

Abstract
Increased Na+-Ca2+ exchange (NCX) activity in heart failure and hypertrophy may compensate for depressed sarcoplasmic reticular Ca2+ uptake, provide inotropic support through reverse-mode Ca2+ entry, and/or deplete intracellular Ca2+ stores. NCX is electrogenic and depends on Na+ and Ca2+ transmembrane gradients, making it difficult to predict its effect on the action potential (AP). Here, we examine the effect of [Na+]i on the AP in myocytes from normal and pacing-induced failing canine hearts and estimate the direction of the NCX driving force using simultaneously recorded APs and Ca2+ transients. AP duration shortened with increasing [Na+]i and was correlated with a shift in the reversal point of the NCX driving force. At [Na+]i ≥ 10 mmol/L, outward NCX current during the plateau facilitated repolarization, whereas at 5 mmol/L [Na+]i, NCX had a depolarizing effect, confirmed by partially inhibiting NCX with exchange inhibitory peptide. Exchange inhibitory peptide shortened the AP duration at 5 mmol/L [Na+]i and prolonged it at [Na+]i ≥ 10 mmol/L. With K+ currents blocked, total membrane current was outward during the late plateau of an AP clamp at 10 mmol/L [Na+]i and became inward close to the predicted reversal point for the NCX driving force. The results were reproduced using a computer model. These results indicate that NCX plays an important role in shaping the AP of the canine myocyte, helping it to repolarize at high [Na+]i, especially in the failing heart, but contributing a depolarizing, potentially arrhythmogenic, influence at low [Na+]i.
Keywords: heart failure, Na+-Ca2+ exchanger, reversal potential, Ca2+ transients
The Na+-Ca2+ exchanger (NCX) catalyzes the electrogenic exchange of Na+ for Ca2+ across the cardiac sarcolemma and is reversible, operating in either forward (Ca2+-efflux) or reverse (Ca2+-influx) modes, depending on the prevailing electrochemical driving forces for Ca2+ and Na+. This complex dependence on transmembrane ion and voltage gradients, which are both rapidly changing during the cellular action potential (AP), makes predictions about the overall influence of NCX current (INCX) on excitation-contraction coupling challenging. NCX is the primary Ca2+ extrusion mechanism in the heart1,2 and is required to remove the increment of Ca2+ entering the myocyte via Ca2+ channels on each beat,3 but the timing of the transition from reverse-mode exchange (outward INCX) to forward-mode exchange (inward INCX) has been difficult to determine. Because the membrane impedance during the AP plateau of large mammals (including humans) is high, the net direction of current flow through NCX is likely to be a fundamental determinant of AP duration (APD).
Early investigations into the net effect of INCX on the AP demonstrated that it contributed net inward current and prolonged the AP in the rat.4 This is expected in this species, because of the rapid AP repolarization brought about by large transient outward K+ currents. In guinea pig myocytes,5 experimental data supported predominantly outward INCX during the AP plateau, but in that study, INCX was measured with sarcoplasmic reticular (SR) Ca2+ release minimized with a Ca2+ channel antagonist. This unphysiological condition would tend to exaggerate the magnitude of reverse-mode NCX. More recently, Weber et al6 have argued that INCX may be inward during most of the AP at physiological [Na+]i, with NCX sensing a higher submembrane concentration of Ca2+ than that reported by intracellular Ca2+ dyes.
In the failing heart, where SR function is impaired, a greater dependence on NCX for Ca2+ removal is expected. Moreover, a prominent increase in NCX function, in both a relative and absolute sense, has been demonstrated in animal models and in human heart failure,7–10 in accord with an increase in NCX mRNA and/or protein.9–12 There are multiple consequences of increased NCX. More forward-mode exchange compensates for defective SR Ca2+ removal, but it is at the expense of depleting the releasable pool of Ca2+ 13,14 and contributing more depolarizing current, increasing the propensity for arrhythmias triggered by delayed afterdepolarizations.11,15–17 On the other hand, more reverse-mode exchange during the AP plateau may provide inotropic support to the failing heart18,19 and could help to accelerate repolarization by carrying more outward current during the AP plateau.20
To address these questions, we investigated the [Na+]i dependence of the NCX driving force and determined its contribution to repolarization of the AP in myocytes from normal and failing canine hearts. Our findings indicate that the APD is strongly influenced by [Na+]i, with NCX contributing a net repolarizing influence at medium to high [Na+]i but a depolarizing influence at low [Na+]i. This repolarizing influence is enhanced in heart failure.
Materials and Methods
Induction of heart failure and ventricular cardiomyocyte isolation were carried out as described previously8,9,21 using protocols approved by the institution’s Animal Care and Use Committee (please see the online data supplement, available at http://www.circresaha.org).
Isolated midmyocardial ventricular myocytes were superfused (at 37 °C) with a physiological salt solution containing (mmol/L) NaCl 140, KCl 5, MgCl2 1, CaCl2 2, glucose 10, and HEPES 10 (pH 7.4 with NaOH). The intracellular solution contained (mmol/L) potassium glutamate 130, KCl 9, NaCl 5 to 15, MgCl2 0.5, MgATP 5, and HEPES 10 (pH 7.2 with KOH), along with 50 μmol/L indo 1 (Molecular Probes). Electrophysiological and fluorescence signals were acquired simultaneously and analyzed offline. Indo 1 transients were calibrated in terms of [Ca2+]i as described previously8,9,21 (also see the online data supplement).
Exchange-inhibitory peptide (XIP) was synthesized and purified by the protein synthesis core facility of The Johns Hopkins University. A small concentration-dependent shift in the pipette-to-bath junction potential was introduced by intracellular XIP, as evidenced by a 3- to 5-mV right shift in the zero current potential for the inward rectifier K+ current (data not shown). This offset was corrected in the AP records.
APs and [Ca2+]i were measured in current-clamp mode pacing at 1 Hz to steady state (after at least 20 stimuli). The driving-force reversal point (RP) of the NCX was measured as the membrane potential (Em) at which the NCX driving force (Em − ENCX) equaled zero, with ENCX defined as 3ENa − 2ECa. ENa and ECa are the equilibrium potentials for Na+ and Ca2+, respectively, with EX=(RT/zF)log([X]o/[X]i, where R is the ideal gas constant; T, absolute temperature; z, valence; and F, Faraday constant, and all ions except [Ca2+]i were assumed to be equivalent to their bulk concentrations (see Discussion). The time to RP (TTRP) was defined as the time from the AP upstroke to the NCX RP and was normalized to the duration of the AP for determination of the fraction of the duty cycle the exchanger was in reverse versus forward mode.
In some experiments, as indicated, a “paired-pipette” approach was used to analyze the effects of changing intracellular Na+ or to introduce XIP into the cytoplasm. In these experiments, recordings were made with a particular pipette composition, and then the patch was rapidly excised. In most cases, the myocyte membrane resealed immediately on excision, and a whole-cell recording could be made with a second (or third) pipette of different composition. This sequential whole-cell patch-clamp method was reasonably efficient, and there was usually no difference in the background current or resting Em of the myocyte. If such signs of myocyte damage were observed, then the data were excluded.
An AP-clamp experiment (Figure 7) was carried out in a freshly isolated guinea pig ventricular myocyte to eliminate a contribution from the Ca2+-activated Cl− current, which is prevalent in the dog but absent in the guinea pig. In this experiment, K+ currents were blocked by Cs+ substitution using an external solution consisting of (mmol/L) NaCl 138, CsCl 5, MgCl2 1, CaCl2 2, and HEPES 10 (pH 7.4 with NaOH) and an internal solution consisting of (mmol/L) glutamate 120, CsCl 19, sodium HEPES 10, MgCl2 0.5, and MgATP 5, along with 50 μmol/L indo 1 (pH 7.2 with CsOH).
Figure 7.
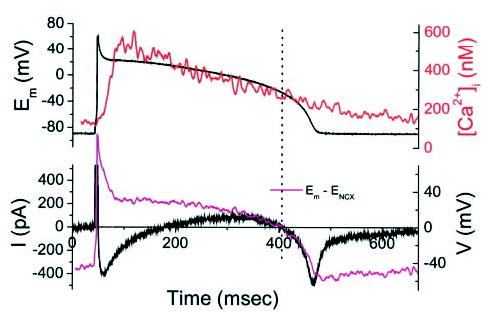
AP-clamp currents with 10 mmol/L [Na+]i and K+ currents blocked. Top panel shows AP waveform used to voltage-clamp a cardiomyocyte with Cs+ substitution of K+ in all solutions (to eliminate contamination by Ca2+ -activated Cl− current; this experiment was carried out in a guinea pig cardiomyocyte). Under these conditions, the predominant membrane currents were ICa,L and INCX. The kinetics and direction of the membrane currents during the AP plateau corresponded to estimates of the NCX driving force (magenta trace) calculated from the simultaneously recorded [Ca2+]i transient (red trace).
Statistical Analysis
Myocytes from four normal and six failing hearts were studied at 5, 10, or 15 mmol/L [Na+]i (n5 = 10/14, n10 = 12/12, and n15 = 16/14 for normal/failing myocytes, respectively). Comparisons between groups were made using the Student unpaired t test. A 95% confidence interval was used to determine statistical significance. Data are expressed as mean ± SE (unless otherwise specified).
Results
Effect of Intracellular Na+ on NCX Driving Force and APD
Figure 1 illustrates the approach used to estimate the driving force for INCX at 5, 10, or 15 mmol/L [Na+]i and shows representative APs and Ca2+ transients for myocytes from normal and failing hearts. ENCX (Figure 1, blue traces) was calculated as 3ENa − 2ECa (3:1 stoichiometry) from the Ca2+ transient, assuming a constant extracellular Ca2+ and transmembrane Na+ gradient. The difference between Em and ENCX was determined and is plotted on the same voltage scale for comparison (Figure 1, magenta traces). The NCX RP was taken as the membrane voltage corresponding to the point at which the NCX driving force reversed the sign (Em = ENCX). It is clear from the figure that this point shifted in the hyperpolarized direction at higher [Na+]i, favoring more outward current during the AP plateau, suggesting that repolarization should be facilitated at higher [Na+]i. Notably, the NCX driving force was more outward in myocytes from failing hearts at any given [Na+]i, as a consequence of both a more depolarized early plateau potential and a smaller cytosolic Ca2+ transient.
Figure 1.
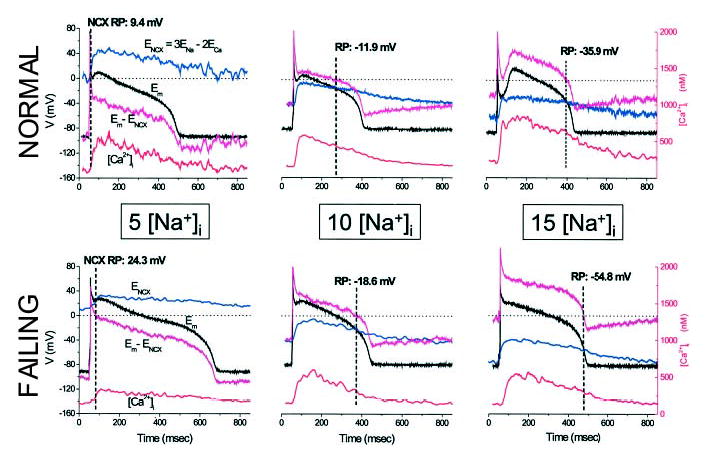
Effect of [Na+]i on AP and NCX driving force. Top, Representative APs (black trace) and Ca2+ transients (red trace) from myocytes isolated from normal hearts dialyzed with 5, 10, or 15 mmol/L [Na+]i. Left panel illustrates the method for calculating the NCX driving force (Em − ENCX, magenta trace) from ENCX (blue trace). Bottom, Analogous records for myocytes from failing hearts with 5, 10, or 15 [Na+]i. RP is the potential at which Em − ENCX=0 (indicated with a dashed line in all panels).
There were no statistically significant differences of the resting potential either between groups or as a function of [Na+]i (Table). Consistent with the shift in NCX driving force, APD at 90% repolarization (APD90) was inversely correlated with [Na+]i (Figure 2A). APD90 decreased on elevating [Na+]i from 5 to 15 mmol/L in myocytes from normal (by ≈ 175 ms) and failing (by ≈ 125 ms) hearts (Table). Hyperpolarization of the NCX RP was also associated with AP shortening (Figure 2B), and the fraction of the APD the NCX spent in the reverse mode (ie, the TTRP normalized to the APD [TTRP/APD90]) was increased at high [Na+]i (Figure 2C).
Summary of Statistics for Normal and Failing Hearts
| Normal (Nn=4; n5=10; n10=12; n15=16)
|
Failing (Nf=6; n5=14; n10=12; n15=14)
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Er, mV | RP, mV | APD90, msec | TTRPn | Er, mV | RP, mV | APD90, msec | TTRPn | |
| 5 Na | −86±2 | 25.8±2.7* | 499.6±53.4 | 0.137±0.039* | −81±2 | 25.7±2.1* | 543.5±33.3* | 0.098±0.022* |
| 10 Na | −81±1 | −20.6±1.2 | 420.8±29.7 | 0.896±0.038 | −86±2 | −22.2±3.5 | 430.0±23.1 | 0.804±0.031 |
| 15 Na | −85±2 | −45±3* | 327.5±22.8* | 0.949±0.053 | −85±1 | −56.9±1.7*† | 419.4±17.0† | 0.975±0.014* |
Summary statistics for the resting potential (Er), driving-force reversal point (RP), action potential duration (APD90), and time-to-RP (TTRP) normalized to the action potential duration (TTRPn) at 5, 10, and 15 mmol/L [Na+]i of myocytes from normal and failing hearts. Nn and Nf indicate number of normal and failing hearts correspondingly; n5, n10, and n15, number of myocytes at 5, 10, and 15 mmol/L [Na+]i correspondingly.
Statistically significant difference (P<0.05) between a given parameter and its value recorded at 10 mmol/L [Na+]i.
statistical significance (P<0.05) between normal and failing groups at the same [Na+]i.
Figure 2.
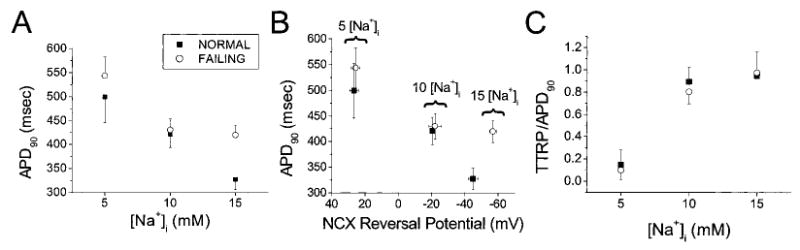
[Na+]i dependence of the APD, NCX driving-force RP, and TTRP in normal (solid square) and failing (open circle) myocytes. A and B, APD90 shortened with increasing [Na+]i (A), in correspondence with a shift in the NCX RP toward more hyperpolarized potentials (B). C, RP occurred later in the AP at higher [Na+]i. TTRP/APD90 is the TTRP normalized to APD90.
The influence of [Na+]i on the AP was further confirmed in a subset of myocytes by acquiring paired data. APD90 was inversely correlated with [Na+]i in 14 of 16 paired-pipette recordings (see Figure 3 and Materials and Methods), regardless of the direction of the change in [Na+]i (indicated by the direction of the arrows in Figure 3).
Figure 3.
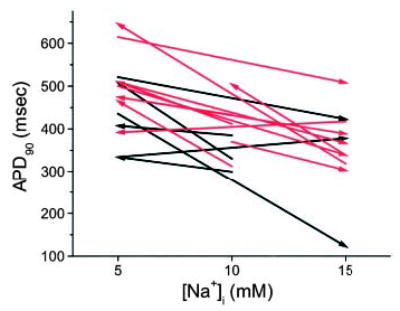
[Na+]i dependence of APD using paired pipette technique. [Na+]i was varied within individual myocytes using sequential pipettes containing different levels of [Na+]i. The direction and amplitude of the change in [Na+]i are indicated by the arrows. Black and red arrows denote myocytes from normal and failing hearts, respectively. In one cell from a normal heart and four cells from failing hearts, a third pipette was used to sequentially patch the same cell with a different [Na+]i.
The qualitative observation that the net balance of NCX driving force was shifted toward more reverse-mode exchange in myocytes from failing hearts was confirmed by calculating the ratio of reverse-mode to forward-mode exchange driving force during the AP. This was determined by integrating the area bounded by the NCX driving-force curve from the upstroke of the AP to the RP (Intrev) divided by the forward-mode integral from the RP to phase 3 repolarization of the AP (Intfor) (Figure 4A). Intrev/Intfor was higher in the failing group at all three [Na+]i levels, reaching statistical significance at 15 mmol/L [Na+]i (Figure 4B). Because this parameter reflects a change in the balance of outward versus inward NCX driving force, it underestimates the difference of the NCX contribution in shaping the AP between groups, inasmuch as the total INCX in the failing heart would also take into account the increase in INCX density in this model, which is approximately twice that of the normal myocyte.8 At 10 and 15 [Na+]i, Intrev/Intfor was >1, suggesting a net repolarizing influence of NCX at these levels of [Na+]i. Inspection of the running integral of NCX shows that the NCX driving force for reverse-mode exchange rises more rapidly and is larger in the failing group at all levels of [Na+]i (Figure 4C). Furthermore, in the context of increased resting [Na+]i in heart failure,22 Intrev/Intfor significantly differed between cells from failing hearts at 10 mmol/L [Na+]i and cells from normal hearts at 5 mmol/L [Na+]i (P=0.01).
Figure 4.
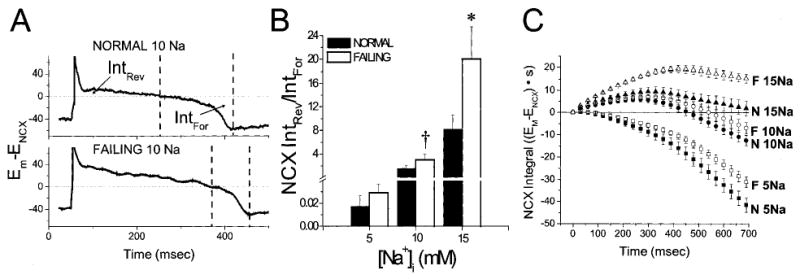
Ratio of integrated reverse-mode to forward-mode NCX driving force during the AP. A, Intrev represents the integral of the Em−ENCX curve from the upstroke of the AP to the NCX RP. Intfor is the integral from the driving-force RP to the point of AP repolarization (taken as the minimum of the NCX driving-force curve). B, Intrev/Intfor increased as a function of [Na+]i and was significantly higher in the failing group. C, Averaged running integrals for all data sets illustrate more rapid reverse-mode NCX activation in myocytes from failing (F) than normal (N) hearts at all levels of [Na+]i. *P<0.05 for normal vs failing myocytes; †P<0.05 for myocytes from failing hearts at 10 mmol/L [Na+]i vs myocytes from normal hearts at 5 mmol/L [Na+]i.
Effect of NCX Inhibition on APD
To test the validity of the assumption that NCX driving force calculated from the Ca2+ transient was a suitable method for estimating the RP and net effect of NCX on the AP waveform, we reasoned that the APD90 should be predictably influenced by partial inhibition of NCX (Figure 5A), depending on [Na+]i. At 5 mmol/L [Na+]i, INCX is inward for almost the entire APD; therefore, inhibition of this depolarizing current should shorten the APD90. In contrast, at 10 or 15 mmol/L [Na+]i, inhibition of NCX should prolong the AP if it is contributing outward current during the mid to late plateau. This was tested using the same paired-pipette method described above for changing [Na+]i (Figure 3), except that in this case, [Na+]i was held constant, and 100 μmol/L XIP was included in the second pipette. As predicted, XIP shortened the AP at 5 mmol/L [Na+]i but prolonged the AP at 10 or 15 mmol/L [Na+]i in both experimental groups (Figure 5B). To avoid the confounding secondary effect of XIP to increase SR Ca2+ load, we matched Ca2+ transient recordings that had similar rise times and amplitudes by selecting records in the presence of XIP during the pulse train before the maximal SR Ca2+ load was attained. The significant (P<0.05) prolongation of the AP by XIP (30 μmol/L) at 10 mmol/L [Na+]i was also confirmed in separate unpaired experiments in myocytes paced at 0.25 Hz (Figure 5C).
Figure 5.
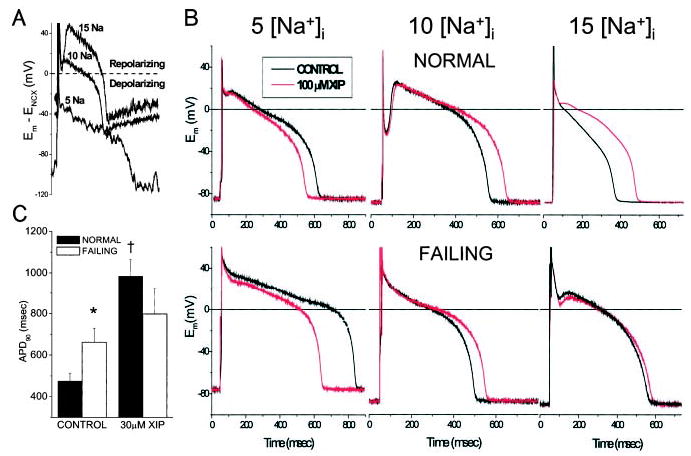
Effect of XIP on APD. A, Block of net inward current at 5 [Na+]i during the AP plateau would be expected to shorten the APD90, whereas the predicted effect at 10 or 15 [Na+]i would be AP prolongation, based on the net repolarizing influence of outward INCX. B, Paired pipette experiments confirmed the predicted effect of XIP on APD, supporting the validity of NCX driving-force estimates. C, Unpaired experiments show the change in mean APD90 induced by 30 μmol/L XIP with 10 mmol/L [Na+]i.*P<0.05 for normal vs failing myocytes; †P<0.05 for XIP vs control in normal myocytes. Myocytes were paced at 1 Hz in panel B and 0.25 Hz in panel C.
Model Simulations
We developed an AP-clamp variation of the canine cardiomyocyte computer model described by Winslow, Greenstein, and colleagues20,23 that allowed the use of an experimentally recorded AP as an input function to simulate the Ca2+ transient and underlying membrane currents.24 By comparing experimental and model results, we reasoned that if a given AP waveform produces a simulated Ca2+ transient and NCX driving force that reproduce experimental estimates, then the assumptions of the model do not grossly misrepresent the behavior of the myocyte.
Model-derived Ca2+ transients in the AP clamp at 5, 10, or 15 mmol/L [Na+]i were compared with experimental transients recorded in current-clamp mode (Figure 6A). The model-derived NCX driving-force trajectories and RPs robustly reproduced the experimental data, and model RPs were correlated over the range of [Na+]i studied (R=0.99, Figure 6B). Importantly, the slope of this relationship was 1.0, indicating that the experimental RP did not deviate significantly from that determined by the model. This would not have been the case if the driving force for NCX was primarily governed by Ca2+ in a microdomain that differs from the bulk Ca2+ reported by indo 1. Thus, when NCX in the model was reformulated to respond to Ca2+ in the subsarcolemmal junctional subspace (by modifying equations A.39 and A.91 of Winslow et al20; see online supplement), a marked reduction in Ca2+ transient amplitude was observed that was due to enhanced forward-mode exchange and Ca2+ extrusion from the myocyte (Figure 6C). If these conditions existed, the result would be a shift in the NCX RP; consequently, the experimentally obtained RP values would have been shifted to more negative values and the slope of the relation in Figure 6B would not have been 1. From a different perspective, when NCX is formulated to respond to Ca2+ within the subspace, the shift in INCX substantially alters the AP waveform when the model is not constrained under AP-clamp conditions (Figure 6D). In this case, the enhanced inward INCX results in overshoot of the AP dome and prolongation of the APD.
Figure 6.
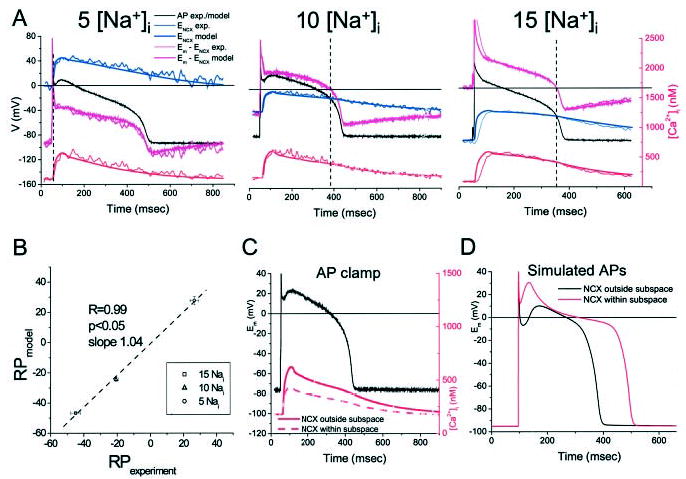
AP-clamp model simulations. A, Examples of model-derived Ca2+ transients and NCX driving forces using experimental AP recordings as inputs at varying levels of [Na+]i. B, Correlation between model-derived and experimental NCX driving-force RPs. C and D, Influence of Ca2+ in the junctional subspace on the Ca2+ transient in AP-clamp mode (C) or free-running AP simulations (D).
INCX During AP Clamp
Direct measurement of INCX during a physiological AP and Ca2+ transient would yield the ultimate answer to the central question of the present study; however, selectively measuring INCX without perturbing other parameters is difficult. For example, inhibiting NCX with Ni2+ or KBR-794325 will also block Na+ and Ca2+ channels (among others), which will profoundly change the AP profile and the Ca2+ transient, as will selective inhibition of L-type Ca2+ current (ICa,L).5 Therefore, we sought confirmation that our estimation of timing of the reversal of the NCX driving force matches that of the INCX by performing an AP-clamp experiment (in 10 mmol/L Na+) with K+ currents blocked by Cs+ substitution and Ca2+-activated Cl− currents eliminated using the guinea pig ventricular myocyte. This limited the major currents flowing during the plateau to ICa,L and INCX (with a minor contribution of the Na+,K+-ATPase current; see Discussion). Under these conditions, a net outward current was recorded beginning ≈ 100 ms after the upstroke of the AP and extending until just before phase 3 repolarization (Figure 7). Consistent with predictions, the kinetics and late RP of the plateau current corresponded well with the RP of the NCX driving force. A notable deviation between the NCX driving-force estimate and the presumed NCX current at the resting potential was evident (see Discussion).
Discussion
The results indicate that the APD is strongly dependent on [Na+]i, with increased [Na+]i resulting in shortening of the APD90. This finding is consistent with a shift in the balance of reverse-mode versus forward-mode NCX. Inhibition of NCX with XIP supports the hypothesis that at high [Na+]i (>10 mmol/L), outward INCX during the plateau contributes to AP repolarization, whereas at low [Na+]i (5 mmol/L), INCX is predominantly inward. The net direction of the NCX driving force during the AP plateau can be reasonably determined from the cytosolic Ca2+ transient.
APD90 decreased significantly in both groups when [Na+]i increased from 5 to 15 mmol/L [Na+]i (Figure 2). This relationship was almost perfectly linear over the whole range of [Na+]i in normal myocytes (R=0.998, P<0.05 for the data in Figure 2A), but in the failing group, the [Na+]i-induced shortening of APD90 appeared to reach a limit at 10 mmol/L [Na+]i. This was true despite the fact that the NCX RP shifted to a more hyperpolarized point (Figure 2B), reached later in the AP (see TTRP/APD90; Figure 2C), when [Na+]i was increased from 10 mmol/L to 15 mmol/L in the failing group. Furthermore, the ratio of integrated reverse-mode to forward-mode exchange very strongly favored repolarization at 15 mmol/L [Na+]i compared with 10 mmol/L [Na+]i (Figure 4). The diminished effect of increasing [Na+]i from 10 to 15 mmol/L on APD90 in the failing group suggests that an upper limit of outward INCX may have been reached at 10 mmol/L [Na+]i or that the contribution of INCX to the APD is overwhelmed by other factors in this range of [Na+]i.
In our hands, blocking Na+,K+-ATPase (10 μmol/L strophanthidin) prolonged the APD (data not shown); thus, a limitation of the present study is that the Na+-K+ pump may partially contribute to [Na+]i-mediated AP shortening. However, the extent of its contribution cannot be easily ascertained using inhibitors, because [Na+]i homeostasis is dramatically altered in their presence. Further investigation will be necessary to determine how much of the [Na+]i-mediated AP shortening might be related to Na+,K+-ATPase current, but we estimate the pump current to be less than ¼ the density of INCX at plateau potentials. Moreover, the pump will always have a hyperpolarizing influence and therefore could not explain the differential effects of XIP at low versus high [Na+]i.
At all levels of [Na+]i, myocytes from failing hearts, compared with cells from normal hearts, displayed a higher ratio of outward/inward NCX driving force (Intrev/Intfor, Figure 4). This difference could be explained by two factors: (1) less early repolarization, presumably due to diminished transient outward K+ current,21 and (2) a reduction in the amplitude of the Ca2+ transient in the failing myocyte.9,26 The result is earlier activation of reverse-mode exchange and more outward INCX during the AP plateau. It is important to note that this conclusion is based solely on the estimate of NCX driving force and would be true whether or not the current density of NCX8,9,21 or [Na+]i27 was increased in heart failure, two factors that would further enhance reverse-mode exchange. This could be considered as an adaptation that would counteract excessive AP prolongation resulting from decreased Ca2+-dependent inactivation of ICa,L in the failing myocyte20 but would result in Ca2+ extrusion occurring primarily on completion of repolarization. Whereas the former effect would be beneficial, the latter could contribute to an enhanced susceptibility to delayed afterdepolarizations, as suggested previously.28
Early afterdepolarizations also could be strongly influenced by the RP of INCX. Once turning inward (which happens very early at low [Na+]i), INCX is a depolarizing force that is enhanced during the vulnerable late phase of the AP, when Ca2+ channels may be recovering from inactivation. In the context of the present study, in paired-pipette experiments, we have observed early after depolarizations triggered in response to lowering [Na+]i from 10 to 5 mmol/L (data not shown).
The results obtained using the AP-clamp variation of the canine myocyte model24 were consistent with experimental results with respect to the profile of the Ca2+ transient and the influence of [Na+]i on the AP. Because the only input to the model was the AP waveform, this implies that the model assumptions for describing the Ca2+ transient waveform and the behavior of INCX were generally correct (Figures 6B through 6D). These include a 3:1 Na+:Ca2+ stoichiometry for NCX, distribution of the exchanger in the nonjunctional compartment, and NCX driving force determined by the bulk cytoplasmic Ca2+ signal.
Recent reports suggest that NCX is exclusively localized in the T tubules29,30 but does not have direct access to the Ca2+ released into the junctional space of the dyad,31 in as much as caffeine-evoked SR Ca2+ release does not activate INCX when [Ca2+]i is buffered to within ≈ 50 nm of the release sites.32 Our XIP and computer simulation results (Figures 6A through 6C) indicate that NCX is unlikely to sense Ca2+ in the dyadic cleft, a conclusion that is in agreement with the results of López-López et al,33 who have shown that reverse-mode NCX cannot directly trigger Ca2+ sparks in the absence of ICa,L. However, the possibility still exists that subsarcolemmal [Na+]i or [Ca2+]i gradients may be present during the AP. Na+ entering the myocyte through the Na+ channel into a hypothetical “fuzzy space”34 has been proposed to account for NCX-triggered Ca2+ release.35 This idea has been challenged,36 but the issue has not been definitively resolved. The increase in [Na+]i due to the Na+ current has been estimated to be on the order of 25 μmol/L,34 an amount that should not have a major impact on our conclusions, because the changes we made were in the millimolar range.
Ca2+ released from the SR into the junctional space does greatly exceed that measured by an evenly distributed Ca2+ indicator in the cytoplasm. NCX in proximity to the release sites could theoretically respond to a local submembrane Ca2+ microdomain, in effect switching the exchanger to forward mode much earlier in the AP. A recent report by Weber et al,6 who used NCX tail currents as an index of submembrane Ca2+ during truncated AP-clamp stimuli, suggested that the driving force for NCX during an AP may be much more inward than had been previously determined (eg, see Grantham and Cannell5) or estimated from our experiments. This would imply that the balance of reverse-mode to forward-mode NCX activity could favor depolarization rather than repolarization at higher [Na+]i, a conclusion that was not supported by the XIP experiments or the AP-clamp experiment. We cannot rule out that early in the AP, subsarcolemmal Ca2+ gradients exist that could cause the exchanger to transiently shift to forward mode during the SR Ca2+ release phase of the Ca2+ transient. Late in the plateau, however, the present findings indicate that the measured [Ca2+]i is a good approximation of that seen by the NCX and is useful in determining the effect of NCX on APD90. This is in partial agreement with the conclusions of Weber et al,6 with the only modification being that the subsarcolemmal gradients probably dissipate earlier in the AP (ie, during the notch phase) than they predicted.
In 10 mmol/L [Na+]i, with K+ currents blocked, total membrane current during the late plateau in the AP-clamp experiment (Figure 7) was outward and had an RP similar to that of the calculated NCX driving force. These data provide additional support for our conclusions but also highlight a limitation of the method, ie, that the NCX current magnitude (but not the direction of current flow) can deviate substantially from the driving force estimate during different phases of the cardiac cycle. This can be seen at the resting Em in 10 mmol/L [Na+]i, where there is substantial driving force for inward INCX but the net inward current diminishes. A reduction in NCX activity at the resting potential, described previously in voltage-clamp experiments,37 could explain this observation; however, further studies will be required to reconcile this aspect of the results.
In summary, these findings underscore the prominent role of NCX in modulating the APD in myocytes from normal and failing hearts and provide support for the hypothesis that INCX can be a significant repolarizing force in ventricular myocytes from large mammals. In the context of heart failure, in which [Na+]i may increase >10 mmol/L,22 this effect could help to prevent excessive AP prolongation and provide inotropic support via reverse-mode exchange.38 At [Na+]i <10 mmol/L, this balance shifts toward more forward-mode exchange, so that INCX contributes to depolarization of the AP plateau and AP prolongation, factors that may contribute to arrhythmogenesis.
Acknowledgments
This study was supported by the National Institutes of Health (NIH grant R01 HL-61711 to Dr O’Rourke), American Heart Association Fellowships (0020257U to Dr Armoundas and 0020155U to Dr Hobai), and the SCOR on Sudden Cardiac Death (NIH grant P50 HL-52307 to Drs Tomaselli, Winslow, and O’Rourke). We would like to thank Richard Tunin and Dr Nazareno Paolocci for helping with the dog preparation.
Footnotes
This manuscript was sent to Michael R. Rosen, Consulting Editor, for review by expert referees, editorial decision, and final disposition.
References
- 1.Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev. 1999;79:763–854. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- 2.Shigekawa M, Iwamoto T. Cardiac Na+-Ca2+ exchange: molecular and pharmacological aspects. Circ Res. 2001;88:864–876. doi: 10.1161/hh0901.090298. [DOI] [PubMed] [Google Scholar]
- 3.Choi HS, Trafford AW, Eisner DA. Measurement of calcium entry and exit in quiescent rat ventricular myocytes. Pflugers Arch. 2000;440:600–608. doi: 10.1007/s004240000295. [DOI] [PubMed] [Google Scholar]
- 4.duBell WH, Boyett MR, Spurgeon HA, Talo A, Stern MD, Lakatta EG. The cytosolic calcium transient modulates the action potential of rat ventricular myocytes. J Physiol. 1991;436:347–369. doi: 10.1113/jphysiol.1991.sp018554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grantham CJ, Cannell MB. Ca2+ influx during the cardiac action potential in guinea pig ventricular myocytes. Circ Res. 1996;79:194–200. doi: 10.1161/01.res.79.2.194. [DOI] [PubMed] [Google Scholar]
- 6.Weber CR, Piacentino V, III, Ginsburg KS, Houser SR, Bers DM. Na+-Ca2+ exchange current and submembrane [Ca2+] during the cardiac action potential. Circ Res. 2002;90:182–189. doi: 10.1161/hh0202.103940. [DOI] [PubMed] [Google Scholar]
- 7.Flesch M, Putz F, Schwinger RH, Bohm M. Functional relevance of an enhanced expression of the Na+-Ca2+ exchanger in the failing human heart. Ann N Y Acad Sci. 1996;779:539–542. doi: 10.1111/j.1749-6632.1996.tb44832.x. [DOI] [PubMed] [Google Scholar]
- 8.Hobai IA, O’Rourke B. Enhanced Ca2+-activated Na+-Ca2+ exchange activity in canine pacing-induced heart failure. Circ Res. 2000;87:690–698. doi: 10.1161/01.res.87.8.690. [DOI] [PubMed] [Google Scholar]
- 9.O’Rourke B, Kass DA, Tomaselli GF, Kääb S, Tunin R, Marbán E. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, I: experimental studies. Circ Res. 1999;84:562–570. doi: 10.1161/01.res.84.5.562. [DOI] [PubMed] [Google Scholar]
- 10.Reinecke H, Studer R, Vetter R, Holtz J, Drexler H. Cardiac Na+/Ca2+ exchange activity in patients with end-stage heart failure. Cardiovasc Res. 1996;31:48–54. [PubMed] [Google Scholar]
- 11.Pogwizd SM, Qi M, Yuan W, Samarel AM, Bers DM. Upregulation of Na+/Ca2+ exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ Res. 1999;85:1009–1019. doi: 10.1161/01.res.85.11.1009. [DOI] [PubMed] [Google Scholar]
- 12.Studer R, Reinecke H, Bilger J, Eschenhagen T, Bohm M, Hasenfuss G, Just H, Holtz J, Drexler H. Gene expression of the cardiac Na+-Ca2+ exchanger in end-stage human heart failure. Circ Res. 1994;75:443–453. doi: 10.1161/01.res.75.3.443. [DOI] [PubMed] [Google Scholar]
- 13.Pieske B, Sutterlin M, Schmidt-Schweda S, Minami K, Meyer M, Olschewski M, Holubarsch C, Just H, Hasenfuss G. Diminished post-rest potentiation of contractile force in human dilated cardiomyopathy: functional evidence for alterations in intracellular Ca2+ handling. J Clin Invest. 1996;98:764–776. doi: 10.1172/JCI118849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hasenfuss G, Reinecke H, Studer R, Pieske B, Meyer M, Drexler H, Just H. Calcium cycling proteins and force-frequency relationship in heart failure. Basic Res Cardiol. 1996;91:17–22. doi: 10.1007/BF00795357. [DOI] [PubMed] [Google Scholar]
- 15.Koster OF, Szigeti GP, Beuckelmann DJ. Characterization of a [Ca2+]i-dependent current in human atrial and ventricular cardiomyocytes in the absence of Na+ and K+ Cardiovasc Res. 1999;41:175–187. doi: 10.1016/s0008-6363(98)00202-8. [DOI] [PubMed] [Google Scholar]
- 16.Sipido KR, Volders PG, de Groot SH, Verdonck F, Van de Werf F, Wellens HJ, Vos MA. Enhanced Ca2+ release and Na/Ca exchange activity in hypertrophied canine ventricular myocytes: potential link between contractile adaptation and arrhythmogenesis. Circulation. 2000;102:2137–2144. doi: 10.1161/01.cir.102.17.2137. [DOI] [PubMed] [Google Scholar]
- 17.Schlotthauer K, Bers DM. Sarcoplasmic reticulum Ca2+ release causes myocyte depolarization: underlying mechanism and threshold for triggered action potentials. Circ Res. 2000;87:774–780. doi: 10.1161/01.res.87.9.774. [DOI] [PubMed] [Google Scholar]
- 18.Mattiello JA, Margulies KB, Jeevanandam V, Houser SR. Contribution of reverse-mode sodium-calcium exchange to contractions in failing human left ventricular myocytes. Cardiovasc Res. 1998;37:424–431. doi: 10.1016/s0008-6363(97)00271-x. [DOI] [PubMed] [Google Scholar]
- 19.Flesch M, Schwinger RH, Schiffer F, Frank K, Sudkamp M, Kuhn-Regnier F, Arnold G, Bohm M. Evidence for functional relevance of an enhanced expression of the Na+-Ca2+ exchanger in failing human myocardium. Circulation. 1996;94:992–1002. doi: 10.1161/01.cir.94.5.992. [DOI] [PubMed] [Google Scholar]
- 20.Winslow RL, Rice J, Jafri S, Marbán E, O’Rourke B. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, II: model studies. Circ Res. 1999;84:571–586. doi: 10.1161/01.res.84.5.571. [DOI] [PubMed] [Google Scholar]
- 21.Kääb S, Nuss HB, Chiamvimonvat N, O’Rourke B, Pak PH, Kass DA, Marbán E, Tomaselli GF. Ionic mechanism of action potential prolongation in ventricular myocytes from dogs with pacing-induced heart failure. Circ Res. 1996;78:262–273. doi: 10.1161/01.res.78.2.262. [DOI] [PubMed] [Google Scholar]
- 22.Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation. 2002;105:2543–2548. doi: 10.1161/01.cir.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- 23.Greenstein JL, Wu R, Po S, Tomaselli GF, Winslow RL. Role of the calcium-independent transient outward current Ito1 in shaping action potential morphology and duration. Circ Res. 2000;87:1026–1033. doi: 10.1161/01.res.87.11.1026. [DOI] [PubMed] [Google Scholar]
- 24.Armoundas AA, O’Rourke B, Greenstein JL, Winslow RL, Tomaselli GF. A robust test of a cardiac cell model: correlation between action potential clamp-driven Ca2+ transients and experimentally-obtained ones. Biophys J. 2001;80:325. Abstract. [Google Scholar]
- 25.Tanaka H, Nishimaru K, Aikawa T, Hirayama W, Tanaka Y, Shigenobu K. Effect of SEA0400, a novel inhibitor of sodium-calcium exchanger, on myocardial ionic currents. Br J Pharmacol. 2002;135:1096–1100. doi: 10.1038/sj.bjp.0704574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hobai IA, O’Rourke B. Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation. 2001;103:1577–1584. doi: 10.1161/01.cir.103.11.1577. [DOI] [PubMed] [Google Scholar]
- 27.Pieske B, Maier LS, Piacentino V, III, Weisser J, Hasenfuss G, Houser S. Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation. 2002;106:447–453. doi: 10.1161/01.cir.0000023042.50192.f4. [DOI] [PubMed] [Google Scholar]
- 28.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res. 2001;88:1159–1167. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- 29.Kieval RS, Bloch RJ, Lindenmayer GE, Ambesi A, Lederer WJ. Immunofluorescence localization of the Na-Ca exchanger in heart cells. Am J Physiol. 1992;263:C545–C550. doi: 10.1152/ajpcell.1992.263.2.C545. [DOI] [PubMed] [Google Scholar]
- 30.Yang Z, Pascarel C, Steele DS, Komukai K, Brette F, Orchard CH. Na+-Ca2+ exchange activity is localized in the T-tubules of rat ventricular myocytes. Circ Res. 2002;91:315–322. doi: 10.1161/01.res.0000030180.06028.23. [DOI] [PubMed] [Google Scholar]
- 31.Scriven DR, Dan P, Moore ED. Distribution of proteins implicated in excitation-contraction coupling in rat ventricular myocytes. Biophys J. 2000;79:2682–2691. doi: 10.1016/S0006-3495(00)76506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adachi-Akahane S, Cleemann L, Morad M. Cross-signaling between L-type Ca2+ channels and ryanodine receptors in rat ventricular myocytes. J Gen Physiol. 1996;108:435–454. doi: 10.1085/jgp.108.5.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lopez-Lopez JR, Shacklock PS, Balke CW, Wier WG. Local calcium transients triggered by single L-type calcium channel currents in cardiac cells. Science. 1995;268:1042–1045. doi: 10.1126/science.7754383. [DOI] [PubMed] [Google Scholar]
- 34.Lederer WJ, Niggli E, Hadley RW. Sodium-calcium exchange in excitable cells: fuzzy space. Science. 1990;248:283. doi: 10.1126/science.2326638. [DOI] [PubMed] [Google Scholar]
- 35.Leblanc N, Hume JR. Sodium current-induced release of calcium from cardiac sarcoplasmic reticulum. Science. 1990;248:372–376. doi: 10.1126/science.2158146. [DOI] [PubMed] [Google Scholar]
- 36.Sipido KR, Maes M, Van de Werf F. Low efficiency of Ca2+ entry through the Na+-Ca2+ exchanger as trigger for Ca2+ release from the sarcoplasmic reticulum: a comparison between L-type Ca2+ current and reverse-mode Na+-Ca2+ exchange. Circ Res. 1997;81:1034–1044. doi: 10.1161/01.res.81.6.1034. [DOI] [PubMed] [Google Scholar]
- 37.Convery MK, Hancox JC. Comparison of Na+-Ca2+ exchange current elicited from isolated rabbit ventricular myocytes by voltage ramp and step protocols. Pflugers Arch. 1999;437:944–954. doi: 10.1007/s004240050866. [DOI] [PubMed] [Google Scholar]
- 38.Dipla K, Mattiello JA, Margulies KB, Jeevanandam V, Houser SR. The sarcoplasmic reticulum and the Na+/Ca2+ exchanger both contribute to the Ca2+ transient of failing human ventricular myocytes. Circ Res. 1999;84:435–444. doi: 10.1161/01.res.84.4.435. [DOI] [PubMed] [Google Scholar]