

Abstract
Over the past decade, genetically encoded fluorescent proteins have become widely used as noninvasive markers in living cells. The development of fluorescent proteins, coupled with advances in digital imaging, has led to the rapid evolution of live-cell imaging methods. These approaches are being applied to address biological questions of the recruitment, co-localization, and interactions of specific proteins within particular subcellular compartments. In the wake of this rapid progress, however, come important issues associated with the acquisition and analysis of ever larger and more complex digital imaging data sets. Using protein localization in the mammalian cell nucleus as an example, we will review some recent developments in the application of quantitative imaging to analyze subcellular distribution and co-localization of proteins in populations of living cells. In this report, we review the principles of acquiring fluorescence resonance energy transfer (FRET) microscopy measurements to define the spatial relationships between proteins. We then discuss how fluorescence lifetime imaging microscopy (FLIM) provides a method that is independent of intensity-based measurements to detect localized protein interactions with spatial resolution. Finally, we consider potential problems associated with the expression of proteins fused to fluorescent proteins for FRET-based measurements from living cells.
INTRODUCTION
Genetically encoded fluorescent proteins have transformed studies in cell biology by allowing the behavior of proteins to be tracked in their natural environment within the living cell. Over the past decade, fluorescent proteins have become widely used as noninvasive markers in living cells because their fluorescence does not require the addition of cofactors, and they are very stable and well tolerated by most cell types. The successful integration of these proteins into living systems is illustrated by the many examples of healthy transgenic mice that carry the fluorescent protein markers (1–3). The extensive mutagenesis of the jellyfish green fluorescent protein (GFP), combined with the cloning of new fluorescent protein variants from corals, has yielded fluorescent proteins that emit light from the blue to the red range of the visible spectrum (4–7). The full spectrum of fluorescent protein color variants is being exploited in multicolor fluorescence microscopy experiments to track the distribution of different proteins in the same living cells, allowing for the direct visualization of subcellular protein recruitment, co-localization, and transcription (8–13). Through the combination of fluorescent proteins and advanced digital imaging technologies, it is now possible to visualize diverse biological processes inside the living cell, providing an important complement to the biochemical methods that are traditionally used in this analysis (14–17).
With these advances in live-cell imaging, however, come increasingly complex digital imaging data sets that must be accurately analyzed. Individual digital images may contain more than one million data points, and multidimensional imaging experiments may produce hundreds of images (18–20). In addition, there is often substantial cell-to-cell heterogeneity in the distribution of proteins of interest, making any analysis based on “representative images” difficult, if not impossible. Using protein localization in the mammalian cell nucleus as an example, we will review some recent developments in the application of quantitative imaging to analyze subcellular distribution and co-localization of proteins in populations of living cells. We will discuss the use of computer vision algorithms for the extraction of information from large digital imaging data sets, and bioinformatics tools to manage these data sets.
These quantitative imaging approaches are being used to monitor the co-localization of proteins within different subcellular compartments, providing critical information about cell physiology and pathophysiology. The problem is that the detection of protein co-localization alone cannot distinguish proteins with overlapping distribution from those proteins that are interacting in significant ways. Importantly, the spectral properties of fluorescent proteins also allow them to be used as probes in fluorescence resonance energy transfer (FRET) microscopy, which can provide information about the spatial relationships of proteins on the scale of angstroms (21–24). Generally, FRET microscopy methods are classified into intensity-based and fluorescence decay kinetics-based approaches (14–17). We will review some recent applications of intensity-based FRET microscopy techniques to define the spatial relationships between proteins in living cells and then discuss how measurements based on fluorescence decay kinetics can confirm and extend these observations. Finally, we will discuss potential problems associated with the expression of proteins fused to fluorescent proteins for FRET-based measurements from living cells.
IMAGING PROTEIN BEHAVIOR IN THE LIVING CELL NUCLEUS
Here we use protein localization in the mammalian cell nucleus as an example to illustrate some recent developments in digital imaging. The mammalian cell nucleus contains a variety of subnuclear domains where proteins with specialized functions are localized. These domains range from spherical bodies to diffuse and irregular speckles and have been visualized by both indirect immunofluorescence microscopy and by labeling of the constituent proteins with fluorescent proteins (25–27). For example, subunits of the mRNA splicing machinery localize in domains called nuclear speckles (28), p80 coilin is assembled in Cajal bodies (29), and the promyelocytic leukemia protein (PML) is localized in PML nuclear bodies (30). The partitioning of these different subcompartments in the nucleus without intervening membranes indicates that they likely formed by a process of self-assembly (27,31). The mechanisms that regulate and maintain these higher order protein assemblies in intact cells must be defined to understand the function of these subnuclear domains.
This objective is being achieved through a variety of different imaging techniques to characterize the behavior of proteins within these subcompartments in the living cells nucleus. For example, various classes of nuclear bodies have been visualized by the expression of fluorescent protein-labeled component proteins, allowing their positioning and movement to be observed over time in living cells. This approach was used to demonstrate the energy-dependent movement of both PML and Cajal bodies within the nuclei of living cells and showed that Cajal bodies could merge and bud from one another (10,30). By imaging proteins labeled with different color fluorescent proteins, Platani and colleagues (32) showed that two different protein components, coilin and fibrillarin, were co-localized in the Cajal bodies. Importantly, this co-localization approach also revealed that the protein composition of the Cajal bodies changed over time (32). Thus, measurements of co-localization by multicolor imaging (Figure 1A) can supply important information about the molecular composition of these subnuclear domains.
Figure 1. Different quantitative imaging approaches.
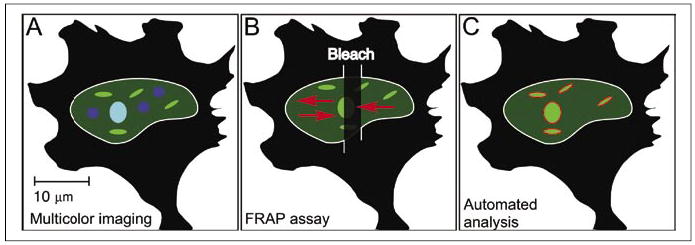
(A) Multicolor imaging to reveal protein co-localization can supply important information about the molecular composition of subnuclear domains. (B) The FRAP technique uses photobleaching of the labeled proteins within a ROI to measure the kinetics of the redistribution of the population of fluorescent-labeled proteins over space and time. (C) The analysis of subcellular structures using computer algorithms to automate the detection and measurement of subcellular features in large sets of high-resolution images. YFP, yellow fluorescent protein; NCoR, nuclear receptor corepressor protein; FRAP, fluorescence recovery after photobleaching; ROI, region of interest.
These observations indicated that these nuclear bodies are active structures, and this was confirmed by measurements of the exchange of proteins within these subnuclear compartments using the technique of fluorescence recovery after photobleaching (FRAP). The FRAP technique uses photobleaching of the labeled proteins in a region of interest (ROI) to measure the kinetics of the redistribution of the population of fluorescent-labeled proteins over space and time (Figure 1B). The influx of labeled proteins from outside the bleached area is monitored, and plotting the recovery of fluorescence in the ROI provides an estimate of the mobile fraction of the labeled protein population (Figure 1B). It is important to note that proteins interact with varying affinity with other molecules in the cell, so diffusion constants determined from FRAP experiments must be carefully interpreted (33). The FRAP technique was recently used to monitor the flux of fluorescent protein-labeled component proteins through Cajal bodies. Studies by Sleeman et al. (34) showed that p80 coilin was rapidly exchanged from Cajal bodies. Dundr et al. (35) then analyzed many different proteins known to associate with Cajal bodies using a modified FRAP approach, and these studies revealed several distinct kinetic classes of protein exchange from these nuclear bodies. Together, these results demonstrated that these nuclear bodies are very dynamic structures.
A third quantitative imaging approach takes the analysis of subcellular structures in single cells to the cell population level. This approach uses computer algorithms to automate the detection and measurement of subcellular features in large sets of high-resolution images (Figure 1C). These automated approaches are designed to automatically segment images into ROI, and then apply the same set of rules to acquire measurements of those ROI in each image within the data set 20). These automated approaches are important because they allow consistent and rigorous analysis of subcellular features in each of the high-resolution images in the data set. We discuss in more detail the application of computer-based approaches to the analysis of nuclear protein distribution in the next section.
THE SELECTION OF TRANSFECTED CELLS FOR IMAGE ANALYSIS
Cell biologists are faced with a dilemma when using live-cell imaging to define the mechanisms that regulate the subcellular distribution of proteins. The problem is that the inherent heterogeneity in subcellular distribution prevents one from using the protein that is under investigation as the criterion for the selection of the cells to be imaged and analyzed. For example, the transcriptional corepressor proteins, including nuclear receptor corepressor protein (NCoR) and the silencing mediator of retinoid and thyroid hormone receptors, are organized with their histone deacetylase partners in discrete nuclear bodies called matrix-associated deacetylase bodies (36,37). Images of different cells transiently transfected with plasmids encoding NCoR labeled with yellow fluorescent protein (YFP) illustrates the heterogeneity in the organization of these subnuclear bodies, ranging from a diffuse distribution to an arrangement of highly concentrated focal bodies (Figure 2). This may partly reflect observations made by mRNA expression profiles that revealed extreme variability in transcriptional activity between individual cells in clonal populations; results that argue against the widely held notion of the “average” cell (38). Additionally, the heterogeneity in subnuclear organization may reflect cells that are in different phases of the cell cycle (25) and can be compounded further by differences in protein expression levels within the transfected cell population (39).
Figure 2. Heterogeneity of NCoR subnuclear organization in a transfected cell population.
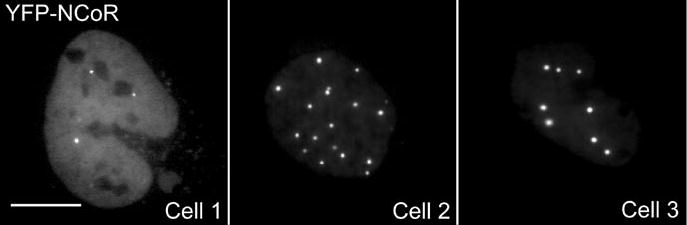
Digital images of three different cells expressing the nuclear localized YFP-NCoR were obtained to demonstrate the variability in the subnuclear organization of NCoR. Scale bar indicates 10 μm. YFP, yellow fluorescent protein; NCoR, nuclear receptor corepressor protein; FRAP, fluorescence recovery after photobleaching.
The heterogeneity that results from differences in protein expression levels in the transiently transfected cells can be reduced by using cloned cell lines that stably express the fluorescent protein-fusion proteins. However, the generation of stable cell lines is time-consuming and may not be conducive to screening approaches to characterize protein function that may require many different combinations of the fluorescent protein-labeled proteins. Unfortunately, using transiently transfected cells that express heterogeneous levels of labeled protein(s), as illustrated in Figure 2, creates a problem for the image analysis because no single image will adequately represent the population of cells expressing the labeled protein. This is complicated further when protein organization is influenced by changes in the experimental conditions, such as treatments that affect cell-signaling pathways. Even in situations where the investigator is blinded to treatment protocols, cell selection is subjected to unintentional user bias during the image acquisition, and the sampled cells may not accurately represent the population.
One solution is to use staining with fluorescent dyes to identify the cells for automated image analysis (40). However, this approach is not an effective way to choose cells that have taken up the plasmids for protein expression because transient transfection can be an inefficient process. We observed that cells co-transfected with plasmids encoding two or more of the fluorescent protein color variants predictably express each of the different color proteins. Taking advantage of this observation, we developed an approach for the unbiased selection of transfected cells using the rapid maturing, monomeric variant of Discosoma sp. red fluorescent protein (mRFP; Reference 41). Because mRFP is among the most redshifted of the fluorescent proteins yet described, it is very suitable for multi-spectral imaging applications. The approach is to co-transfect cells with expression plasmids encoding the protein(s) of interest fused to fluorescent proteins along with a plasmid encoding mRFP. This allows for the selection of cells for imaging based on the expression of the diffuse cellular mRFP, with no prior knowledge of the subnuclear organization of the co-expressed fluorescent proteinfusion proteins. The examination of hundreds of randomly selected mRFP-expressing cells revealed that over 95% also contained a detectable fluorescence signal from the co-expressed fluorescent protein-fusion proteins. The detection of mRFP provides an unbiased selection of cells expressing the labeled protein (39,41a).
QUANTITATIVE ANALYSIS OF PROTEIN ORGANIZATION IN CELLS
Following the unbiased collection of cell images, it is necessary to use a consistent and rigorous method to analyze the subcellular features in each of the high-resolution images in the data set. Here again, user bias in the selection and measurement of specific subcellular features will introduce inaccuracies into the morphometric data, and this problem becomes more pronounced when multiple features within sets of digital images are subjectively measured (42). This problem is being addressed through the development of automated computer algorithms, called computer vision approaches, which are designed for the segmentation of images into ROI using concisely defined rules (20).
These computer vision algorithms have been used in a variety of image analysis applications to extract information from large data sets. Although they were not originally developed for use in cell biology, many of these computational techniques have been adapted for the analysis of fluorescent protein-fusion proteins concentrated in subcellular compartments. For instance, edge detection techniques and adaptive threshold calculations can automatically separate bright ROI from surrounding regions (43,44). These methods are less effective for fluorescence microscopy images containing low signal-to-noise ratios, and additional image smoothing techniques can be used to attenuate this problem (45). Automated systems have also been designed to recognize and select ROI with more complex combinations of subcellular features (46–48). Despite the power of these automated segmentation and measurement techniques, there are currently only a few examples where the approach has been used to address specific problems in cell biology. For instance, a combination of smoothing and edge detection techniques was used to identify and measure RNA splicing factor compartments, and an adaptive thresholding strategy was employed to select Cajal bodies for quantitative analysis (10,49,50).
We used a similar approach to analyze the distribution of the YFP-NCoR in the nuclei of over 100 cells selected from the transfected population using mRFP (Figure 3A). Within the cell population, individual cells expressed different relative levels of both mRFP and YFP-NCoR. We observed only a weak correlation between the expression of the mRFP selection marker and the YFP-labeled NCoR in the co-transfected cells (Figure 3B). This weak correlation allowed the selection of cells that expressed a wide range of YFP-NCoR, including cells that would not have been detected by eye through the fluorescence microscope. Therefore, the population of cells used for the analysis contained cells expressing very low levels of the YFP that would not have been captured using conventional qualitative imaging approaches. The population analysis of YFP-NCoR subnuclear distribution shown in Figure 3A precisely quantifies the significant relationship between fusion protein expression level and higher order protein organization. Similar concentration-dependent behavior was observed for several transcriptional corepressor proteins, as well as the nuclear receptor coactivator glucocorticoid receptor interacting protein (GRIP; Reference 51), indicating that fusion protein expression levels must be considered when comparing protein organization in different experimental cell populations. The application of these methods effectively reduces many millions of data points into a few thousand morphometric measurements. However, even these simplified morphometric data sets contain many interrelated parameters, which require statistical modeling to define the relationships between parameters. Fortunately, bioinformatics tools are being developed to manage the large amount of data generated by automated image analysis methods (23). The integration of automated analysis routines and customized database software will allow high-resolution imaging techniques to rigorously quantify the behavior of large cell populations.
Figure 3. Cell population studies using the computer-assisted image analysis protocol.
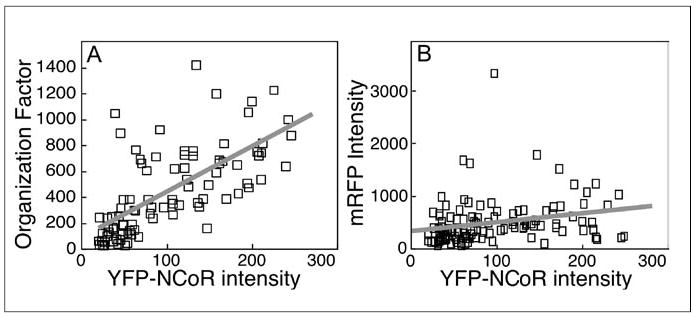
Images of over 100 cells were acquired and analyzed using an automated protocol. In the plots, each square represents data from a single cell, and a best-fit line is shown in gray. (A) The relationship for the cell population between YFP-NCoR subnuclear organization and relative YFP-NCoR expression level. (B) The relationship for the cell population between YFP-NCoR and mRFP expression levels. The R2 value (coefficient of determination) and the ANOVA F significance value estimate the correlation between the parameters, as calculated by linear regression analysis. (Adapted with permission from Reference 28). YFP, yellow fluorescent protein; NCoR, nuclear receptor corepressor protein; mRFP, monomeric variant of Discosoma sp. red fluorescent protein; ANOVA, analysis of variance.
PROTEIN RECRUITMENT AND CO-LOCALIZATION
An important application of these quantitative imaging methods is the analysis of the functional associations between specific protein partners in living cells, complementing observations made using in vitro techniques. For instance, the use of biochemical approaches to characterize multi-protein complexes that function to modify chromatin structure and control the gene expression has been invaluable in describing mechanisms underlying gene regulation (51–53). What is missed by the in vitro analysis, however, is the role that the organized microenvironment within the nucleus plays in the formation of these complexes. The distributions of gene regulatory factors in the nuclei of living cells are dynamic, and their recruitment to particular intranuclear sites reflects the balance of their interactions with other protein partners and their association with the chromatin (53). This is illustrated by the studies of Rivera and et al. (54) who used the cyan [cyan fluorescent protein (CFP)] and yellow (YFP) color variants to visualize the androgen receptor and the steroid receptor coactivator protein 1 in living cells and to characterize their association with the nuclear bodies formed by PML. These observations revealed the ligand-dependent translocation of CFP-labeled androgen receptor into the nucleus, where it functioned to redistribute the YFP-steroid receptor coactivator protein 1 away from the PML bodies (54). A similar approach was used to demonstrate the recruitment of transcription factors and coactivator proteins by transcription factor CAATT enhancer binding protein α (9,11). These changes in subnuclear organization likely reflect direct protein-protein interactions, but the optical resolution of the light microscope is not sufficient to detect this. Fortunately, there are imaging techniques available that allow us to further define the spatial relationships between specific protein partners in living cells.
DEFINING THE SPATIAL RELATIONSHIPS BETWEEN PROTEINS IN LIVING CELLS
The ability to define the spatial relationships between proteins labeled with the different color fluorescent proteins using fluorescence microscopy is limited by the diffraction of light to a resolution of approximately 200 nm. Objects that are closer together will appear as a single object, so considerable distances may actually separate proteins that appear co-localized by fluorescence microscopy (Figure 4A). Importantly, the spectral properties of the fluorescent proteins allow them to be used as donor and acceptor fluorophores in FRET microscopy (21–24). FRET microscopy detects the transfer of excitation energy from donor to acceptor fluorophores that is the result of their direct electromagnetic interactions; there is no intermediate photon involved. When energy transfer occurs, the donor fluorescence is quenched, and there is a concomitant increase in acceptor fluorescence, which is called sensitized emission (Figure 4B). Because the distance over which energy transfer can occur between fluorescent proteins is limited to less than about 8 nm, the detection of FRET, provides measurements of the spatial relationship of the fluorophores on the scale of angstroms.
Figure 4. Improving spatial resolution using FRET microscopy.
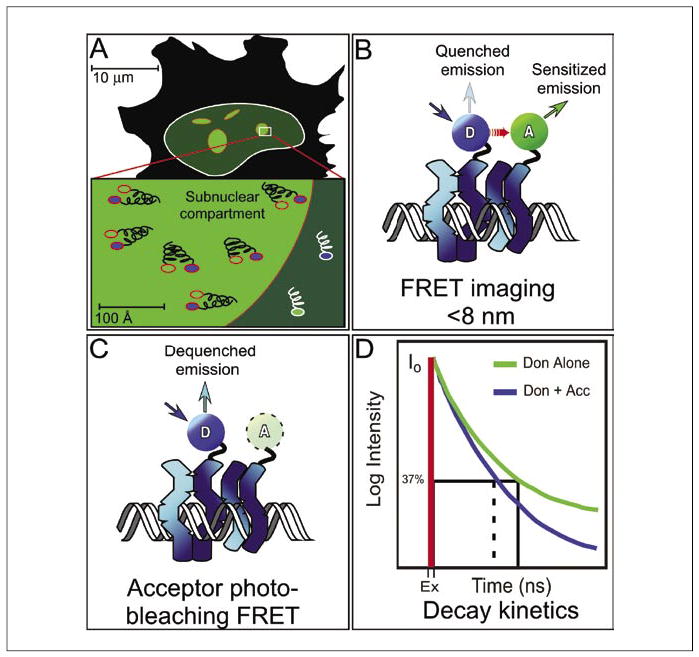
(A) Fluorescence microscopy is limited by the diffraction of light to a resolution of approximately 200 nm, and objects that are closer to-gether will appear as a single object, so considerable distances may actually separate proteins that appear co-localized by fluorescence microscopy. (B) FRET microscopy detects the direct transfer of excitation energy (red arrow) from a donor (D) fluorophore to an acceptor (A) fluorophore that is limited to distances of less than about 8 nm. When energy transfer occurs, the donor fluorescence signal is quenched, and there is sensitized emission from the acceptor. (C) Acceptor photobleaching FRET measures donor quenching by destroying the acceptor, resulting in the elimination of FRET and an increase in the donor signal. (D) Fluorescence decay kinetic measurements determine the time that a probe spends in the excited state prior to returning to the ground state. The excited-state lifetime measurements can be separated into different decay components that provide more detailed information about the environment surrounding the probe. FRET, fluorescence resonance energy transfer; Ex, excited-state lifetime; Don, donor; Don + Acc, donor and acceptor.
The efficient transfer of energy from donor to acceptor fluorescent proteins requires a significant overlap of the donor emission and acceptor absorption spectra. The Förster distance (R0), the distance at which energy transfer is 50% efficient, is derived based on the spectral overlap of the fluorophore pair used for the FRET experiments (55,56). Provided that the donor fluorophore has a high quantum yield, fluorophore pairs that share more spectral overlap will be more efficient FRET partners. However, the increased spectral overlap also leads to a substantial increase in the background noise because of spectral bleed-through (SBT) from both donor and acceptor fluorophores. This is the case for the CFP and YFP color variants that are commonly used as a donor-acceptor pair for live-cell FRET imaging (17). Their broad excitation and emission spectra and relatively small Stokes’ shifts make the separation of sensitized acceptor emission (FRET) from the SBT background signals challenging. The continued discovery and modification of the fluorescent proteins promise the development of new FRET probes with improved characteristics. For instance, the recent cloning of new cyan and orange color variants may provide fluorescent proteins with improved characteristics for FRET imaging (57). Until these are generally available and well characterized, however, we must take advantage of the fluorescent proteins that are currently at hand.
MEASUREMENT OF SENSITIZED EMISSION
The SBT background signals result from the excitation of the acceptor at the wavelengths used to excite the donor, and from donor emission into the channel used to detect the sensitized acceptor emission. The accurate measurement of sensitized acceptor emission requires methods to identify and remove sources of noise and SBT signals, and several different computer algorithms have been designed for this purpose (58–60). A comprehensive comparison of these and other correction methods was recently published (61). The common approach is to acquire reference images of control cells expressing either the donor- or the acceptor-labeled proteins alone and then to use these data to define the SBT components in the FRET channel. These methods assume a linear relationship between the double-labeled experimental cells and single-labeled reference cells imaged under the same conditions and use the information from the reference cells for the correction of donor and acceptor bleed-through signal. Both SBT and fluorophore expression level corrections are incorporated in mathematical calculations, allowing corrections based on variable donor and acceptor levels in individual cells within a transfected population (60). The contribution of background and SBT signals is then removed from the FRET data on a pixel-by-pixel basis to obtain a corrected FRET signal. These correction approaches work best when the donor and acceptor concentrations are similar because estimates of sensitized emission following subtraction of large SBT contributions are more susceptible to noise (61).
Measurements of sensitized emission using SBT correction methods have provided important insights into the subcellular distribution of interacting protein complexes within living cells. For example, Jiang and Sorkin (62) used SBT correction FRET measurements to demonstrate the interaction of epidermal growth factor receptor with cell signaling adaptor proteins. These studies revealed that the internalized receptor protein complex was associated with signaling proteins in endosomes, which resulted in prolonged receptor signaling (62). Similarly, Oliveria and colleagues (63) used an SBT correction FRET approach to detect the interactions of the catalytic subunits of protein kinases with scaffold-anchoring proteins located at cell membranes. Their application of SBT correction FRET measurements demonstrated for the first time the formation of a ternary complex involving two different protein kinase catalytic subunits with the scaffold-anchoring proteins (63).
METHODS BASED ON ACCEPTOR PHOTOBLEACHING
Importantly, the measurements of sensitized acceptor emission obtained using SBT correction methods can be verified by using the technique of acceptor photobleaching FRET (pbFRET). This approach, first described by Bastiaens and Jovin (64, 65) uses photobleaching to selectively destroy the acceptor fluorophores. When energy transfer occurs, the donor emission is partially quenched by the direct transfer of excitation energy to the acceptor. If the acceptor fluorophore is destroyed, FRET will be eliminated and the donor signal will increase (i.e., dequench), with the change in the donor signal being a measure of the efficiency of FRET (Figure 4C). Acceptor pbFRET offers a direct method to assess donor quenching. The efficiency of energy transfer is calculated by direct comparison of donor fluorescence in the presence and absence of the acceptor:
| [Eq. 1] |
where Dpre and Dpost are the donor fluorescence intensity before and after photobleaching the acceptor (66,67).
The photobleaching approach requires the selective bleaching of the acceptor because any bleaching of the donor fluorophore will lead to an under-estimation of the dequenching (67–69). A method to correct for decreased energy transfer efficiency resulting from donor bleaching during acceptor pbFRET has been recently described (70). Likewise, it is important that the selective bleaching of the acceptor be as complete as possible because incomplete acceptor bleaching can give rise to an underestimation of energy transfer efficiency (61).
The major limitation of acceptor photobleaching measurements is that they represent an end-point experiment, but they can serve to verify FRET results obtained by other methods at earlier time points. For example, Wouters and Bastiaens (71) used acceptor pbFRET to calibrate their FRET measurements of epidermal growth factor receptor phosphorylation. Elongovan et al. (60) substantiated their SBT correction method with the acceptor bleaching method and found that the average efficiencies of FRET estimated using the acceptor bleaching method were somewhat lower than average efficiencies estimated by software correction, which may reflect incomplete acceptor photobleaching. In their studies, Riven et al. (72) used both SBT correction and acceptor bleaching methods to characterize the interactions of potassium channel subunits in membranes of living cells. These authors then applied the technique of fluorescence anisotropy to demonstrate that the CFP and YFP linked to the membrane proteins adopted random orientations on the time scale of FRET, allowing them to accurately estimate the distance separating the fluorophores labeling the potassium channel subunits (72).
Cell movement and focal plane drift during the measurements are a common source of error in acceptor pbFRET measurements. Such movement artifacts appear as regions of either very high or negative FRET in energy transfer efficiency images (67). While there are many recent examples of the successful application of acceptor pbFRET to living cell preparations (35, 72–74), protein movement during image acquisition and photobleaching can be a significant obstacle. For example, both the studies by Jiang and Sorkin (62) and Oliveria et al. (63), which were described above, reported that movement of the membrane-associated protein complexes prevented the use of acceptor pbFRET in living cells. These authors were able, however, to make these measurements in fixed cell preparations. For live-cell measurements, it is advisable to bleach half the field of cells to provide an internal control for overall photobleaching or focus shift during the measurement (64, 65, 75). For single cell measurements, it is also possible to use acceptor photobleaching measurements on a defined ROI. This technique, called FRET-FRAP, monitors the dequenching of the donor signal after acceptor photobleaching within a particular ROI. The kinetics of requenching of the donor within the ROI is then measured as the bleached acceptor is exchanged for the nonbleached acceptor (76). This approach has the advantage of measuring FRET in two ways (donor dequenching and requenching) and has the added benefit of providing information about the stability of the protein complex being investigated.
As with all experimental approaches, it is important to make many independent FRET measurements from a population of cells and to use statistical approaches to verify differences between samples or treatments (77). Careful analysis of the control samples is also important to rule out other potential artifacts. The inclusion of donor and acceptor-labeled proteins that are known to dimerize or that are fused directly to one another can be used as positive controls (73, 78). Similarly, a sample containing co-localized but noninteracting donors and acceptors expressed at levels similar to the experimental proteins can serve as a negative control. Using this control, Karpova and co-workers (73) identified a low level of FRET signal from the negative control cells. This result may be related to the weak interactions between the fluorescent proteins themselves. Zacharias and colleagues (74) identified a hydrophobic patch at the carboxyl-terminus of the Aequorea-based fluorescent proteins that allows the β-barrels to associate. Studies using fluorescence fluctuation spectroscopy measurements of GFP expressed in living cells demonstrated that when GFP is free to diffuse within the volume of the cell, there does not appear to be significant self-association, even at high concentrations (79). However, the potential for fluorescent proteins to form dimers becomes a significant concern when fusion proteins are expressed at high concentrations in a restricted volume, such as inside a cellular organelle or in diffusion-limited compartments, such as in the two-dimensional space of biological membranes (80). Zacharias et al. (74) showed that the substitution of alanine206 with lysine blocked dimer formation without changing any other characteristic of the fluorescent proteins. Given the concerns for potential artifacts in FRET studies, especially when measuring dynamic protein interactions in restricted volumes inside the living cell, it seems sensible to use the monomeric fluorescent protein mutants.
FLUORESCENCE DECAY KINETICS MEASUREMENTS
Conventional fluorescence microscopy uses differences in the intensity of the probes to reveal microscopic morphology and report the location of particular molecular components. In contrast, FLIM measures the fluorescence lifetime of the probes—the time that a probe spends in the excited state prior to returning to the ground state (81–86). The fluorescence lifetime is an inherent property of a probe that is sensitive to environmental and physical processes that influence the excited state (85, 86). The advantage of FLIM over fluorescence intensity measurements is that the excited-state lifetime measurements can be separated into different decay components that provide more detailed information about the environment surrounding the probe (Figure 4D). Thus, where conventional microscopy detects fluorescent proteins with similar fluorescence intensity distribution throughout a cell, FLIM may detect regional differences in the fluorescence lifetimes, which would indicate different local microenvironments.
There are two different microscope-based methods that are most commonly used to acquire fluorescence lifetime measurements made in the frequency domain and those made in the time domain. The time-domain methods determine probe lifetimes using extremely fast excitation pulses (femtosecond) and fast-gated detection circuits that are synchronized to the excitation source. In this case, the fluorescence signal emitted after the short excitation pulse is integrated in two or more time windows. The relative intensity captured in the time windows is used to calculate the decay of fluorescence as a function of time. Alternatively, fluorescence lifetime can be measured using frequency-domain methods. Here, the specimen is excited by a sinusoidally modulated source at frequencies typically between 20 and 200 Mhz (to resolve nanosecond lifetimes). The resulting fluorescence emission will also be sinusoidally modulated, but with a different phase and amplitude than the excitation waveform. This is because the excited-state lifetime of the fluorophores imposes a time lag, and the resulting phase shift and modulation can be used to determine the lifetime composition of the sample volume. There are several review articles that provide a comprehensive discussion of these different methods (16,85–88).
The strength of the FLIM approach is that it can detect probes residing in different subcellular environments that would not be revealed by fluorescence intensity measurements. Because the fluorescence lifetime of a fluorophore is sensitive to environmental and physical processes that influence the excited state, it also provides a method to detect FRET (16, 71, 87). In the presence of an acceptor, the mean lifetime for the donor population is shifted to shorter lifetimes because energy transfer dissipates the donor excited-state energy, and measurements made using decay kinetics can detect this (Figure 4C). These measurements can potentially provide more detailed information about molecular interactions in living cells. For instance, measuring fluorescence lifetimes of the protein population within the living cell may expose several different molecular species, each with characteristic lifetimes. Repeated measurements may reveal dynamic changes in fluorescence lifetimes within the protein population and can be used to evaluate temporal changes in resonance energy transfer.
In practice, however, the fluorescence decay of fluorophores, especially in complex environments within living cells, is most often multi-exponential. For example, most of the GFP mutants examined in living cells exhibit multi-exponential fluorescence decays, and this complicates the interpretation of fluorescence lifetime measurements (89–91). In this regard, CFP is known to exhibit different fluorescent states that are reflected in lifetime measurements from cells that express CFP (92,93). Recently, Rizzo et al. (94) reported that the substitution of two hydrophobic residues on the solvent-exposed surface of CFP stabilized the protein in a single conformation. Lifetime measurements determined for the purified recombinant protein were best fit to a single exponential decay, indicating that this mutant CFP may be a more suitable probe for FLIM studies (94), but measurements in the context of living cells will be necessary to support this.
For some biological applications, it may be adequate to assume that FRET measurements obtained from lifetime decay kinetics conform to a simple two-component model, which describes the donor lifetimes as a population quenched by FRET and an unquenched population (95). However, this assumption may not be valid in other complex cellular systems because fluorescent protein lifetimes and FRET efficiencies may vary locally in unpredictable ways, resulting in an unknown number of lifetime components (96). Again, it is possible to verify the effect of FRET on the donor population by using the acceptor photobleaching method described above. Here the selective photobleaching of the acceptor should lead to a shift in the donor lifetime distribution to that of the unquenched donor population (97). The information obtained using fluorescence decay kinetic measurements can both complement intensity-based FRET imaging approaches and extend the analysis to the mapping of specific angstrom-level interactions that are localized in distinct regions of the living cell.
CONCLUSIONS
Our goal here was to introduce the basic concepts behind the quantitative techniques and statistical methods used to analyze the functional recruitment, co-localization, and interactions of proteins within subcellular compartments. We outlined an approach for the unbiased selection of cells and the consistent quantitative analysis of protein distribution within those cells. While these approaches can quantify the co-localization of specific protein partners in the same subcellular domains, the optical resolution of the light microscope is not sufficient to define precise spatial relationships between proteins. In this report, we reviewed the use of the FRET technique for acquiring more precise information about the distances separating proteins expressed in living cells. These intensity-based imaging methods detect localized concentrations of the probes and are prone to problems associated with out-of-focus signal from outside the focal plane. We discussed how the technique of FLIM overcomes these problems, and how the FLIM-FRET approach can provide more detailed information about molecular interactions in living cells. Despite the power of these FRET-based approaches, it is important to point out that by themselves, they cannot prove the direct interaction of the labeled proteins. Other techniques, such as co-immunoprecipitation, epitope-tagged protein pull-down, and two-hybrid assays are needed to substantiate the direct protein-protein interactions between specific protein partners. However, these approaches are also subject to potential artifacts due to the nonphysiological conditions of protein extraction and analysis. Despite its limitations, live-cell FRET-based imaging provides the most physiological relevant method for studying protein interactions currently available.
Finally, FRET results from single cells are, by themselves, not sufficient to determine whether, or how, proteins interact in living cells. In this regard, it is important to realize that the detection of FRET provides information about the spatial relationship of the fluorophores and not necessarily the proteins to which they are linked. FRET results by themselves cannot prove the direct interaction of the labeled proteins, but rather the fluorescent proteins serve as surrogates for the relative spatial positions of specific protein domains. Although the FRET measurements, when collected and quantified properly, are remarkably robust, there is still heterogeneity in the measurements. Further, there may also be substantial cell-to-cell heterogeneity for some interactions. Therefore, data must be collected and statistically analyzed from multiple cells to prevent the user from reaching false conclusions from a nonrepresentative measurement. The correlation of data from FRET imaging techniques and quantitative morphometric analysis methods reviewed here will establish further connections between protein interactions, subcellular protein localization, and biological function in the context of the living cell.
Acknowledgments
The authors wish to thank Cindy Booker for expert technical assistance and our colleagues Dr. Fred Schaufele (University of California at San Francisco) and Dr. Ammasi Periasamy (W.M. Keck Center for Cellular Imaging, University of Virginia) for help with these studies. This work was supported by grant nos. NIH DK47301 (to R.N.D.) and F32 DK60315 (to T.C.V.).
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare no competing interests.
References
- 1.Hadjantonakis AK, Nagy A. The color of mice: in the light of GFP-variant reporters. Histochem Cell Biol. 2001;115:49–58. doi: 10.1007/s004180000233. [DOI] [PubMed] [Google Scholar]
- 2.Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 3.Walsh MK, Lichtman JW. In vivo time-lapse imaging of synaptic takeover associated with naturally occurring synapse elimination. Neuron. 2003;37:67–73. doi: 10.1016/s0896-6273(02)01142-x. [DOI] [PubMed] [Google Scholar]
- 4.Patterson G, Day RN, Piston D. Fluorescent protein spectra. J Cell Sci. 2001;114:837–838. doi: 10.1242/jcs.114.5.837. [DOI] [PubMed] [Google Scholar]
- 5.Zhang J, Campbell RE, Ting AY, Tsien RY. Creating new fluorescent probes for cell biology. Nat Rev Mol Cell Biol. 2002;3:906–918. doi: 10.1038/nrm976. [DOI] [PubMed] [Google Scholar]
- 6.Matz MV, Fradkov AF, Labas YA, Savitsky AP, Zaraisky AG, Markelov ML, Lukyanov SA. Fluorescent proteins from nonbioluminescent Anthozoa species. Nat Biotechnol. 1999;17:969–973. doi: 10.1038/13657. [DOI] [PubMed] [Google Scholar]
- 7.Matz MV, Lukyanov KA, Lukyanov SA. Family of the green fluorescent protein: journey to the end of the rainbow. Bioessays. 2002;24:953–959. doi: 10.1002/bies.10154. [DOI] [PubMed] [Google Scholar]
- 8.Tsukamoto T, Hashiguchi N, Janicki SM, Tumbar T, Belmont AS, Spector DL. Visualization of gene activity in living cells. Nat Cell Biol. 2000;2:871–878. doi: 10.1038/35046510. [DOI] [PubMed] [Google Scholar]
- 9.Schaufele F, Enwright 3rd JF, Wang X, Teoh C, Srihari R, Erickson R, Mac-Dougald OA, Day RN. CCAAT/enhancer binding protein alpha assembles essential cooperating factors in common subnuclear domains. Mol Endocrinol. 2001;15:1665–1676. doi: 10.1210/mend.15.10.0716. [DOI] [PubMed] [Google Scholar]
- 10.Platani M, Goldberg I, Lamond AI, Swedlow JR. Cajal body dynamics and association with chromatin are ATP-dependent. Nat Cell Biol. 2002;4:502–508. doi: 10.1038/ncb809. [DOI] [PubMed] [Google Scholar]
- 11.Enwright JF, 3rd, Kawecki-Crook MA, Voss TC, Schaufele F, Day RN. A PIT-1 homeodomain mutant blocks the intranuclear recruitment of the CCAAT/enhancer binding protein alpha required for prolactin gene transcription. Mol Endocrinol. 2003;17:209–222. doi: 10.1210/me.2001-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerlich D, Beaudouin J, Kalbfuss B, Daigle N, Eils R, Ellenberg J. Global chromosome positions are transmitted through mitosis in mammalian cells. Cell. 2003;112:751–764. doi: 10.1016/s0092-8674(03)00189-2. [DOI] [PubMed] [Google Scholar]
- 13.Janicki SM, Tsukamoto T, Salghetti SE, Tansey WP, Sachidanandam R, Prasanth KV, Ried T, Shav-Tal Y, et al. From silencing to gene expression: real-time analysis in single cells. Cell. 2004;116:683–698. doi: 10.1016/s0092-8674(04)00171-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lippincott-Schwartz J, Snapp E, Kenworthy A. Studying protein dynamics in living cells. Nat Rev Mol Cell Biol. 2001;2:444–456. doi: 10.1038/35073068. [DOI] [PubMed] [Google Scholar]
- 15.van Roessel P, Brand AH. Imaging into the future: visualizing gene expression and protein interactions with fluorescent proteins. Nat Cell Biol. 2002;4:E15–E20. doi: 10.1038/ncb0102-e15. [DOI] [PubMed] [Google Scholar]
- 16.Wouters FS, Verveer PJ, Bastiaens PI. Imaging biochemistry inside cells. Trends Cell Biol. 2001;11:203–211. doi: 10.1016/s0962-8924(01)01982-1. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J, Campbell RE, Ting AY, Tsien RY. Creating new fluorescent probes for cell biology. Nat Rev Mol Cell Biol. 2002;3:906–918. doi: 10.1038/nrm976. [DOI] [PubMed] [Google Scholar]
- 18.Andrews PD, Harper IS, Swedlow JR. To 5D and beyond: quantitative fluorescence microscopy in the postgenomic era. Traffic. 2002;3:29–36. doi: 10.1034/j.1600-0854.2002.30105.x. [DOI] [PubMed] [Google Scholar]
- 19.Swedlow JR, Goldberg I, Brauner E, Sorger PK. Informatics and quantitative analysis in biological imaging. Science. 2003;300:100–102. doi: 10.1126/science.1082602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eils R, Athale C. Computational imaging in cell biology. J Cell Biol. 2003;161:477–481. doi: 10.1083/jcb.200302097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Periasamy A, Day RN. Visualizing protein interactions in living cells using digitized GFP imaging and FRET microscopy. Methods Cell Biol. 1999;58:293–314. doi: 10.1016/s0091-679x(08)61962-7. [DOI] [PubMed] [Google Scholar]
- 22.Pollok BA, Heim R. Using GFP in FRET-based applications. Trends Cell Biol. 1999;9:57–60. doi: 10.1016/s0962-8924(98)01434-2. [DOI] [PubMed] [Google Scholar]
- 23.Truong K, Ikura M. The use of FRET imaging microscopy to detect protein-protein interactions and protein conformational changes in vivo. Curr Opin Struct Biol. 2001;11:573–578. doi: 10.1016/s0959-440x(00)00249-9. [DOI] [PubMed] [Google Scholar]
- 24.Jares-Erijman EA, Jovin TM. FRET imaging. Nat Biotechnol. 2003;21:1387–1395. doi: 10.1038/nbt896. [DOI] [PubMed] [Google Scholar]
- 25.Spector DL. Nuclear domains. J Cell Sci. 2001;114:2891–2893. doi: 10.1242/jcs.114.16.2891. [DOI] [PubMed] [Google Scholar]
- 26.Carmo-Fonseca M. The contribution of nuclear compartmentalization to gene regulation. Cell. 2002;108:513–521. doi: 10.1016/s0092-8674(02)00650-5. [DOI] [PubMed] [Google Scholar]
- 27.Lamond AI, Spector DL. Nuclear speckles: a model for nuclear organelles. Nat Rev Mol Cell Biol. 2003;4:605–612. doi: 10.1038/nrm1172. [DOI] [PubMed] [Google Scholar]
- 28.Misteli T, Caceres JF, Spector DL. The dynamics of a pre-mRNA splicing factor in living cells. Nature. 1997;387:523–527. doi: 10.1038/387523a0. [DOI] [PubMed] [Google Scholar]
- 29.Gall JG, Bellini M, Wu Z, Murphy C. Assembly of the nuclear transcription and processing machinery: Cajal bodies (coiled bodies) and transcriptosomes. Mol Biol Cell. 1999;10:4385–4402. doi: 10.1091/mbc.10.12.4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muratani M, Gerlich D, Janicki SM, Gebhard M, Eils R, Spector DL. Metabolic-energy-dependent movement of PML bodies within the mammalian cell nucleus. Nat Cell Biol. 2002;4:106–110. doi: 10.1038/ncb740. [DOI] [PubMed] [Google Scholar]
- 31.Misteli T. The concept of self-organization in cellular architecture. J Cell Biol. 2001;155:181–185. doi: 10.1083/jcb.200108110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Platani M, Goldberg I, Swedlow JR, Lamond AI. In vivo analysis of Cajal body movement, separation, and joining in live human cells. J Cell Biol. 2000;151:1561–1574. doi: 10.1083/jcb.151.7.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Phair RD, Misteli T. Kinetic modelling approaches to in vivo imaging. Nat Rev Mol Cell Biol. 2001;2:898–907. doi: 10.1038/35103000. [DOI] [PubMed] [Google Scholar]
- 34.Sleeman JE, Trinkle-Mulcahy L, Prescott AR, Ogg SC, Lamond AI. Cajal body proteins SMN and Coilin show differential dynamic behaviour in vivo. J Cell Sci. 2003;116:2039–2050. doi: 10.1242/jcs.00400. [DOI] [PubMed] [Google Scholar]
- 35.Dundr M, Hebert MD, Karpova TS, Stanek D, Xu H, Shpargel KB, Meier UT, Neugebauer KM, et al. In vivo kinetics of Cajal body components. J Cell Biol. 2004;164:831–842. doi: 10.1083/jcb.200311121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Söderström M, Vo A, Heinzel T, Lavinsky RM, Yang WM, Seto E, Peterson DA, Rosenfeld MG, et al. Differential effects of nuclear receptor corepressor (N-CoR) expression levels on retinoic acid receptor-mediated repression support the existence of dynamically regulated corepressor complexes. Mol Endocrinol. 1997;11:682–692. doi: 10.1210/mend.11.6.0018. [DOI] [PubMed] [Google Scholar]
- 37.Downes M, Ordentlich P, Kao HY, Alvarez JG, Evans RM. Identification of a nuclear domain with deacetylase activity. Proc Natl Acad Sci USA. 2000;97:10330–10335. doi: 10.1073/pnas.97.19.10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levsky JM, Singer RH. Gene expression and the myth of the average cell. Trends Cell Biol. 2003;13:4–6. doi: 10.1016/s0962-8924(02)00002-8. [DOI] [PubMed] [Google Scholar]
- 39.Voss TC, Demarco IA, Booker CF, Day RN. A computer-assisted image analysis protocol that quantitatively measures subnuclear protein organization in cell populations. BioTechniques. 2004;36:240–247. doi: 10.2144/04362BI01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elliott JT, Tona A, Plant AL. Comparison of reagents for shape analysis of fixed cells by automated fluorescence microscopy. Cytometry . 2003;52A:90–100. doi: 10.1002/cyto.a.10025. [DOI] [PubMed] [Google Scholar]
- 41.Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci USA. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41a.Voss, T.C., I.A. Demarco, C.F. Booker, and R.N. Day. Quantitative methods analyze sub-nuclear protein organization in cell populations with varying degrees of protein expression. J. Biomed. Optics (In press). [DOI] [PMC free article] [PubMed]
- 42.Webb D, Hamilton MA, Harkin GJ, Lawrence S, Camper AK, Lewandowski Z. Assessing technician effects when extracting quantities from microscope images. J Microbiol Methods. 2003;53:97–106. doi: 10.1016/s0167-7012(02)00228-2. [DOI] [PubMed] [Google Scholar]
- 43.Otsu N. A threshold selection method from gray-level histogram. IEEE Trans Systems Man Cybernet. 1979;9:62–66. [Google Scholar]
- 44.Canny J. A computational approach to edge detection. IEEE Trans Pattern Anal Mach Intell. 1986;8:679–698. [PubMed] [Google Scholar]
- 45.Gerlich D, Mattes J, Eils R. Quantitative motion analysis and visualization of cellular structures. Methods. 2003;29:3–13. doi: 10.1016/s1046-2023(02)00287-6. [DOI] [PubMed] [Google Scholar]
- 46.Boland MV, Murphy RF. Automated analysis of patterns in fluorescence-microscope images. Trends Cell Biol. 1999;9:201–202. doi: 10.1016/s0962-8924(99)01550-0. [DOI] [PubMed] [Google Scholar]
- 47.Boland MV, Murphy RF. A neural network classifier capable of recognizing the patterns of all major subcellular structures in fluorescence microscope images of HeLa cells. Bioinformatics. 2001;17:1213–1223. doi: 10.1093/bioinformatics/17.12.1213. [DOI] [PubMed] [Google Scholar]
- 48.Danckaert A, Gonzalez-Couto E, Bollondi L, Thompson N, Hayes B. Automated recognition of intracellular organelles in confocal microscope images. Traffic. 2002;3:66–73. doi: 10.1034/j.1600-0854.2002.30109.x. [DOI] [PubMed] [Google Scholar]
- 49.Eils R, Gerlich D, Tvarusko W, Spector DL, Misteli T. Quantitative imaging of pre-mRNA splicing factors in living cells. Mol Biol Cell. 2000;11:413–418. doi: 10.1091/mbc.11.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tvarusko W, Bentele M, Misteli T, Rudolf R, Kaether C, Spector DL, Gerdes HH, Eils R. Time-resolved analysis and visualization of dynamic processes in living cells. Proc Natl Acad Sci USA. 1999;96:7950–7955. doi: 10.1073/pnas.96.14.7950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lefstin JA, Yamamoto KR. Allosteric effects of DNA on transcriptional regulators. Nature. 1998;392:885–888. doi: 10.1038/31860. [DOI] [PubMed] [Google Scholar]
- 52.Cosma MP. Ordered recruitment: gene-specific mechanism of transcription activation. Mol Cell. 2002;10:227–236. doi: 10.1016/s1097-2765(02)00604-4. [DOI] [PubMed] [Google Scholar]
- 53.Alvarez M, Rhodes SJ, Bidwell JP. Context-dependent transcription: all politics is local. Gene. 2003;313:43–57. doi: 10.1016/s0378-1119(03)00627-9. [DOI] [PubMed] [Google Scholar]
- 54.Rivera OJ, Song CS, Centonze VE, Lechleiter JD, Chatterjee B, Roy AK. Role of the promyelocytic leukemia body in the dynamic interaction between the androgen receptor and steroid receptor co-activator-1 in living cells. Mol Endocrinol. 2003;17:128–140. doi: 10.1210/me.2002-0165. [DOI] [PubMed] [Google Scholar]
- 55.Förster, V.T. 1948. Zwischenmolekulare energiewanderung und fluoreszenz. Annalen der Physik (Leipzig) 2:55–75. Translated: 1993. p. 148–160. In E.V Mielczarek, E. Greenbaum, and R.S. Knox (Eds.), Biological Physics. American Institute of Physics, New York.
- 56.Patterson GH, Piston DW, Barisas BG. Förster distances between green fluorescent protein pairs. Anal Biochem. 2000;284:438–440. doi: 10.1006/abio.2000.4708. [DOI] [PubMed] [Google Scholar]
- 57.Karasawa S, Araki T, Nagai T, Mizuno H, Miyawaki A. Cyan-emitting and orange-emitting fluorescent proteins as a donor/acceptor pairfor fluorescence resonance energy transfer. Biochem J. 2004;381:307–312. doi: 10.1042/BJ20040321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gordon GW, Berry G, Liang XH, Levine B, Herman B. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys J. 1998;74:2702–2713. doi: 10.1016/S0006-3495(98)77976-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xia Z, Liu Y. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys J. 2001;81:2395–2402. doi: 10.1016/S0006-3495(01)75886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Elangovan M, Wallrabe H, Chen Y, Day RN, Barroso M, Periasamy A. Characterization of one- and two-photon excitation fluorescence resonance energy transfer microscopy. Methods. 2003;29:58–73. doi: 10.1016/s1046-2023(02)00283-9. [DOI] [PubMed] [Google Scholar]
- 61.Berney C, Danuser G. FRET or no FRET: a quantitative comparison. Biophys J. 2003;84:3992–4010. doi: 10.1016/S0006-3495(03)75126-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jiang X, Sorkin A. Coordinated traffic of Grb2 and Ras during epidermal growth factor receptorendocytosis visualized in living cells. Mol Biol Cell. 2002;13:1522–1535. doi: 10.1091/mbc.01-11-0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oliveria SF, Gomez LL, Dell’Acqua ML. Imaging kinase—AKAP79—phosphatase scaffold complexes at the plasma membrane in living cells using FRET microscopy. J Cell Biol. 2003;160:101–112. doi: 10.1083/jcb.200209127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bastiaens PI, Jovin TM. Micro-spectroscopic imaging tracks the intracellular processing of a signal transduction protein: fluorescent-labeled protein kinase C beta I. Proc. Natl Acad Sci USA. 1996;93:8407–8412. doi: 10.1073/pnas.93.16.8407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bastiaens PI, Majoul IV, Verveer PJ, Soling HD, Jovin TM. Imaging the intracellular trafficking and state of the AB5 quaternary structure of cholera toxin. EMBO J. 1996;15:4246–4253. [PMC free article] [PubMed] [Google Scholar]
- 66.Kenworthy AK, Edidin M. Distribution of a glycosylphosphatidylinositol-anchored protein at the apical surface of MDCK cells examined at a resolution of <100 Å using imaging fluorescence resonance energy transfer. J Cell Biol. 1998;142:69–84. doi: 10.1083/jcb.142.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kenworthy AK. Imaging protein-protein interactions using fluorescence resonance energy transfer microscopy. Methods. 2001;24:289–296. doi: 10.1006/meth.2001.1189. [DOI] [PubMed] [Google Scholar]
- 68.Day RN, Periasamy A, Schaufele F. Fluorescence resonance energy transfer microscopy of localized protein interactions in the living cell nucleus. Methods. 2001;25:4–18. doi: 10.1006/meth.2001.1211. [DOI] [PubMed] [Google Scholar]
- 69.Miyawaki A, Tsien RY. Monitoring protein conformations and interactions by fluorescence resonance energy transfer between mutants of green fluorescent protein. Methods Enzymol. 2000;327:472–500. doi: 10.1016/s0076-6879(00)27297-2. [DOI] [PubMed] [Google Scholar]
- 70.Zal T, Gascoigne NR. Photobleaching-corrected FRET efficiency imaging of live cells. Biophys J. 2004;86:3923–3939. doi: 10.1529/biophysj.103.022087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wouters FS, Bastiaens PI. Fluorescence lifetime imaging of receptor tyrosine kinase activity in cells. Curr Biol. 1999;9:1127–1130. doi: 10.1016/s0960-9822(99)80484-9. [DOI] [PubMed] [Google Scholar]
- 72.Riven I, Kalmanzon E, Segev L, Reuveny E. Conformational rearrangements associated with the gating of the G protein-coupled potassium channel revealed by FRET microscopy. Neuron. 2003;38:225–235. doi: 10.1016/s0896-6273(03)00193-4. [DOI] [PubMed] [Google Scholar]
- 73.Karpova TS, Baumann CT, He L, Wu X, Grammer A, Lipsky P, Hager GL, McNally JG. Fluorescence resonance energy transfer from cyan to yellow fluorescent protein detected by acceptor photobleaching using confocal microscopy and a single laser. J Microsc. 2003;209:56–70. doi: 10.1046/j.1365-2818.2003.01100.x. [DOI] [PubMed] [Google Scholar]
- 74.Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- 75.Wouters FS, Bastiaens PI, Wirtz KW, Jovin TM. FRET microscopy demonstrates molecular association of non-specific lipid transfer protein (nsL-TP) with fatty acid oxidation enzymes in peroxisomes. EMBO J. 1998;17:7179–7189. doi: 10.1093/emboj/17.24.7179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vermeer JE, Van Munster EB, Vischer NO, Gadella, Jr TW. Probing plasma membrane microdomains in cow-pea protoplasts using lapidated GFP-fusion proteins and multimode FRET microscopy. J Microsc. 2004;214:190–200. doi: 10.1111/j.0022-2720.2004.01318.x. [DOI] [PubMed] [Google Scholar]
- 77.Schaufele F, Wang X, Liu X, Day RN. Conformation of CCAAT/enhancer-binding protein alpha dimers varies with intranuclear location in living cells. J Biol Chem. 2003;278:10578–10587. doi: 10.1074/jbc.M207466200. [DOI] [PubMed] [Google Scholar]
- 78.Day RN, Voss TC, Enwright 3rd JF, Booker CF, Periasamy A, Schaufele F. Imaging the localized protein interactions between Pit-1 and the CCAAT/enhancer binding protein alpha in the living pituitary cell nucleus. Mol Endocrinol. 2003;17:333–345. doi: 10.1210/me.2002-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen Y, Wei LN, Muller JD. Probing protein oligomerization in living cells with fluorescence fluctuation spectroscopy. Proc Natl Acad Sci USA. 2003;100:15492–15497. doi: 10.1073/pnas.2533045100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kenworthy A. Peering inside lipid rafts and caveolae. Trends Biochem Sci. 2002;27:435–437. doi: 10.1016/s0968-0004(02)02178-3. [DOI] [PubMed] [Google Scholar]
- 81.Gadella WJJ, Jovin TM, Clegg RM. Fluorescence lifetime imaging microscopy (FLIM): spatial resolution of microstructures on the nanosecond time scale. Biophys Chem. 1993;48:221–239. [Google Scholar]
- 82.Clegg, R.M. 1996. Fluorescence resonance energy transfer, p. 179–252. In X.F. Wang and B. Herman (Eds.), Fluorescence Imaging Spectroscopy and Microscopy. John Wiley & Sons, New York.
- 83.Periasamy A, Wodnicki P, Wang XF, Kwon S, Gordon GW, Herman B. Time-resolved fluorescence lifetime imaging microscopy using a picosecond pulsed tunable dye laser system. Rev Sci Inst. 1996;67:3722–3731. [Google Scholar]
- 84.Lakowicz, J.R. 1999. Principles of Fluorescence Spectroscopy, 2nd edition. Kluwer Academic/Plenum, New York.
- 85.Clegg RM, Holub O, Gohlke C. Fluorescence lifetime-resolved imaging: measuring lifetimes in an image. Methods Enzymol. 2003;360:509–542. doi: 10.1016/s0076-6879(03)60126-6. [DOI] [PubMed] [Google Scholar]
- 86.Dong CY, French T, So PT, Buehler C, Berland KM, Gratton E. Fluorescence-lifetime imaging techniques for microscopy. Methods Cell Biol. 2003;72:431–464. doi: 10.1016/s0091-679x(03)72021-4. [DOI] [PubMed] [Google Scholar]
- 87.Bastiaens PI, Squire A. Fluorescence lifetime imaging microscopy: spatial resolution of biochemical processes in the cell. Trends Cell Biol. 1999;9:48–52. doi: 10.1016/s0962-8924(98)01410-x. [DOI] [PubMed] [Google Scholar]
- 88.Centonze VE, Sun M, Masuda A, Gerritsen H, Herman B. Fluorescence resonance energy transfer imaging microscopy. Methods Enzymol. 2003;360:542–560. doi: 10.1016/s0076-6879(03)60127-8. [DOI] [PubMed] [Google Scholar]
- 89.Striker G, Subramaniam V, Seidel CAM, Volkmer A. Photochromicity and fluorescence lifetimes of green fluorescent protein. J Phys Chem B. 1999;103:8612–8617. [Google Scholar]
- 90.Heikal AA, Hess ST, Webb WW. Multiphoton molecular spectroscopy and excited-state dynamics of enhanced green fluorescent protein (EGFP): acid-base specificity. Chem Phys. 2001;275:37–55. [Google Scholar]
- 91.Suhling K, Siegel J, Phillips D, French PM, Leveque-Fort S, Webb SE, Davis DM. Imaging the environment of green fluorescent protein. Biophys J. 2002;83:3589–3595. doi: 10.1016/S0006-3495(02)75359-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tramier M, Kemnitz K, Durieux C, Coppey-Moisan M. Picosecond time-resolved microspectrofluorometry in live cells exemplified by complex fluorescence dynamics of popular probes ethidium and cyan fluorescent protein. J Microsc. 2002;213:110–118. doi: 10.1111/j.1365-2818.2004.01271.x. [DOI] [PubMed] [Google Scholar]
- 93.Hyun Bae J, Rubini M, Jung G, Wiegand G, Seifert MH, Azim MK, Kim JS, Zumbusch A, et al. Expansion of the genetic code enables design of a novel “gold” class of green fluorescent proteins. J Mol Biol. 2003;328:1071–1081. doi: 10.1016/s0022-2836(03)00364-4. [DOI] [PubMed] [Google Scholar]
- 94.Rizzo MA, Springer GH, Granada B, Piston DW. An improved cyan fluorescent protein variant useful for FRET. Nat Biotechnol. 2004;22:445–449. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- 95.Clayton AH, Hanley QS, Verveer PJ. Graphical representation and multicomponent analysis of single-frequency fluorescence lifetime imaging microscopy data. J Microsc. 2004;213:1–5. doi: 10.1111/j.1365-2818.2004.01265.x. [DOI] [PubMed] [Google Scholar]
- 96.Subramaniam V, Hanley QS, Clayton AH, Jovin TM. Photophysics of green and red fluorescent proteins: implications for quantitative microscopy. Methods Enzymol. 2003;360:178–201. doi: 10.1016/s0076-6879(03)60110-2. [DOI] [PubMed] [Google Scholar]
- 97.Chen Y, Periasamy A. Characterization of two-photon excitation fluorescence lifetime imaging microscopy for protein localization. Microsc Res Tech. 2004;63:72–80. doi: 10.1002/jemt.10430. [DOI] [PubMed] [Google Scholar]