to 12.5 min PT) of the third transient phase correlates with cell fate, i.e., most transected cells that exclude dye (restore a barrier) and die have a significantly higher (p<0.005) SCC in this Critical Interval than transected cells that exclude dye and survive at 24h PT. Loading BAPTA (chelation of somal Ca2+) before, but not after, the Critical Interval increases the percentage of cells that survive compared to cells transected without BAPTA loading. Furthermore, most transected cells that die, despite successful barrier restoration, exhibit characteristics consistent with apoptosis initiated during the Critical Interval of the SCC, including caspase activation and plasmalemmal phosphatidylserine translocation. These data suggest that decreased cell survival for injuries near the soma is due to Ca2+-initiated apoptosis during the Critical Interval of the third phase of the SCC transient.
Keywords: plasmalemmal injury, calcium-initiated cell degeneration, injury-induced apoptosis, injured cell mortality, cell death prevention
INTRODUCTION
Plasmalemmal repair by Ca2+-induced accumulation of membranous vesicles shortly after a plasmalemmal disruption is necessary for cell survival in many types of injured cells from invertebrates to mammals (see, Fishman and Bittner, 2003). After severance of crayfish giant axons, a seal is restored (i.e., ionic injury-current decays to preinjury levels) at the damage site and axonal electrical function recovers (i.e., action potential propagation returns) about 1h posttransection (PT) (Eddleman et al., 1997). Similarly, after neurite transection, mouse spinal neurons (Lucas et al., 1985), rat septal neurons (Xie and Barrett, 1991), and rat neuroblastoma (B104) cells (Yoo et al., 2004) in cultures form a membrane barrier (exclude extracellular dye) and survive for 24 h.
Plasmalemmal repair is necessary for cell survival, but is not sufficient to ensure cell survival after plasmalemmal damage because other factors such as animal age (O’Connor et al., 1974), axonal size (Lucas et al., 1985), and injury location (Lucas et al., 1985; Yoo et al., 2004) affect survival after plasmalemmal damage. As an example of the effect of location on B104 cell survival, neurite transection near the cell body decreases survival compared to severance farther from the cell body, even though both lesion sites exclude external dye (restore a barrier) with equal probability and at the same rate (Yoo et al., 2004). Cell survival following plasmalemmal damage very likely also depends on the regulation of intracellular Ca2+ (Ca2+ homeostasis), the loss of which is also associated with a number of neuropathologies. For example, a transient increase in [Ca2+]i coincides with increased neuronal death following cerebral ischemia (Stys and Ashby, 1990; Cheung et al., 1986), excitotoxicity (Hyrc et al., 1997), spinal cord injury (Balentine et al., 1982), and neurodegenerative diseases (e.g., Alzheimer’s, Parkinson’s, and Huntington’s {Vito et al., 1996}). In these and other in vivo and in vitro studies of neuronal survival after injury, quantification and correlation of changes in somal Ca2+ concentration (SCC) with neuronal fate were not done or were complicated by interactions with glia and other cells.
This work was undertaken to determine the intrinsic SCC response of individual neurons to injury and to relate with minimal complication the response to neuronal fate. To quantify the SCC transient in individual cells after a neurite transection and to correlate the transient with cell fate, we identified Ca2+-initiated effects during the time course of the SCC transient that affected the survival of a neuronal cell line (Schubert et al., 1974) that was sustainable without neurotrophic factors and glial interactions. Decreased cell survival following injury near the soma is explained by data, which indicate that apoptosis is initiated by Ca2+ during the SCC transient.
MATERIALS AND METHODS
Solutions, Media, and Chemicals
A phosphate buffered saline (PBS) (Cellgro, Herndon, VA) containing Ca2+ and Mg2+ (Ca/Mg -PBS, 0.9 mM Ca2+ and 0.5 mM Mg2+) and lacking Ca2+ and Mg2+ (PBS=) were used for cell passaging and washing. PBS= containing 0.25% Trypsin (Invitrogen, Carlsbad, CA) was used to dissociate cultured cells from the surface of culture flasks. Control medium consisted of Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented 1:1 with Ham’s F12 (no antibiotics or sera). Cells were grown in culture medium, i.e., control medium containing fetal bovine serum (Invitrogen, Carlsbad, CA) and antibiotics (penicillin/streptomycin (10,000 units penicillin/ml and 10 mg streptomycin/ml in PBS=). Other chemicals used were: culture substrate, poly-D-Lysine (mw 30,000 – 70,000) [Sigma, St. Louis, MO]; a broad caspase inhibitor, benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (Z VAD FMK), a Caspase-9 inhibitor, N-benzyloxycarbonyl-Leu-Glu-His-Asp-fluoromethylketone (Z LEHD FMK), and a Ca2+ chelator, 1,2-bis (2-Aminophenoxy) ethane-N, N, N’, N’-tetraacetic acid tetrakis (acetoxymethyl) ester (BAPTA-AM) [BD Biosciences, San Jose, CA]; ratiometric, Ca2+-reporter fluorchromes, Fura-2K+ and Fura-2-AM (Tef Labs, Austin, TX); hydrophilic fluorochromes, Fluorescein-dextran (3kD), Rhodamine green-dextran (3kD), Tetramethyrhodamine-dextran (3kD), and an apoptotic affinity assay for phosphatidylserine at the extracellular plasmalemmal surface, Annexin V conjugated to Alexa Fluor 647 together with nuclear stain, Sytox green to assess plasmalemmal integrity (Molecular Probes, Eugene, OR). [Ca2+] standards (0–40μM) were used to calibrate intensity ratios obtained from Ca2+-reporter fluorochromes (Molecular Probes, Eugene, OR). Dimethyl-sulfoxide (DMSO) was used to solubilize chemical agents (Sigma, St. Louis, MO). Rhodamine green was dissolved as concentrated stock (1% w/v) solutions in Ca/Mg-PBS and diluted for experiments in control medium to yield a final concentration of 0.01% w/v.
Transmission and Fluorescence Microscopy
Individual B104 cells (see next section) in a plastic culture dish were assessed using transmission {phase and differential interference contrast (DIC)} and fluorescence microscopy. Images analyzed for quantitative SCC determinations were acquired using a Nikon epifluorescence TE 200 inverted microscope, equipped with a Cool Snap HQ Digital Camera, a 10X/0.3NA phase or a 40X/0.9NA Superfluar objective, a 150-W Xenon Arc Lamp Controlled by a Lambda DG-4 excitation filter wheel switch (Sutter Instruments CO., CA, 94949) and Fura-2 compatible filter sets (bandpass 340 and 380nm excitation/400nm Dichroic/bandpass 510nm emission, Chroma Technology Corp., VT). For apoptosis/necrosis determinations, we used a Zeiss 510 laser-scanning confocal, inverted microscope equipped with the following objective lenses: 10X/0.30NA (Numerical Aperture), 20X/0.4NA phase objective, and 2 water-immersion lenses, 40X/1.2NA and 40X/0.75NA. All images were digitized and analyzed at 12-bit resolution using MetaFluor imaging software (Universal Imaging, Downingtown, PA).
Cell Culture Preparation
A cell line (B104) derived from neuroblastoma of rat brain (Schubert et al., 1974) was a gift from Dr. Jane Bottenstein (Department of Neuroscience and Cell Biology, University of Texas Medical Branch, Galveston, TX). B104 cells display specific functional characteristics, including propagation of action potentials, expression of plasmalemmal ion channels and transporters, and secretion of neurotransmitter that qualify them as a neuronal model (Tan et al., 2003, Dib-Haji et al., 1996, Gu and Waxman, 1996, Luo et al., 1999, Luo et al., 1997, Tyndale et al., 1994, Bottenstein et al., 1979, Schubert et al., 1975 and 1974).
B104 cells were grown in a humidified incubator (37°C, 5% CO2) on 25cm2 tissue culture flasks containing control medium (see Solutions etc.) supplemented with heat-inactivated 10% fetal bovine serum and 1% penicillin/streptomycin (culture medium) sterile-filtered through a 0.22μm-diameter pore cellulose acetate filter) that was replaced every other day. Upon reaching 70–80% confluency, the cells were subcultured onto new 25cm2 tissue-culture flasks or seeded onto 35mm tissue-culture dishes.
B104 cells were seeded at a density of ~2000 cells/cm2 onto 35mm (8cm2) plastic, culture dishes coated with poly-D-Lysine as described previously (Yoo et al., 2004). Sometimes, for greater microscopic resolution, cells were seeded onto an Aclar (plastic) coverslip that was attached with silicone epoxy (Sylgard 185, Dow Corning Corp., MI) in the overlapping region beyond the periphery of a drilled hole at the bottom of a 35 mm tissue culture dish. Before plating cells onto the bottom plastic coverslip, these dishes were placed in an oven at 60° C overnight to cure the epoxy, and then sterilized by washing the dishes with 80% ethanol and illuminating for 20min with an ultraviolet-germicidal lamp. After 24h of proliferation, the culture medium was replaced with control medium. B104 neurites rapidly elongated (to >100μm from the cell body) 24h after serum deprivation, and the cells could be maintained for nearly a week in the presence of control medium.
Neurite Transection, Dye Barrier Assessment, and Cell Viability Assessment
We used the sharp edge of the tip (10–15μm diameter) of a micropipette, to transect neurites as described previously (Yoo et al., 2004). Successfully transected cells (neurites) showed separation of the proximal and distal stumps at the site of transection (marked by the line scored in the plastic culture dish by the pipette tip). When this separation occurred, 100% of transected cells (40) took up hydrophilic dye (fluorescein dextran) present in the culture medium at the time of transection. Dye barrier identification 20 min after neurite transection was assessed by quantifying the ratio of the average fluorescence intensity of a different hydrophilic dye, added extracellularly at 20 min PT in a transected cell, to the average intensity in a neighboring, untransected cell. When this ratio, calculated as a percentage, was >5%, the injured cell was counted as “taking up dye.” Transected cells with a ratio of <5% were tallied as “excluding dye.” The viability of B104 cells was evaluated using morphological criteria at 2, 4, and 8h PT, and lastly at 12h PT, a time that accurately predicts B104 cell survival at 24h PT (Yoo et al., 2004).
SCC Determinations
To measure SCC quantitatively, Fura-2 (Grynkiewicz et al., 1985) was loaded into cells by incubating them for 10 min at 37° C, 5% CO2 in control medium containing 1μM Fura-2 AM [Fura-2 conjugated to an acetoxymethyl ester (AM) group]. Then, the cells were rinsed twice and replaced with control medium. To ensure complete cleavage of the AM group by intracellular esterases, the cells were incubated for another 10min at 37° C and rinsed twice more with control medium. Fura 2 was contained largely in the somal cytosol and nucleus, as indicated by uniform rather than punctate fluorescence intensities in the soma and by a substantially decreased (<5%) somal fluorescence intensity following permeabilization of the plasma membrane with 50 μM digitonin.. Further permeabilization of the intracellular organelles with Triton (0.1% v/v) lowered the fluorescence intensity to background levels. To prevent UV-induced cell mortality, we determined empirically a UV level sufficient for Fura-2 excitation/emission at which the survival of untransected, exposed cells was indistinguishable from untransected, unexposed cells.
An area of the culture dish containing Fura-2-loaded cells, which were well dispersed and easily identified, was selected using the 10X/0.3NA phase objective of the Nikon TE 200 microscope. An SCC transient was determined at sample times (beginning just before neurite transection and during the transient’s time course PT) from a pair of 12-bit gray-scaled images of emission intensities at 510nm for separate excitations at 340 and 380nm. Image pairs were acquired in <2s through the 40x/0.9NA Superfluar (Nikon) objective and intensities digitized by MetaFluor (Universal Imaging) software.
For each image pair, intensities were measured exclusively within the somal region of the image field. After subtracting the background intensity of a region lacking cells adjacent to the soma of interest, the F340/F380 emission intensities ratio (R) was used to determine the SCC from the equation (Grynkiewicz et al., 1985): [Ca2+] = {Kd x {(R-Rmin)/(Rmax-R)} x (F380min/F380max)}, where Kd is the dissociation constant of Fura-2 in the soma. Such a ratiometric determination of SCC effectively normalizes differences in cell thickness and dye concentration (Tsien et al., 1985). The Kd of Fura-2 was determined from an in vitro calibration using the salt form of Fura-2 and a set of known [Ca2+] standards (0 to 75μM, Molecular Probes). Emission (at 510nm) image pairs (for excitations at 340 and 380nm) were recorded from a drop of Ca2+ buffer at each [Ca2+] and from an adjacent area devoid of solution. The background intensity of the adjacent area was substracted from the total fluorescence emission intensity, and the corrected 340nm and 380nm fluorescence intensities were plotted and fitted according to the equations: Log (F340 – F340min)/(F340max-F340) vs. Log [Ca2+] and Log (F380 – F380min)/(F380max-F380) vs. Log [Ca2+]. Calibration pairs were obtained early {Kds of 331nM (340nm) and 323nM (380nm)} in the course of experiments and much later {Kd values of 335nM (340nm) and 322nM (380nm)} to ensure that the Kd of Fura-2 was time invariant. The Kd values of both sets of calibrations were averaged to yield 328 nM; a value that is in the range [200–600 nM] reported for Fura-2 (Grynkiewicz et al., 1985) and that we used to calculate SCC.
Statistical Analyses
The mean SCC (<SCC>) was compared among different experimental groups at various time points using the Kruskal-Wallis test, a non-parametric statistical analysis (Montgomery, 2001). Viability was calculated as the ratio (expressed as a percentage) of the number of cells with a transected neurite that were viable at any given time to the total number of cells with a transected neurite. To determine whether a difference in viability existed at 12h Post-Transection (PT), comparisons were analyzed using Kaplan-Meier-Survival, Log Rank. Similarly, the viability of untransected cells was calculated as a percentage of the number of visible cells, at any given time, to the total number of untransected cells, as determined at the time of transection. All statistical tests of data for significance were examined at the 0.05 level. For each sample, we recorded the % survival, n = number of cells, N = number of dishes.
RESULTS
The SCC transient as an indicator of cell survival after injury
To examine whether the SCC transient after neurite transection near the soma is an indicator of cell survival, we recorded the SCC of individually identified, Fura-2-loaded B104 cells from just before injury to 60min PT (Figure 1, 2, n=37, N =21) (where n = # cells, N = # dishes). To determine SCC ratiometrically (see, Methods), pairs of images were obtained at 510nm emission following 340nm and 380nm excitation. A neurite was transected near the soma (<50μm) while acquiring image pairs (samples) of the SCC every 2s until 20s PT; thereafter, samples were obtained at longer intervals varying from 20s to 60min. This protocol allowed detailed capture of the rapid phases (rising and early decay) of the SCC transient and reduced photodamage to the cell and fluorochrome during the slow decay phase. Occasionally, the micropipette was positioned to transect the neurite <50μm from the cell body of two or more adjacent cells oriented in parallel in a single continuous stroke of the micropipette. In such cases, the SCC measured separately in all cells in the field of view was tracked simultaneously. Neighboring untransected cells served as controls in all experiments.
Figure 1.
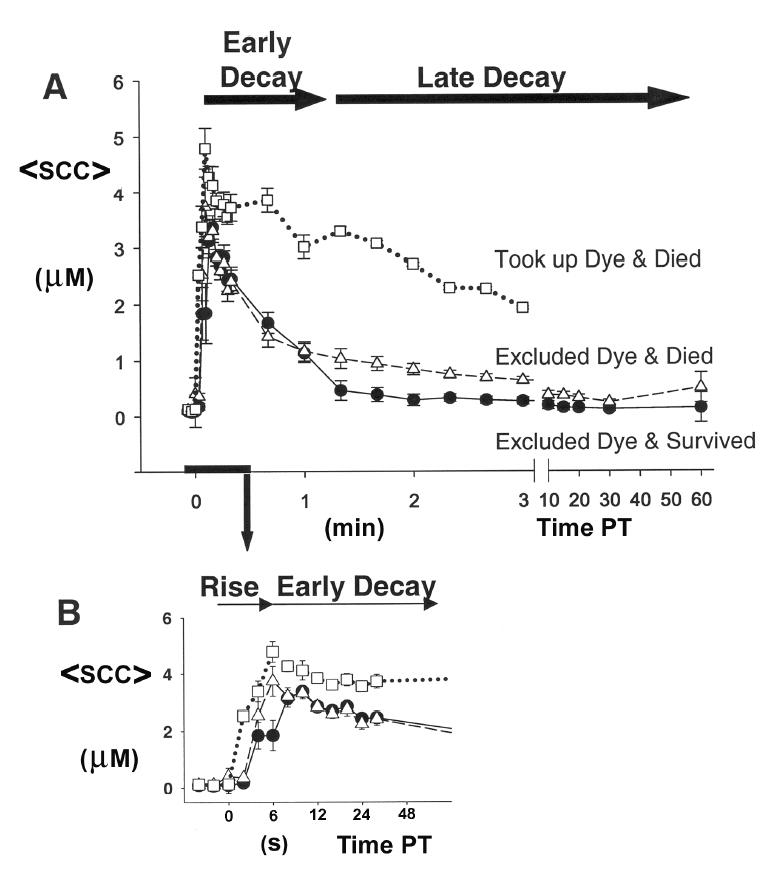
(A) Mean (±SE) somal [Ca2+] (<SCC>) before transection and posttransection (PT) of cells (n [cells] =37, N [cultures] =21; single neurite transection near the soma of each cell). (B) Expanded time scale for the first 0.5min (30s) PT of data shown in (A). Dye exclusion for each uniquely identified and tracked transected cell was assessed at 20min PT, and its survival assessed for up to 12h PT. Data are plotted for groups of cells that exhibited one of three possible outcomes: (1) excluded dye at 20min PT and survived at 12h PT (51%, filled circles), (2) excluded dye at 20min PT and died within 12hPT (30%, unfilled triangles), and (3) took up dye at 20min PT and died within 12hPT (19%, unfilled squares).
Figure 2.
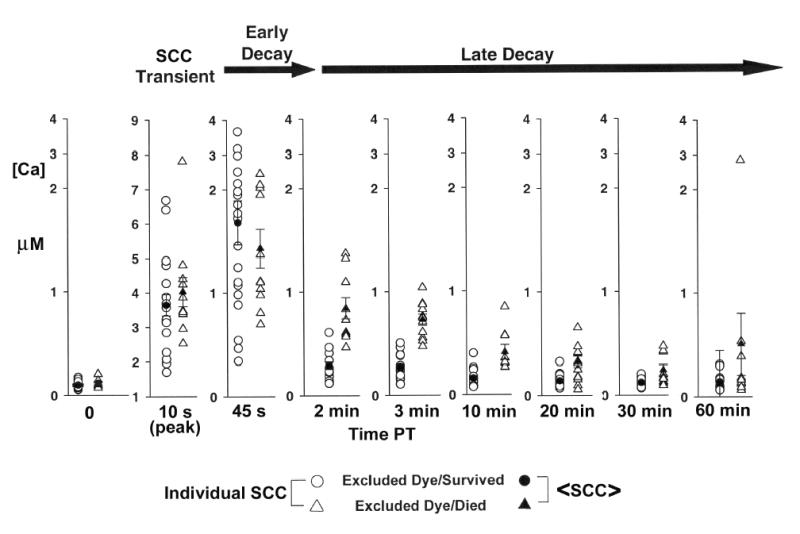
Individual somal [Ca2+] (unfilled symbols) and mean somal [Ca2+] ±SE (filled symbols) at various times (min) before and after neurite transection near the cell body for groups of B104 cells exhibiting the outcomes: (1) excluded dye at 20min PT and survived at 12h PT (circles), (2) excluded dye at 20min PT and died within 12h PT (triangles). Data are the same as in Figure 1 and were obtained as described in that legend. Note: Vertical scale change for data at 10s PT.
Following transection, the <SCC> responded transiently with three distinct phases (Figure 1). Initially, the <SCC> rapidly increased (initial rising phase) and peaked within 0.15min PT. The <SCC> then rapidly decayed from the peak (early decay phase) until 1.5min PT, when it subsequently returned more slowly to control levels at 60min PT (late decay phase).
After uniquely identifying and tracking each transected cell, assessing exclusion of extracellular dye at 20min PT, and assessing viability at 12h PT, the SCC transient associated with each transected cell was assigned to one of three groups: (1) (Figure 1, filled circles) cells that excluded dye and survived, (2) (Figure 1, unfilled triangles) cells that excluded dye and died, or (3) (Figure 1, unfilled squares) cells that took up dye and died. Before transection, the SCC of all cells in each group ranged from 50–150nM and the <SCC> was not significantly different (Kruskal-Wallis, p>0.05) among any of the three groups. Following transection, the peak of the initial rising phase of the SCC for all cells in each group ranged from 2–10μM. In transected cells that took up dye and died, the <SCC> at the peak (4.68μM) was significantly higher (p<0.05, Kruskal-Wallis test) than in transected cells that excluded dye and survived (3.27μM) and in cells that excluded dye and died (3.63μM). The <SCC> (1.92μM) in the late decay phase at 3min PT of cells that took up dye and died was significantly higher (p<0.05, Kruskal-Wallis test) than either of the other 2 groups that excluded dye (0.62 μM excluded dye and died and 0.26 μM excluded die and survived). Beyond 3min PT, the SCC for cells that took up dye and died could not be followed because of substantial dye leakage. Furthermore, since all transected cells that took up dye died, we focused our studies on transected cells that excluded dye, some of which died and some of which survived. Specifically, we examined whether an elevated <SCC> during the late decay phase might account for the death of some cells that excluded dye after neurite transection.
The <SCC> (3.63μM) at the peak of the initial rising phase of transected cells (Figure 1) that excluded dye and died was not significantly different (Kruskal-Wallis, p>0.05) from transected cells that excluded dye and survived (3.27μM). Although the time-to-peak of the <SCC> transient of these two groups of transected cells appears different (Figure 1B), the difference was not statistically significant (p>0.05). The <SCC> during the early rapid decay phase of these two groups was also not significantly different until 1.4min PT. Although the <SCC> exhibited a slow late decay phase for both groups, the <SCC> (1.00μM) at 1.4 min PT of transected cells that excluded dye and died was significantly higher (Kruskal-Wallis, p<0.05) than the <SCC> (0.45μM) of cells that excluded dye and survived. The <SCC> in the late decay phase remained significantly higher for those cells that excluded dye and died until 30min PT (Kruskal-Wallis, p<0.05). The <SCC> (0.49μM) of transected cells that excluded dye and died at 60min PT was not significantly different (p>0.05, Kruskal-Wallis) from transected cells that excluded dye and survived (0.14μM). That is, the <SCC> returned to baseline levels at 60min PT for all cells that excluded dye after neurite transection.
To differentiate changes in SCC among B104 cells in each of the two groups (Figure 2, excluded dye and survived, circles; excluded dye and died, triangles) at various times before and after neurite transection , we plotted the SCC of each transected cell (Figure 2, unfilled symbols) and the <SCC> (Figure 2, filled symbols). In the interval from 1.4min to 30min PT, the <SCC> of transected cells that excluded dye and died was significantly greater (p<0.05, Kruskal Wallis test) than the <SCC> in transected cells that excluded dye and survived. Examination of the data for individual values of SCC obtained at 2min, 3min, and 10min PT (Figure 2), showed that individual values for cells that excluded dye and died (Figure 2, unfilled triangles) and cells that excluded dye and survived (Figure 2, unfilled circles) overlapped less compared to the overlap of individual values obtained at 20min and 30min PT. Nevertheless, the <SCC> of cells that excluded dye and died (Figure 2, filled triangle) compared to those that excluded dye and survived (Figure 2, filled circles) was significantly (Kruskal Wallis) different at 2min PT (p<0.001), 3min PT (p<0.001), 10min PT (p =0.001), 20min PT (p =0.006) and 30min PT (p =0.027). From these data, we determined a Critical Interval (1.5 to 12.5min PT) that best predicted B104 cell survival following neurite transection near the cell body based on the PT times during which the statistical significance of the difference between the <SCC> of cells that died and those that survived was highest (i.e., the PT interval during which p<0.005).
We controlled for several dye-related artefacts that could affect the data. To prevent leakage of Fura-2 from loaded, transected cells contributing to the observed decay in SCC, Fura-2 was briefly reloaded just after measuring Fura-2 fluorescence at 20min PT. In addition, induced elevation of the SCC in transected cells by application of ionomycin (permeabilizes plasmalemma to Ca2+), carbonylcyanide m-chlorophenylhydrazone (CCCP, induces Ca2+ release from mitochondria), and Thapsigargin (depletes ER Ca2+ stores) treatment in separate sets of transected cells at 20min PT elicited changes in Fura-2 intensity that were similar to those changes observed after induced elevation of SCC in untransected cells (data not shown). Likewise, induced elevation of SCC in transected cells at 60min PT (after briefly reloading Fura-2 at 20minPT), elicited changes in Fura-2 intensity similar to the intensity changes elicited in untransected cells (data not shown).
Somal Ca2+ chelation shortly after transection increases long-term cell survival
Since an increased SCC in the Critical Interval from 1.5–12.5min PT predicted decreased cell survival, we examined whether increasing intracellular Ca2+-buffering capacity could enhance the survival of transected B104 cells. After quickly (within 5s) transecting 1–3 neighboring cells, a membrane-permeant Ca2+-chelator precursor (BAPTA-AM: 30μM) was added to the control medium at 30s PT. Intracellular esterases removed the AM moiety to produce membrane-impermeant BAPTA. After loading the cells with BAPTA for 5min, the cells were washed with fresh control medium. Dye exclusion of each transected cell was assessed at 20min PT, and the viability of transected cells and neighboring, untransected cells was assessed at 2, 4, 8, and 12h PT.
At 12h PT, the survival (Figure 3A, filled circles, 67%, n =17, N=8) of transected cells loaded with BAPTA at 30s PT was significantly greater (p<0.05, Kaplan-Meier survival, Log Rank) than the survival (Figure 3A, unfilled squares, 38%, n = 19, N =8) of transected cells treated with additional control medium lacking BAPTA-AM added at 30s PT. In contrast, when BAPTA was loaded into cells at 20min PT, the survival (Figure 3B, filled circles, 62%, n=29, N =4) of transected cells loaded with BAPTA at 20min PT was not significantly greater than the survival (Figure 3B, unfilled squares, 60%, n =20, N =2) of transected cells with additional control medium added at 20min PT. The increased survival of transected cells with BAPTA-AM added at 30s PT was consistent with the hypothesis that increased SCC in the Critical Interval 2–10min PT determines cell survival.
Figure 3.
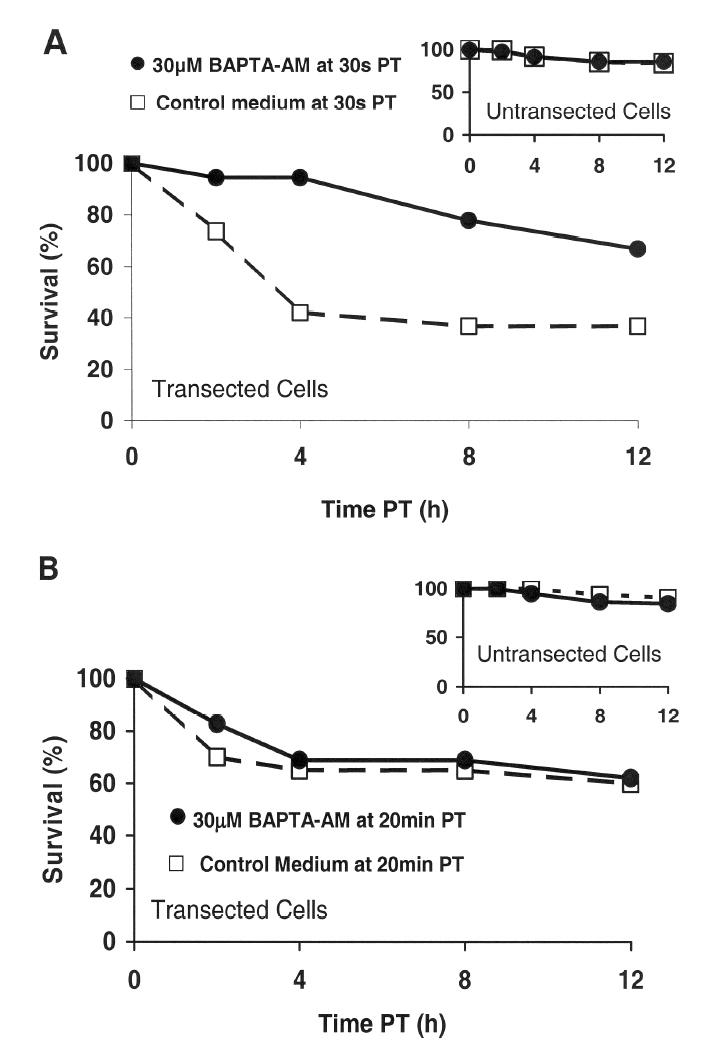
Effect of application of BAPTA-AM at 30s (A) or 20min PT (B) on the % survival within 12h PT of transected and neighboring, untransected (inset, A, B) B104 cells.
Control medium containing BAPTA-AM (filled circles) or control medium lacking BAPTA-AM (unfilled squares) was added to cells at 30s PT (A) or at 20min PT (B) (see Methods). After incubating in BAPTA-AM for 5min, the B104 cells were washed with control medium lacking BAPTA-AM, and the viability of transected and neighboring, untransected cells was assessed morphologically at 2, 4, 8, and 12h PT.
To exclude BAPTA effects on cell survival unrelated to injury, we determined that the survival of neighboring, untransected cells was not affected by loading BAPTA at 30s or 20min PT. That is, at 12h PT, the survival of untransected cells loaded with BAPTA at 30s PT (inset, Figure 3A, filled circles, 86%, n =83, N =8) or 20min PT (inset, Figure 3B, filled circles, 84%, n =58, N =4) was not significantly different (p>0.05, Kruskal-Wallis) than the survival of untransected cells treated with additional control medium lacking BAPTA (at 30s PT: inset, Figure 3A, unfilled squares, 84%, n =106, N =7; at 20min PT: inset, Figure 3B, 91%, unfilled squares, n =33, N =2). These data indicate that effects of AM cleavage (e.g., production of formalin) on cell survival are negligible and that chelating intracellular Ca2+ in untransected cells does not increase the percentage of surviving, untransected cells. The lack of an effect of BAPTA on the 12h PT survival of untransected cells suggests that the small percentage (10%) of untransected cells that died at 12h PT degenerated through a Ca2+-independent mechanism unrelated to injury. Furthermore, the percentage of transected cells that survived after BAPTA loading (Figure 3A, filled circles, 67%, n =17, N=8) did not recover to the level of survival of surrounding untransected cells (inset, Figure 3A, filled circles, 86%, n =83, N =8). The lack of a complete rescue of transected cells from death at 12h PT by BAPTA application at 30s PT (19% less than the survival level of untransected cells at 12h PT) implies that other Ca2+ independent causes of degeneration may occur.
Death of transected cells that exclude dye occurs mainly by apoptosis
To ascertain the relationship among transected cells that i) excluded dye, ii) had an elevated SCC during the Critical Interval, and iii) died, we examined the onset and mode of cell death following the Critical Interval. To differentiate between transected cells that die through apoptosis vs. necrosis, we used an Annexin-V-affinity-assay (Engeland et al., 1998) as follows. Transected cells that restore a barrier but are in early apoptosis, when plasmalemmal integrity is still maintained, exclude dyes such as Rhodamine green (a plasmalemal-impermeant hydrophilic dye) or Sytox green (a plasmalemmal-impermeant nuclear staining dye). Moreover, phospholipid (phosphatidylserine) translocation from the cytoplasmic to the extracellular plasmalemmal surface occurs early in apoptosis and is indicated by the selective binding of Annexin V conjugated to a fluorochrome (Alexa Fluor 647). Cells that first label with Annexin V-Alexa Fluor 647 but not Sytox green after simultaneous application of both would indicate apoptosis. In contrast, necrotic cells would label simultaneously with Annexin V-Alexa Fluor 647 and Sytox green, i.e., when the integrity of the plasma membrane is compromised, both dyes can enter the cell and bind to their specific epitopes (Engeland et al., 1998).
To assess the mode of cell death, neurites of B104 cells (8–15 per dish within 2min) were transected near the soma (n =34, N =3), and the cells were incubated at 37° C/5%CO2 for 20min in control medium. At 20min PT, the cells were assessed for exclusion of Rhodamine green-dextran. Subsequently, at 40min, 2h, 4h, 8h, 12h, 16h, and 24h PT, the cells were incubated in control medium containing both Annexin V (5μl/100μl) and Sytox green (50nM) for 10min, and then washed 3 times with control medium lacking dye. A transected cell (Figure 4A’, “Tr”) that received this treatment initially (at 40min PT) labeled with Annexin V, but not Sytox green, (Figure 4A’, red), consistent with the time course of a cell undergoing apoptosis at 40min PT or earlier, and much later (at 12h PT) co-labeled (Figure 4B’) with both Annexin V-Alexa Fluor 647 (red) and Sytox green (green), indicating that the cell had become necrotic. In contrast, a neighboring, untransected cell (Figure 4A’,”Un”) remained unlabeled with either dye for 12h PT. In a set of such experiments to assess the mode of cell death, most cells (Figure 5, unfilled bar, 77%, n =13, N=3) that excluded dye at 20min PT and died within 24h PT, labeled with only Annexin V-Alexas Fluor 647 in the interval 40min to 8h PT and labeled with both Annexin V-Alexas Fluor 647 and Sytox green in the interval 8h to 24h PT. These data indicated that these cells were apoptotic. A smaller percentage of transected cells that excluded dye at 20min PT and died within 24h PT (Figure 5, filled bar, 23%, n =4, N =3,) were labeled simultaneously by Annexin V-Alexas Fluor 647 + Sytox green in the interval 40min to 4h PT, indicating that these cells were necrotic. All transected cells that took up dye at 20min PT subsequently died within 24h PT (100%, Figure 5, hatched bar). The apoptotic or necrotic mode of death for these cells could not be distinguished by this assay since any labeling by these indicators was almost certainly due to diffusion of the indicators at the injury site. Such assays showed that most transected cells that excluded dye (e.g., Sytox green at 40min PT) and died at 12h PT were already undergoing degeneration by apoptosis shortly after the Critical Interval; consistent with the notion that apoptosis was initiated during the Critical Interval.
Figure 4.
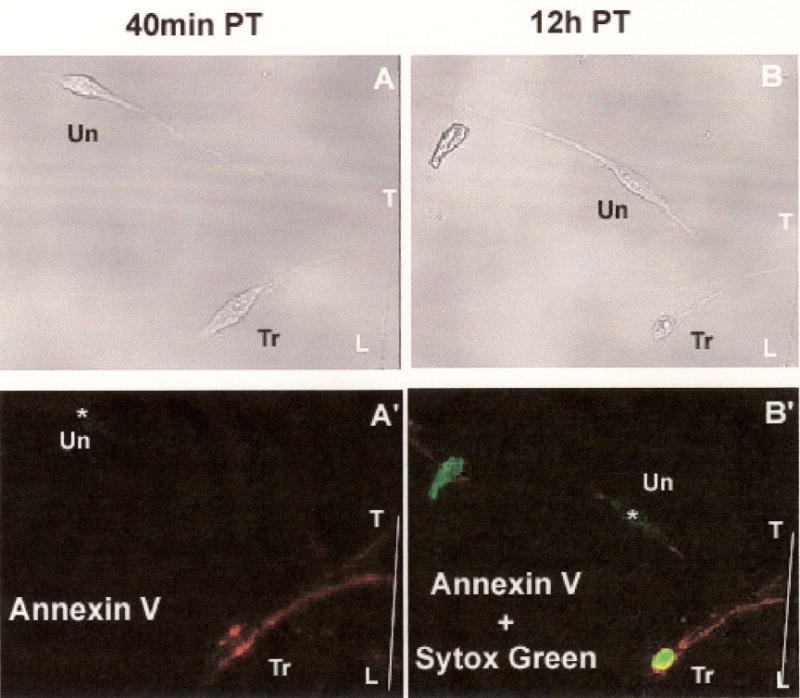
Transmission (A, B) and fluorescence (A’B’) images used to detect apoptosis with the Annexin-V-affinity assay (see Methods). Scored line in plastic culture dish labeled “T-L” marks the site of neurite transection produced by the sharp edge of the pipette tip.
(A, A’) Images of transected (“Tr”) B104 cell and a neighboring untransected (“Un”) B104 cell after incubation in control medium containing Annexin V + Sytox green at 40min PT, followed by subsequent washes with control medium lacking fluorchromes, The transected cell (A’, “Tr”) was labeled by Annexin V (Red) but not Sytox green. The untransected cell, marked as “*” (A’, “Un”), was not labeled by either Annexin V or Sytox green.
(B, B’) Images of a transected (“Tr”) B104 cell and a neighboring untransected (“Un”) B104 cell after incubation in control medium containing Annexin V-Alexa Fluor 647 + Sytox green at 12h PT, followed by subsequent washes with control medium lacking fluorchromes. The transected cell (A’, “Tr:) was labeled by Annexin V-Alexa Fluor 647 (Red) and Sytox green. The untransected cell, marked as “*” (B’, “Un”), remained unlabeled by either Annexin V-Alexa Fluor 647 or Sytox green.
Figure 5.
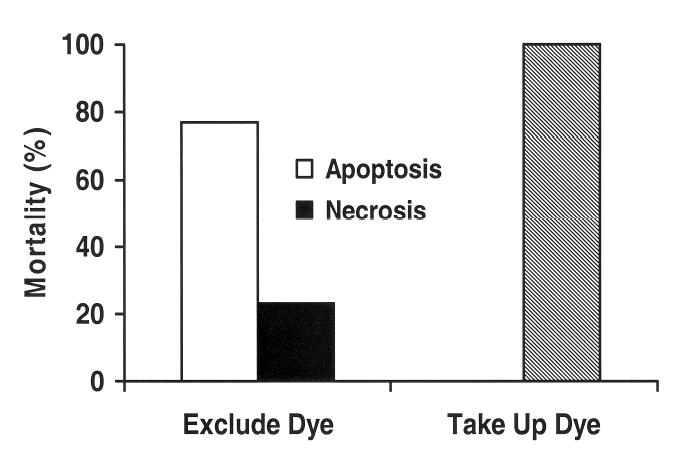
Percentage of transected cells that died (% mortality) within 12h PT for cells that took up dye and died (hatched bar, n =2, N =3) or excluded dye and died (n =17, N =3) via apoptosis (unfilled bars) or necrosis (filled bars) as indicated by the Annexin-V-affinity assay. (see, Figure 4 Legend and Results)
Caspase activation is involved in many important neurodegenerative pathways (Chan and Mattson, 1999). To examine whether apoptosis in transected B104 cells is mediated by caspases, cell survival was morphologically assessed following neurite transection in control medium containing or lacking the broad caspase inhibitor N-benzylcarbonyl-Val-Ala-Asp-fluromethylketone (Z VAD FMK), which is plasma membrane permeant (Chan and Mattson, 1999). Cells were incubated in control medium containing 200μM Z VAD FMK for 60min before transection (10–14 cells per dish within 2min) and then remained in the control medium with inhibitor. At 12h PT, a significantly (p<0.05, Kaplan-Meier survival analysis) larger percentage of transected cells maintained in control medium with Z VAD FMK survived (Figure 6A, filled circles, 73%, n=55, N =5) compared to transected cells maintained in control medium lacking caspase inhibitor (Figure 6A, unfilled squares, 43%, n=60, N =6). That is, broad caspase inhibition before transection increased cell survival by 30%.
Figure 6.
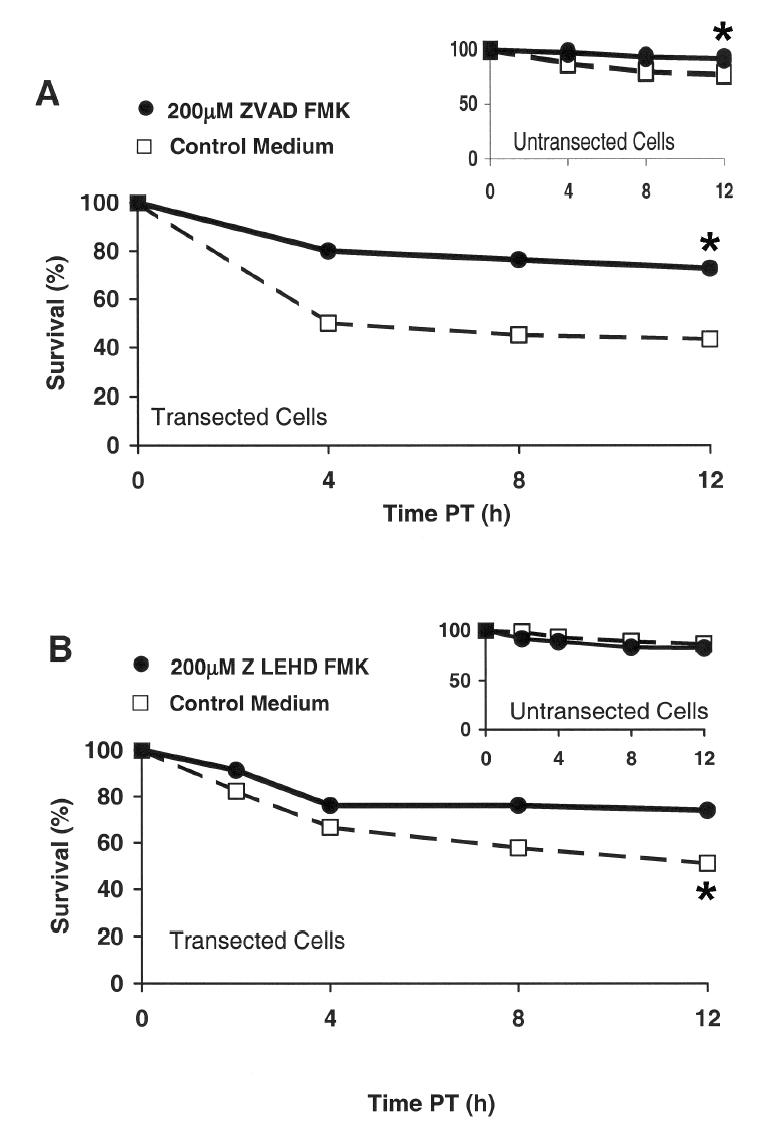
Effect of a caspase inhibitor (A, Z VAD fmk or B, Z LEHD fmk) on the % survival within 12h PT of transected and neighboring, untransected (inset, A, B) B104 cells.
B104 cells were preincubated for 60min, transected, and maintained for 12h PT in control medium, (A, B unfilled squares) or in control medium containing a broad caspase inhibitor (Z VAD fmk; A, filled circles), or a specific Caspase-9 inhibitor (Z LEHD fmk; B, filled circles). The viability of transected and neighboring, untransected cells (insets in A, B) was assessed morphologically at 2, 4, 8, and 12h PT.
To assess mitochondrial involvement in the initiation of apoptosis during the Critical Interval of the SCC transient, we used a membrane-permeant inhibitor (Z LEHD FMK [N-benzyloxycarbonyl-Val-Ala-Asp-fluoromehtylketone], Thornberry and Lazebnik, 1998) of Caspase-9, which is activated when cytochrome C is released from mitochondria (Liu et al., 1996)). Cells were preincubated in control medium containing 200μM Z LEHD FMK for 60min, and then transected (10–14 cells per dish within 2min) and maintained in control medium with the inhibitor. At 12h PT, a significantly (p<0.05, Kaplan-Meier survival analysis) larger percentage of transected cells maintained in control medium with Z LEHD FMK survived (Figure 6B, filled circles, 74%, n=46, N =4) compared to transected cells maintained in control medium lacking the Caspase-9 inhibitor (Figure 6B, unfilled squares, 51%, n= 45, N =4). That is, Caspase 9 inhibition before transection increased cell survival by 23%, which was less than the increased survival (30%) with broad caspase inhibition, suggesting that other apoptotic pathways in addition to the Caspase-9 pathway are involved.
Since unhealthy intact cells undergoing apoptosis before transection could be rescued by caspase inhibition, such untreated cells could produce an overestimate of the percentage of untreated cells that died due to injury. To assess this possibility, intact, untransected cells were incubated in control medium containing and lacking the broad caspase inhibitor (inset, Figure 6A). At 12h PT, the percentage of surviving intact, untransected cells was significantly (Kaplan-Meier survival, Log Rank, p<0.05) increased (15% at 12h PT, inset, Figure 6A, filled circle, 92%, n=73, N=5 vs unfilled square, 77%, n=78, N=6). In contrast, the same comparison in intact, untransected cells in control medium containing and lacking Caspase-9 inhibitor showed no significant (p>0.05) increase (inset, Figure 6B, filled circle, 86%, n=139, N=4 vs unfilled square, 82%, n=114, N=4). These data on intact, untransected cells suggest that the 43% of transected cells that survived in the broad caspase-inhibitor experiment (Figure 6A, unfilled square) included some cells that were undergoing apoptosis before injury, and, consequently, the survival due to injury alone was likely higher. The underestimation of the percentage of injured cells that survived transection (15% less than expected if all cells were healthy before transection) would virtually eliminate the difference between the increase in survival (rescue) produced by the broad vs Caspase-9 inhibitor, suggesting that transected cells that exclude dye and die from injury are likely to degenerate via the Caspase 9 pathway. A similar effect of BAPTA in rescuing cells from death, when applied before or during the Critical Interval (Figure 3A), and the fact that the Caspase-8 pathway is not known to be activated by an increase in SCC also suggest that degeneration following injury is predominantly via the Caspase 9 pathway.
DISCUSSION
Survival of transected cells is determined during the Critical Interval of the SCC transient
The main finding of this report is consistent with a previous qualitative study that indicated Fluo-3-loaded B104 cells with neurites also transected near the cell body experienced a substantially larger transient increase in SCC, which correlated with an increased mortality, compared to the much smaller SCC transient and mortality of cells with neurites transected far from the cell body (Yoo et al., 2004). However, while the previous study recognized that some cells survived neurite transection near the cell body despite a substantial SCC transient, the use of non ratiometric Ca2+ imaging precluded determination of differences among the SCC transients of transected cells that could account for injured cells that died vs. those that survived. Similarly, cell death correlated with [Ca2+]i following axotomy at various distances from the soma of cultured dorsal-root ganglion cells (George et al., 1995) and concussive injury to the rat spinal cord (i.e., injury induced by a weight drop) (Balentine et al., 1988). However, neither study examined SCC following injury. Thus, despite correlations between [Ca2+]i and cell death following traumatic injury to neuronal-type cells (Balentine et al., 1982; George et al., 1995; Shi et al., 2000; Yoo et al., 2004), a causal relationship between a change in [Ca2+]i after traumatic neuronal injury and cell death previously was inconclusive. In contrast, the quantitative data presented here show that the <SCC> transient in the Critical Interval from 1.5 to 12.5min PT is a key indicator of transected cell fate and is consistent with the interpretation that a significant increase of <SCC> in this interval initiates apoptosis. While the <SCC> during the late decay phase (i.e., Critical Interval) predicted cell death, the <SCC> at the peak and throughout the early decay phase of the <SCC> transient was not a reliable indicator of transected cell fate. These results are consistent with findings in embryonic spinal neurons following glutamate excitotoxicity, in which the plateau [Ca2+]i (at ~5min or later following glutamate application), but not the peak [Ca2+]i, corresponded to cell death (Tymianski et al., 1994). While glutamate exposure produced a [Ca2+]i transient that recovered to a steady-state plateau [Ca2+] i that remained greater than preinjury levels, neurite transection produced a SCC transient that continued to recover towards preinjury levels after neurite transection. Such a difference in recovery behavior could signify differences in Ca2+ regulation as a result of the different modes of injury and differences in B104 cells in each of the two groups (Fig. 2, excluded dye and survived, circles; excluded dye and died, triangles) at various times before and after neurite transection.
The main source of Ca2+ that produces the SCC transient appears to arise from Ca2+ entry through the injury site from the extracellular medium (Yoo et al., 2004) rather than through plasma membrane ion channels (George et al., 1995). The latter effect should produce a Ca2+- indicator fluorescence intensity distribution that increases with time but relatively independent of location. Instead, the observed spatial-temporal fluorescence pattern after injury showed a steep gradient moving from the injury site toward the soma, which is indicative of Ca2+ inflow. Some contribution to the SCC transient from Ca2+-induced Ca2+ release from internal stores cannot be excluded (Yoo et al, 2004). Furthermore, internal sources of Ca2+ released or sequestered by the interplay between endoplasmic reticulum and mitochondria may play a key role in survival, as indicated by the Caspase-9 involvement in transected cell degeneration.
In adult in vivo models, cell death was more likely following axotomy near the soma of adult rat spinal dorsal root ganglion (Hines and Tessler, 1989), cranial motoneurons (Snider and Thaneder, 1989), retinal ganglion cells (Villegas-Perez, 1993; Kermer et al., 1998; Chaudhary et al., 1999), corticospinal tract neurons (Giehl and Tetzlaff, 1996), and axons of Clarkes’ nucleus (Sanner et al., 1993). Similar to the outcome of B104 cells with transected neurites, a percentage (20–50%) of axotomized neurons in vivo survived, but the increased mortality of neurons axotomized near the soma was attributed to the loss of neurotrophic factors delivered by untransected collaterals (of neighboring cells) more proximal to the soma (Berkelaar et al., 1994, Villegas-Perez et al., 1993). These previous in vivo studies did not examine the association between SCC and cell death. In addition, those in vivo studies examined myelinated tissue preparations that were axotomized in a heterogeneous network of neuronal and non-neuronal type cells that had neurotrophic factor support. In contrast, the present experiments involved transected neurites of single, isolated cells within a homogenous population that did not have exogenous trophic support. Thus, although this study does not rule out a role for trophic factors, the data indicate that the SCC transient following neurite transection initiates an intrinsic process of cell death independent of trophic factor withdrawal.
Compared to axotomized neurons in vivo, we found that transected B104 cells died much more rapidly. For example, most axotomized retinal ganglion cells (Villegas-Perez, 1993; Kermer et al., 1998; Chaudhary et al., 1999) survive until 5 days post-axotomy. Many of these cells then died from 5 days to 2 weeks post-axotomy. In contrast, B104 cells with transected neurites died within 12h PT. Trophic support of in vivo models may have prolonged survival through various actions, such as the activation of novel pathways of survival, maintenance of processes required for survival, or inhibition of cell death pathways. Often the roles of trophic factors in vivo are difficult to ascertain because of variable environmental factors, such as multiple cell types and multiple trophic factors. Although it is uncertain whether B104 cells can be sustained in the presence of trophic support for periods equivalent to neurons in vivo, because of their homogeneity and independence of trophic support, B104 cells could be useful in future studies of trophic factors (e.g. BDNF, CNTF, bFGF) (Ingoglia and Murray, 2001) on cell survival.
These data also showed that the survival of transected cells can be increased by loading cells with BAPTA at 30s PT to enhance intracellular Ca2+-buffering capacity before the Critical Interval. However, loading cells with BAPTA at 20min PT to enhance the intracellular Ca2+-buffering capacity after the Critical Interval did not increase the survival of transected B104 cells. These results are consistent with the interpretation that the SCC transient initiates Ca2+-dependent cell death within the fate-determining Critical Interval. Enhancing intracellular Ca2+-buffering capacity before neurite transection of B104 cells (Yoo et al., 2004) and before excitotoxic glutamate exposure in cultured mouse spinal neurons (Tymianski et al., 1994) was shown to enhance cell survival. However, these studies did not examine the ability of enhancing intracellular Ca2+-buffering capacity after injury to increase cell survival. Our data indicate that enhancement of intracellular Ca2+-buffering capacity before but not after the Critical Interval of the SCC can prevent Ca2+-dependent cell death.
Cell death following neurite transection near a B104 soma occurs by apoptosis
Most cells in these experiments died following neurite transection near a B104 cell body and degenerated in an organized manner characteristic of apoptosis. Within 12h PT, most transected B104 cells that exclude dye and die displayed translocation of phosphatidylserine from the inner to outer leaflet of the plasmalemma, as indicated by the Annexin-V-affinity assay, followed by rounding and shrinking of the soma. Only a small number of transected cells showed early Sytox green labeling and presumably die by necrosis.
The death of most transected B104 cells is mediated by caspases. Preloading cells before injury with a broad caspase inhibitor (Z VAD FMK) and incubating the cells in the inhibitor during and after neurite transection increased significantly the percentage of transected cells that survived at 12h PT. This result further supports the conclusion that most transected cells died through apoptosis, since apoptosis is a degenerative process that is most typically mediated by the organized activation of caspases, a group of cysteine proteases. As commonly reported, caspase-mediated apoptosis begins with the sequential activation of Caspase-9 and/or Caspase-8 (initiator caspases) leading to activation of effector caspases (e.g., 3, 6, 7) that execute apoptosis (Chan et al., 1999; Yakovlov et al., 1997; Franz et al., 2000). Caspase-8 is activated by the association or dissociation of ligands to “death” receptors located on the plasmalemma (Chan et al., 1999). Caspase-9 is typically activated through a mitochondrial-dependent pathway following mitochondrial cytotoxicity (Chan et al., 1999). Since selective inhibition of Caspase-9 also increased survival of transected B104 cells, our results suggest that the SCC transient in some transected cells may initiate caspase-mediated apoptosis via mitochondrial-dependent activation of Caspase-9. Caspase-9 activation was also previously implicated following axotomy of rat retinal ganglion in vivo, and Caspase-9 inhibition increased the percentage of retinal ganglion that survived axotomy (Kermer et al., 2000).
These results from B104 cells are consistent with a growing number of studies indicating that apoptosis mediates cell death following a variety of injuries. Apoptosis is induced after in vivo axotomy of retinal ganglion, spinal cord, and cortical neurons (Kermer et al., 1998; Al-Abdulla et al., 1998; Chaudary et al., 1999; Martin, et al., 1999) or percussive trauma to the cortex or spinal cord (Yakovlev et al., 1997). Neurons also die via apoptosis following growth factor withdrawal (Cardone et al., 1998), glutamate excitotoxicity (Portera-Cailliau et al., 1997), cell exposure to reactive oxygen species (Volbracht et al., 2001; Leist et al., 1997), ischemia (Namura et al., 1998; Velier et al., 1999), and normal development (O’Connor et al., 1974). Other previous reports indicate that caspases are activated following axotomy of CNS retinal ganglion cells (Kermer et al., 1998; Chaudary et al., 1999) or axotomy of the dorsal lateral geniculate nucleus (Al-Abdulla et al., 1998). In addition, broad caspase inhibition increased the percentage of cells that survived in vivo following traumatic (percussive) brain injury in rat (Yakovlev et al., 1997; Franz et al., 2000) and spinal-cord contusions in rat (Chan et al., 2001). However, caspase activation in axotomized retinal ganglion cells has been attributed to the loss of neurotrophic factors from the collaterals of neighboring, untransected cells and the effects of a rise in SCC has not been previously examined.
The enhanced survival of transected B104 cells due to caspase inhibition in these experiments suggests that transected B104 cells may be rescued from death by intervention downstream from the Critical Interval of the SCC transient. Presumably, Caspase-9 is activated after mitochondial-initiated apoptosis during the Critical Interval of the SCC transient in most transected B104 cells that excluded dye and die, and activation of other caspases in the cascade occurs much later. Such long-term post injury caspase activation is consistent with reports of other injuries that transiently elevated [Ca2+]i, such as glutamate excitotoxicity (Ko et al., 1998; Namura et al., 1998; Velier et al., 1999), ionomycin addition (Ko et al., 1998), or Thapsigargin addition in which caspases were activated several hours to even days after the injury and the transient elevation in [Ca2+]i, The initiation of apoptosis by Ca2+ during the Critical Interval and the caspase execution of apoptosis long after the Critical Interval would explain the ineffectiveness in preventing cell death of enhancing intracellular Ca2+-buffering capacity after the Critical Interval. However, the prevention of cell death subsequent to neurite transection and after the Critical Interval may still be possible by inhibition of caspases downstream. Future studies in B104 cells of therapeutic interest should consider this possibility by systematically examining the time of activation of various caspases in the cascade to determine when (at which points and PT times) intervention can prevent cell death. Furthermore, an explanation of what causes an elevated SCC during the Critical Interval in some transected cells but not in others is also likely to be significant.
Acknowledgments
We thank Dr. Leoncio Vergara for the use of the Optical Imaging Laboratory at UTMB. This work was supported by NIH grant NS31256.
References
- Al- Abdulla NA, Portera-Cailliau C, Martin LJ. Occipital cortex ablation in adult rat causes retrograde neuronal death in the lateral geniculate nucleus that resembles apoptosis. Neuroscience. 1998;86(1):191–209. doi: 10.1016/s0306-4522(98)00014-1. [DOI] [PubMed] [Google Scholar]
- Balentine JD, Dean DL., Jr Calcium-induced spongiform and necrotizing myelopathy. Laboratory Investigation. 1982;47(3):286–295. [PubMed] [Google Scholar]
- Balentine JD. Spinal cord trauma: in search of the meaning of granular axoplasm and vesicular myelin. Journal of Neuropathology and Experimental Neurology. 1988;47(2):77–92. doi: 10.1097/00005072-198803000-00001. [DOI] [PubMed] [Google Scholar]
- Berkelaar M, Clarke DB, Wang YC, Bray GM, Aguayo AJ. Axotomy results in delayed death and apoptosis of retinal ganglion cells in adult rats. Journal of Neuroscience. 1994;14(7):4368–74. doi: 10.1523/JNEUROSCI.14-07-04368.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottenstein JE, Sato GH. Growth of a rat neuroblastoma cell line in serum-free supplemented medium. Proceedings of the National Academy of Science. 1979;76(1):514–517. doi: 10.1073/pnas.76.1.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone MH, Roy N, Stennicke HR, Salvese GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- Chan SL, Mattson MP. Caspase and calpain substrates: Roles in synaptic plasticity and cell death. Journal of Neuroscience Research. 1999;58:167–190. [PubMed] [Google Scholar]
- Chan YM, Wu W, Yip HK, So KF, Oppenheim RW. Caspase inhibitors promote the survival of avulsed spinal motoneurons in neonatal rats. Neuroreport. 2001;12(3):541–545. doi: 10.1097/00001756-200103050-00022. [DOI] [PubMed] [Google Scholar]
- Chaudhary P, Ahmed F, Quebada P, Sharma SC. Caspase inhibitors block the retinal ganglion cell death following optic nerve transection. Molecular Brain Research. 1999;67(1):36–45. doi: 10.1016/s0169-328x(99)00032-7. [DOI] [PubMed] [Google Scholar]
- Cheung JY Bonventre JV, Malis CD Leaf A. Calcium and ischemic injury. New England Journal of Medicine. 1986;314(26):1670–1676. doi: 10.1056/NEJM198606263142604. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Hinson AW, Black JA, Waxman SG. Sodium channel mRNA in the B104 neuroblastoma cell line. FEBS Letters. 1996;384:78–82. doi: 10.1016/0014-5793(96)00273-6. [DOI] [PubMed] [Google Scholar]
- Eddleman CS, Ballinger ML, Godell CM, Smyers MS, Fishman HM, Bittner GD. Repair of plasmalemmal lesions by vesicles. Proceedings of the National Academy of Science (USA) 1997;94:4745–4750. doi: 10.1073/pnas.94.9.4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engeland M, Nieland LJW, Ramaekers FCS, Schutte B, Reutelinsperger CPM. Annexin V-affinity assay: A review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry. 1998;31:1–9. doi: 10.1002/(sici)1097-0320(19980101)31:1<1::aid-cyto1>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Fishman HM, Bittner GD. Vesicle-mediated restoration of a plasmalemmal barrier after axonal injury. NIPS. 2003;18:115–118. doi: 10.1152/nips.01429.2002. [DOI] [PubMed] [Google Scholar]
- Franz RB, Srinivasan A, Hayes RL, Pike BR, Newcomb JK, Zhao Z, Schmutzhard E, Poewe W, Kampfl A. Temporal profile of activated caspase-3 following experimental traumatic brain injury. Journal of Neurochemistry. 2000;75:1264–1273. doi: 10.1046/j.1471-4159.2000.0751264.x. [DOI] [PubMed] [Google Scholar]
- Giehl KM, Tetzlaff W. BDNF and NT-3, but not NGF, prevent axotomy-induced death of rat corticospinal neurons in vivo. European Journal of Neuroscience. 1996;8(6):1167–1175. doi: 10.1111/j.1460-9568.1996.tb01284.x. [DOI] [PubMed] [Google Scholar]
- George EB, Glass JD, Griffin JW. Axotomy-induced axonal degeneration is mediated by calcium influx through ion-specific channels. Journal of Neuroscience. 1995;15(10):6445–6452. doi: 10.1523/JNEUROSCI.15-10-06445.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ Indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gu XQ, Waxman SG. Action potential-like responses in B104 cells with low Na+ channel densities. Brain Research. 1996;735:50–58. doi: 10.1016/0006-8993(96)00604-x. [DOI] [PubMed] [Google Scholar]
- Hines BT, Tessler A. Death of some dorsal root ganglion neurons and plasticity of others following sciatic nerve section in adult and neonatal rats. Journal of Comparative Neurology. 1989;284(2):215–230. doi: 10.1002/cne.902840206. [DOI] [PubMed] [Google Scholar]
- Hyrc K, Handran SD, Rothman SM, Goldberg MP. Ionized intracellular calcium concentration predicts excitotoxic neuronal death: Observations with low-affinity fluorescent calcium indicators. Journal of Neuroscience. 1997;17(17):6669–6677. doi: 10.1523/JNEUROSCI.17-17-06669.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingoglia, N.A. and Marion, M. (2001). Axonal Regeneration in the Central Nervous System. Marcel Dekker Inc., New York.
- Kermer P, Ankerhold R, Klocker N, Krajewski S, Reed JC, Bahr M. Caspase-9: involvement in secondary death of axotomized rat retinal ganglion cells in vivo. Molecular Brain Research. 2000;85(1–2):144–150. doi: 10.1016/s0169-328x(00)00256-4. [DOI] [PubMed] [Google Scholar]
- Kermer P, Klocker N, Labes M, Bahr M. Inhibition of CPP32-like proteases rescues axotomized retinal ganglion cells from secondary cell death in vivo. Journal of Neuroscience. 1998;18(12):4656–4662. doi: 10.1523/JNEUROSCI.18-12-04656.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko HW, Park KY, Kim H, Han PL, Kim YU, Gwag BJ, Choi EJ. Ca2+-mediated activation of c-Jun N-terminal kinase and nuclear factor kappa B by NMDA in cortical cell cultures. Journal of Neurochemistry. 1998;71(4):1390–1395. doi: 10.1046/j.1471-4159.1998.71041390.x. [DOI] [PubMed] [Google Scholar]
- Leist M, Volbracht C, Kuhnle S, Fava E, Ferrando-May E, Nicotera P. Caspase-mediated apoptosis in neuronal excitotoxicity triggered by nitric oxide. Molecular Medicine. 1997;3(11):750–764. [PMC free article] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86(1):147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Lucas JH, Gross DG, Emery DG, Gardner CR. Neuronal survival or death after dendritic transection close to the perikariyon: Correlation with electrophysiologic, morphologic, and ultrastructural changes. CNS Trauma. 1985;2:231–255. doi: 10.1089/cns.1985.2.231. [DOI] [PubMed] [Google Scholar]
- Luo J, Miller MW. Basic fibroblast growth factor- and platelet-derived growth factor-mediated cell proliferation in B104 neuroblastoma cells: effect of ethanol on cell cycle kinetics. Brain Research. 1997;770:139–150. doi: 10.1016/s0006-8993(97)00762-2. [DOI] [PubMed] [Google Scholar]
- Luo J, Miller MW. Transforming growth factor beta1-regulated cell proliferation and expression of neural cell adhesion molecule in B104 neuroblastoma cells: differential effects of ethanol. Journal of Neurochemistry. 1999;72:2286–2293. doi: 10.1046/j.1471-4159.1999.0722286.x. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Kaiser A, Price AC. Motor neuron degeneration after sciatic nerve avulsion in adult rat evolves with oxidative stress and is apoptosis. Journal of Neurobiology. 1999;40(2):185–201. [PubMed] [Google Scholar]
- Montgomery, D.C. (2001). Design and Analysis of Experiments, 5th Ed. John Wiley and Sons. Danvers, MA.
- Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. Journal of Neuroscience. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’ Connor TM, Wyttenbach CR. Cell death in the embryonic chick spinal cord. Journal Cellular Biology. 1974;60:448–459. doi: 10.1083/jcb.60.2.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portera-Cailliau C, Price DL, Martin LJ. Excitotoxic neuronal death in the immature brain is an apoptosis-necrosis morphological continuum. Journal of Comparative Neurology. 1997;378:70–87. [PubMed] [Google Scholar]
- Sanner CA, Murray M, Goldberger ME. Removal of dorsal root afferents prevents retrograde death of axotomized Clarke's nucleus neurons in the cat. Experimental Neurology. 1993;123(1):81–90. doi: 10.1006/exnr.1993.1142. [DOI] [PubMed] [Google Scholar]
- Schubert D, Carlisle W, Look C. Putative neurotransmitters in clonal cell lines. Nature. 1975;254:341–343. doi: 10.1038/254341a0. [DOI] [PubMed] [Google Scholar]
- Schubert D, Heinemann S, Carlisle W, Tarikas H, Kimes B, Patrick J, Steinbach JH, Culp W, Brandt BL. Clonal cell lines from the rat central nervous system. Nature. 1974;249:224–227. doi: 10.1038/249224a0. [DOI] [PubMed] [Google Scholar]
- Shi R, Asano T, Vining NC, Blight AR. Control of membrane sealing in injured mammalian spinal cord axons. Journal of Neurophysiology. 2000;84(4):1763–1769. doi: 10.1152/jn.2000.84.4.1763. [DOI] [PubMed] [Google Scholar]
- Snider WD, Thanedar S. Target dependence of hypoglossal motor neurons during development in maturity. Journal of Comparative Neurology. 1989;279(3):489–498. doi: 10.1002/cne.902790312. [DOI] [PubMed] [Google Scholar]
- Stys PK, Ashby P. An automated technique for measuring the recovery cycle of human nerves. Muscle Nerve. 1990;13(8):750–758. doi: 10.1002/mus.880130814. [DOI] [PubMed] [Google Scholar]
- Tan ZY, Chen J, Shun HY, Feng XH, Ji YH. Modulation of Bmk AS, a scorpion neurotoxic polypeptide, on voltage-gated Na+ channels in B104 neuronal cell line. Neuroscience Letters. 2003;340:123–126. doi: 10.1016/s0304-3940(03)00094-6. [DOI] [PubMed] [Google Scholar]
- Thornberry N, Lazebnik Y. Caspases: Enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- Tsien RY, Rink TJ, Poenie M. Measurement of cytosolic free Ca2+ in individual small cells using fluorescence microscopy with dual excitation wavelengths. Cell Calcium. 1985;6(1–2):145–157. doi: 10.1016/0143-4160(85)90041-7. [DOI] [PubMed] [Google Scholar]
- Tymianski M, Charlton MP, Carlen PL, Tator CH. Properties of neuroprotective cell-permeant Ca2+ chelators: effects on [Ca2+]i and glutamate neurotoxicity in vitro. Journal of Neurophysiology. 1994;72(4):1973–1992. doi: 10.1152/jn.1994.72.4.1973. [DOI] [PubMed] [Google Scholar]
- Tyndale RF, Hales TG, Olsen RW, Tobin AJ. Distinctive patterns of GABA receptor mRNAs in 13 cell lines. Journal of Neuroscience. 1994;14:5417–5428. doi: 10.1523/JNEUROSCI.14-09-05417.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velier JJ, Ellison JA, Kikly KK, Spera PA, Barone FC, Feuerstein GZ. Caspase-8 and caspase-3 are expressed in different populations of cortical neurons undergoing delayed cell death after focal stroke in the rat. Journal of Neuroscience. 1999;19:5932–5941. doi: 10.1523/JNEUROSCI.19-14-05932.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villegas-Perez MP, Vidal-Sanz M, Rasminsky M, Bray GM, Aguayo AJ. Rapid and protracted phases of retinal ganglion cell loss follow axotomy in the optic nerve of adult rats. Journal of Neurobiology. 1993;24(1):23–36. doi: 10.1002/neu.480240103. [DOI] [PubMed] [Google Scholar]
- Vito P, Lacana E, D'Adamio L. Interfering with apoptosis: Ca2+ -binding protein ALG-2 and Alzheimer's disease gene ALG-3. Science. 1996;26(2715248):521–525. doi: 10.1126/science.271.5248.521. [DOI] [PubMed] [Google Scholar]
- Volbracht C, Fava E, Leist M, Nicotera P. Calpain inhibitors prevent nitric oxide-triggered excitotoxic apoptosis. Neuroreport. 2001;12(17):3645–3648. doi: 10.1097/00001756-200112040-00008. [DOI] [PubMed] [Google Scholar]
- Xie XY, Barrett JN. Membrane resealing in cultured rat septal neurons after neurite transection: evidence for enhancement by Ca2+-triggered protease activity and cytoskeletal disassembly. Journal of Neuroscience. 1991;11:3257–3267. doi: 10.1523/JNEUROSCI.11-10-03257.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakvolev AG, Knoblacj SM, Fan L, Fox GB, Goodnight R, Faden AI. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. Journal of Neuroscience. 1997;17(19):7415–7424. doi: 10.1523/JNEUROSCI.17-19-07415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S, Bottenstein JE, Bittner GD, Fishman HM. Survival of mammalian B104 cells following neurite transection at different locations depends on somal Ca2+ concentration. Journal of Neurobiology. 2004;60:137–153. doi: 10.1002/neu.20005. [DOI] [PubMed] [Google Scholar]