

Abstract
Tuberculosis (TB), caused by Mycobacterium tuberculosis (MTB), remains a significant global health challenge. Recent advancements in gut microbiota (GM) research have shed light on the intricate relationship between GM and TB, suggesting that GM alterations may influence host susceptibility, disease progression, and response to antituberculosis drugs. This review systematically synthesizes and analyzes the current research progress on the relationship between GM and TB, focusing on six key aspects: (1) bidirectional effects between GM dynamics and TB progression; (2) the interaction between GM and anti‐TB drugs; (3) GM and TB immune response; (4) GM as a potential target for diagnosis and treatment of TB; (5) multi‐omics and artificial intelligence (AI) technologies in GM‐TB research; (6) current challenges and future directions in GM‐TB research. We highlight the bidirectional nature of the GM–TB interaction, where MTB infection can lead to GM dysbiosis, and changes can affect the host's immune response, contributing to TB onset and progression. Advanced molecular techniques, such as next‐generation sequencing and metagenomics, along with AI, play pivotal roles in elucidating these complex interactions. Future research directions include investigating the relationship between GM and TB vaccine efficacy, exploring GM's potential in TB prevention, developing microbiome‐based diagnostic and prognostic tools, and examining the role of GM in TB recurrence. By addressing these areas, we aim to provide a comprehensive perspective on the latest advancements in GM and TB research and offer insights for future studies and clinical applications. Ultimately, the development of novel microbiome‐based strategies may offer new tools and insights for the effective control and management of TB, a disease that continues to pose a significant threat to public health.
Keywords: artificial intelligence, gut microbiota, microbiome‐based diagnostics, Mycobacterium tuberculosis, omics technologies, tuberculosis
This review explores the intricate relationship between gut microbiota (GM) and tuberculosis (TB). The figure illustrates the bidirectional effects between GM dynamics and TB progression, as well as the interactions between the GM and antituberculosis drugs. It also explores the role of GM in the immune response to TB and its potential as a target for the diagnosis and treatment of TB. Finally, the application of multi‐omics and artificial intelligence (AI) technologies in GM‐TB research is highlighted. The review aims to shed light on the complex interplay between GM and TB, paving the way for innovative strategies in TB management.

Highlights
Bidirectional gut microbiota (GM)–tuberculosis (TB) interaction reveals a dynamic interplay where Mycobacterium tuberculosis infection disrupts GM, while GM dysbiosis exacerbates TB progression by modulating host immunity.
Technological innovation integrates next‐generation sequencing, metagenomics, and artificial intelligence (AI) to unravel complex GM‐TB relationships, enabling predictive modeling and precision medicine approaches.
Regarding GM as a diagnostic/therapeutic target, researchers propose GM modulation as a novel strategy to enhance anti‐TB drug efficacy, mitigate side effects, and develop microbiome‐based diagnostics for TB susceptibility and prognosis.
A comprehensive research framework systematically synthesizes six key areas—from GM‐immune crosstalk to recurrence mechanisms—providing a roadmap for future TB management strategies and vaccine optimization.
INTRODUCTION
Tuberculosis (TB), caused by Mycobacterium tuberculosis (MTB), remains a formidable global health challenge despite decades of control efforts [1, 2]. As the leading infectious killer second only to Corona Virus Disease 2019, TB claimed 1.25 million lives in 2023, with 10.8 million new cases reported worldwide [3]. This persistent burden stems from complex host–pathogen interactions and socioeconomic determinants, necessitating novel therapeutic approaches [4, 5].
The gut microbiota (GM) has emerged as a pivotal regulator of systemic immunity, comprising >100 trillion microorganisms that interact with host physiology through metabolic cross‐talk and immune modulation [6, 7]. Beyond its established roles in metabolic disorders and gastrointestinal diseases [8, 9, 10], GM dysbiosis is increasingly implicated in respiratory infections through the gut–lung axis—a bidirectional communication network linking intestinal microbiota with pulmonary immunity [11, 12, 13].
Emerging evidence reveals intricate GM‐TB interplay: (1) Preclinical models demonstrate that GM‐derived metabolites like butyrate enhance macrophage antimicrobial responses via histone deacetylase inhibition [14]; (2) Clinical studies document characteristic GM alterations in TB patients, including reduced Faecalibacterium and elevated Enterobacteriaceae [14, 15, 16, 17]; (3) Anti‐TB chemotherapy induces prolonged GM perturbations, potentially compromising treatment efficacy through immunometabolic disruption [18, 19, 20, 21, 22]. Notably, Mendelian randomization analysis suggests GM composition may causally influence TB susceptibility, with specific taxa exhibiting protective or risk‐enhancing effects [23].
This evolving paradigm raises critical questions: Could GM modulation enhance the efficacy of the Bacille Calmette‐Guérin (BCG) vaccine, a widely used vaccine for TB? How do geographically distinct GM profiles influence TB epidemiology? What biomarkers predict treatment‐induced dysbiosis? Addressing these questions requires integrating multi‐omics approaches with clinical validation—a gap this review seeks to highlight.
We systematically evaluate six key aspects of GM–TB interactions: (1) bidirectional effects between GM dynamics and TB progression; (2) the interaction between GM and anti‐TB drugs (ATD); (3) GM and TB immune response; (4) GM as a potential target for diagnosis and treatment of TB; (5) multi‐omics and artificial intelligence (AI) technologies in GM‐TB research; (6) current challenges and future directions in GM‐TB research. Through an in‐depth exploration of these aspects, we hope to provide readers with a comprehensive perspective on the latest advancements in this emerging research field of GM and TB, as well as offer ideas and insights for future research and clinical applications. As our understanding of the relationship between GM and TB deepens, we anticipate the development of novel microbiome‐based diagnostic, therapeutic, and preventive strategies, providing new weapons for the control of TB.
BIDIRECTIONAL EFFECTS BETWEEN GM DYNAMICS AND TB PROGRESSION
The systemic impact of TB extends beyond pulmonary manifestations, with growing evidence revealing profound GM alterations in TB patients compared to healthy individuals. These compositional changes encompass microbial diversity loss, phylum‐level restructuring, and functional metabolic shifts, which not only emerge as potential biomarkers for disease monitoring and therapeutic targets, but also drive disease progression through immuno–metabolic interactions (Table 1) [16, 24, 25, 26, 27, 28, 29, 30, 31]. The observed GM perturbations in TB patients are characterized by three interconnected phenomena: reduced microbial diversity, specific taxonomic alterations, and bidirectional interactions with host immunity (Figure 1). This section systematically explores the impact of GM on the immune response and barrier function, and illustrates how alterations in GM composition influence the progression of TB. As shown in Figure 2, the mechanisms underlying the bidirectional interactions between GM and TB are also highlighted, showcasing the role of both health‐promoting and pathogenic bacteria in disease progression [15, 32, 33].
TABLE 1.
Overview of changes in GM composition in TB patients and animal models.
| Year* | Species (Sample size) | Analysis methods | Diversity changes | Bacterial changes | Fungal changes | Possible impacts | Refs |
|---|---|---|---|---|---|---|---|
| 2024 | Human (n = 71, including 23 naive TB patients, 48 HCs) | Deep shotgun sequencing; Illumina HiSeq. 2500 platform; Metagenomic analysis | No significant change in α diversity, increase in β diversity | Increase in Actinomycetota, Bacillota, and Pseudomonadota; decrease in Bacteroidota | Unknown | Increased metabolic pathways related to cell division and growth affect gut health. | [16] |
| 2023 | Human (n = 53, including 33 TB patients, 20 HCs) | 16S rRNA sequencing (V3–V4 region); ITS gene sequencing; MiSeq PE300 platform; ASVs | Significant reduction in α diversity | Significant increase in Bacteroides and Prevotella; decrease in Blautia and Bifidobacterium | Increase in Ascomycota; decrease in Basidiomycota | May influence disease pathogenesis or pathology; potential auxiliary strategy for TB treatment | [24] |
| 2022 | Human (n = 135, including 83 PTB patients, 52 HCs) | 16S rRNA sequencing (V3–V4 region); Ion S5TMXL platform; OTUs | Significant reduction in α diversity | Increase in Bacteroidaceae, Tannerellaceae, Fusobacteriaceae, Erysipelotrichaceae; decrease in Bifidobacteriaceae, Lachnospiraceae, Ruminococcaceae, Marinifilaceae, Eggerthellaceae, Barnesiellaceae | Unknown | Changes in GM structure affecting SCFA production | [25] |
| 2022 | Human (n = 142, including 56 PTB patients, 36 LTBI, 50 HCs) | 16S rRNA sequencing (V3‐V4 region); Illumina NovaSeq platform; ASVs | Significant reduction in α diversity | Decrease in Bacillota; increase in Bacteroidota | Unknown | Immune dysregulation leading to impaired immune response against TB | [26] |
| 2019 | Human (n = 77, including 46 TB patients, 31 HCs) | Shotgun metagenomic sequencing; Illumina HiSeq. 2500; Species‐level resolution | Significant reduction in α diversity and abundance | Decrease in Bacillota and Bacteroidota; increase in Pseudomonadota | Unknown | Potential to distinguish TB patients from non‐TB patients | [27] |
| 2018 | Human (n = 24, including 6 HCs, 6 TBZ, 6 TBW, 6 TBM) | 16S rRNA sequencing (V3 region); Whole‐genome shotgun sequencing; Illumina HiSeq. 2000 and NextSeq. 500 platforms; OTUs and KOs | Higher α diversity and species richness | Decrease in Prevotella, Faecalibacterium, Roseburia, Eubacterium, and Bacteroidota; increase in Bacillota | Unknown | Reduced SCFA production with enriched propionate‐ and butyrate‐producing bacteria leads to impaired host immune response | [28] |
| 2017 | Mouse (n = 3–5/group, group = 11) | 16S rRNA sequencing (V3–V4 region); Illumina MiSeq platform; ASVs | Temporary reduction in diversity but significant and persistent structural changes | Changes in Clostridiales, Bacteroidota, and Tenericutes | Unknown | Structural changes in GM affecting host disease resistance and immune function | [29] |
| 2017 | Human (n = 57, including 19 new TB patients, 18 recurrent TB patients, 20 HCs) | 16S rRNA sequencing (V3–V4 region); Illumina MiSeq platform; OTUs | Increase in α diversity | Decrease in Prevotella; increase in Escherichia and Streptococcus | Unknown | It affects immune status and correlates with patient prognosis and outcomes | [30] |
| 2014 | Mouse (n = 5/group, group = 4) | 16S rRNA sequencing (V1‐V2 region); 454 FLX pyrosequencing platform; OTUs | Initial decrease in diversity followed by recovery | Decrease in Lachnospiraceae and Ruminococcaceae | Unknown | Cross‐talk between resident microbiota and mucosal immune system | [31] |
Abbreviations: ASVs, amplicon sequence variants; GM, gut microbiota; HCs, healthy controls; ITS, internal transcribed spacer; KO, KEGG Orthology; LTBI, latent TB infection; OTUs, operational taxonomic units; PTB, pulmonary tuberculosis; Refs, references; SCFA, short‐chain fatty acid; TB, tuberculosis; TBM, tuberculous meningitis; TBW, TB patients after week treatment; TBZ, TB patients at day zero; *Year, the year the study was published.
FIGURE 1.

Relationship between gut microbiota (GM) dysbiosis and tuberculosis (TB) progression. The figure illustrates the association between GM dysbiosis and TB pathogenesis. Dysbiosis is characterized by a loss of biodiversity, particularly a depletion of key short‐chain fatty acid (SCFA)‐producing taxa such as Blautia spp., Roseburia spp., Ruminococcus spp., Bifidobacterium spp., and Eubacterium spp. This is accompanied by an overrepresentation of potentially pathogenic genera, including Bacteroides spp., Prevotella spp., Enterococcus spp., and Fusobacterium spp. These compositional shifts contribute to increased intestinal permeability, disruption of mucosal immune homeostasis, and impaired host immune responses against Mycobacterium tuberculosis, ultimately facilitating disease progression. ATD, antituberculosis drugs.
FIGURE 2.

Comparative characterization of healthy versus pathogenic gut microbiota states. This figure highlights the contrasting features of a healthy versus dysbiotic gut microbiota. In healthy state, commensal bacteria such as Lactobacilli and Bifidobacteria reinforce intestinal barrier function through the production of bacteriocins and short‐chain fatty acids (SCFAs), particularly butyrate. These microbes preferentially colonize the mucosal layer, prevent pathogenic colonization, and modulate host immunity by promoting dendritic cell migration and the release of gut‐derived hormones. In pathogenic state, microbial diversity is reduced (1), with a concomitant overrepresentation of mucin‐degrading bacteria (2) and a loss of beneficial SCFA‐producing species (3). This altered microbial composition compromises intestinal epithelial integrity and enables translocation of pathogens such as Mycobacterium tuberculosis (MTB), which contribute to tissue damage via toxin‐mediated mechanisms (4). The reduction in SCFA‐producing bacteria and rise in pathogenic Enterobacteriaceae promote immune dysregulation through lipopolysaccharides (LPS)‐mediated activation of Toll‐like receptor 4 (TLR4) and G‐protein signaling. This cascade triggers a chronic inflammatory response characterized by elevated cytokine production, neutrophil degranulation, and impaired regulatory T cell function, ultimately disrupting immune homeostasis.
Reduced microbial diversity and TB progression
The GM of TB patients exhibits marked biodiversity loss compared to healthy individuals. Metagenomic sequencing of 46 TB patients and 31 controls revealed significant reductions in α‐diversity indices (species richness and abundance), with Haemophilus parainfluenzae, Roseburia inulinivorans, and Roseburia hominis depletion forming a diagnostic signature (AUC = 0.846) [27]. This finding is corroborated by longitudinal studies showing persistent GM structural changes posttreatment [34]. The significantly reduced GM diversity is not only directly related to the severity and progression of the disease, but also closely associated with the treatment effect. Clinical studies have further confirmed that patients with low GM diversity have an increased risk of treatment failure and a faster disease progression rate [35, 36]. Mechanistically, diminished diversity compromises gut barrier integrity, increasing permeability for MTB metabolites to trigger systemic inflammation [22]. The clinical relevance of diversity loss is further evidenced by reduced short‐chain fatty acids (SCFAs)‐producing genera (such as Faecalibacterium, Roseburia, Eubacterium, and Phascolarctobacterium). These genera are critical for maintaining immune homeostasis through butyrate‐mediated macrophage regulation [28], and their reduction impair macrophage bactericidal activity and antimicrobial peptide synthesis [37].
The GM of healthy individuals produces SCFAs and other metabolites that maintain the integrity of the gut epithelial barrier and promote immune tolerance [38]. In contrast, GM imbalance in TB patients, such as reducing Lactobacilli and Bifidobacteria, can impair gut barrier function [39]. This impairment increases gut permeability, allowing pathogens and inflammatory mediators to enter the bloodstream more easily and triggering systemic inflammation [22]. This “leaky gut” phenomenon can activate the innate immune system, increasing the production of inflammatory cytokines such as tumor necrosis factor‐α (TNF‐α) and interleukin‐6 (IL‐6), which inhibit macrophage antibacterial activity and enhance susceptibility to MTB [40, 41]. For example, Madeleine R. Wood et al. found that the increase in specific microbial communities in the gut of TB patients, such as Prevotella and Veillonella, are associated with disease progression and poor prognosis [42].
The systemic inflammatory response and metabolic changes triggered by MTB infection can also further exacerbate the loss of GM diversity through immune and neuroendocrine pathways, forming a vicious cycle. A large number of pro‐inflammatory cytokines released by the chronic inflammation caused by MTB infection, such as interferon‐γ (IFN‐γ) and IL‐17, can directly or indirectly affect the composition of GM, further reducing the types and quantities of microorganisms that have already decreased due to TB [32, 43]. These findings indicate that the GM ecosystem balance in TB patients is disrupted, potentially exerting profound effects on the host's immune function.
Changes in GM composition and TB progression
MTB infection induces characteristic restructuring of GM at various taxonomic levels. Proteobacteria (particularly Enterobacteriaceae) and Bacillota show marked expansion, while Bacteroidota (especially Prevotella) and Actinobacteria (including Bifidobacterium) populations decline [16, 25, 28]. Parallel alterations are observed in fungal communities, with Ascomycota dominance replacing Basidiomycota [24]. At the genus level, the abundance of Bacteroides and Prevotella increases, whereas Blautia and Bifidobacterium decrease in TB patients [24]. In terms of specific species, Phocaeicola dorei, Escherichia coli, Prevotella copri clade C, and Akkermansia muciniphila are enriched in TB patients, while Phocaeicola vulgatus, Alistipes putredinis, Prevotella copri clade B, and Prevotella SGB1589 are more abundant in healthy controls [16, 25, 28]. Additionally, in TB patients, the abundance of Saccharomyces increases while Aspergillus decreases [24]. Notably, Escherichia and Streptococcus enrichment correlates with impaired immune status, whereas Lachnospiraceae depletion is associated with disrupted gut barrier function [25, 30]. These compositional shifts may establish a pro‐inflammatory milieu through multiple pathways: (1) Reduced mucin‐degrading Prevotella compromises intestinal epithelial integrity; (2) Enterobacteriaceae overgrowth promotes lipopolysaccharide (LPS)‐mediated Toll‐like receptor (TLR) 4 activation; (3) Bifidobacterium deficiency attenuates regulatory T cell (Tregs) responses.
These changes in GM also have a reverse effect on the infection process of MTB. For example, the alterations in the intestinal microenvironment caused by the decrease of Prevotella and the excessive proliferation of Enterobacteriaceae make it easier for MTB to breach the intestinal barrier and enter the bloodstream, thereby exacerbating the systemic infection. Moreover, the systemic inflammation caused by the infection of MTB will continuously disrupt the composition of the GM. For instance, it will further inhibit the growth of beneficial bacteria such as Bifidobacterium, reinforcing this vicious cycle [2, 30, 44].
Decrease in SCFA‐producing bacteria and mutualistic populations and TB progression
SCFA‐producing bacteria, including Faecalibacterium, Roseburia, and Eubacterium, are markedly reduced in TB patients, paralleled by Bacteroidota depletion and Bacillota expansion [28]. These metabolic shifts critically impact host immunity, as SCFAs (acetate, propionate, butyrate) typically enhance anti‐inflammatory responses in monocytes and macrophages through G protein‐coupled receptor activation [45]. Namasivayam et al. identified butyrate reduction as a key severity marker, with levels inversely correlating with cavitary lesion development [29]. Li et al. further elucidated that SCFA deficiency disrupts MTB clearance mechanisms by attenuating immune regulation pathways [45]. The collective depletion of SCFA producers creates a dysbiotic environment favoring MTB persistence and systemic inflammation. Collectively, these findings suggest that the decrease in SCFA‐producing bacteria may significantly affect TB progression.
The GM influences TB progression through immunometabolic crosstalk and systemic immune modulation. Segal and Lachmandas et al. identified SCFA‐mediated suppression of IFN‐γ and IL‐17A as a critical susceptibility mechanism, impairing T helper cell (Th)1/Th17 responses essential for MTB containment [46, 47]. Yu et al. highlighted gut–lung axis interactions, where Bacillota reduction and Pseudomonadota expansion alter pulmonary microbiota and immune dynamics [48]. Khan et al. further demonstrated that GM dysbiosis skews Th17 differentiation, compromising alveolar macrophage function and perpetuating MTB survival [49]. Mayer‐Barber's review synthesizes these mechanisms, emphasizing GM's role in regulating inflammatory cytokine cascades and granuloma formation [50]. Collectively, these insights underscore GM modulation as a strategic target for adjunctive TB therapies.
Commensal species such as Lactobacillus and Bifidobacterium are significantly diminished in TB patients, weakening gut defense mechanisms against MTB invasion [30]. Yang et al. revealed that Bacteroides fragilis enhances anti‐TB immunity via long noncoding RNA regulation, while broader mutualistic depletion compromises pathogen exclusion through reduced lactic acid and hydrogen peroxide production [51, 52]. This loss impairs dendritic cell antigen presentation and effector T cell activation, critical for adaptive immunity [53, 54]. Additionally, mutualistic bacteria reduction elevates luminal pH, facilitating pathogen colonization and systemic dissemination of MTB through compromised mucosal barriers [55].
In addition, during MTB infection, changes in the host's nutritional status and metabolic state, such as malabsorption and metabolic disorders, will significantly reduce the quantity and function of SCFA‐producing bacteria and symbiotic flora. The decrease in the quantity of these flora, in turn, weakens the intestine's ability to resist MTB, promoting the growth and spread of MTB, thus forming a relationship of mutual influence [56]. Thus, this reduction in probiotic populations may lead to impaired adaptive immune responses, exacerbating the progression of TB.
Increase in pathogenic bacteria and TB progression
TB progression is associated with pathogenic bacterial expansion, including Pseudomonadota and Actinomycetota in sputum and fecal samples [30, 57]. Cheung et al. observed phylum‐level shifts favoring Pseudomonadota/Bacteroidota in TB patients versus Bacillota dominance in controls [57]. Pathobiont overgrowth exacerbates disease through toxin‐mediated gut epithelial damage, niche competition suppressing beneficial flora, and gut–lung axis interactions amplifying pulmonary inflammation [58, 59]. For instance, Luo et al. linked Streptococcus enrichment to recurrent TB, while Dumas et al. demonstrated GM modulation of pulmonary immunity via bone marrow‐derived immune cell reprogramming [30, 60]. These pathogens further disrupt immune homeostasis through LPS‐driven TLR4 activation and bile acid metabolism alteration [32, 44].
Moreover, the changes in the immune response triggered by MTB infection will also create a more favorable environment for the proliferation of pathogenic bacteria. For example, the disruption of immune homeostasis caused by the infection allows the pathogenic bacteria that were originally suppressed in the intestine to multiply in large numbers. The increase in the number of pathogenic bacteria, through various pathways such as toxin‐mediated damage to the intestinal epithelium and alteration of the function of immune cells, further promotes the infection of MTB and the progression of the disease [58, 59]. These findings reveal the close relationship between GM and TB progression, providing new insights for disease prognosis assessment and personalized treatment.
THE INTERACTION BETWEEN GM AND ATD
The interplay between GM and TB treatment has emerged as a critical research focus, driven by its dual role in modulating host immunity and interacting with ATD. Emerging evidence indicates that GM not only responds to ATD exposure but also actively influences drug metabolism and treatment outcomes. Additionally, ATD‐induced GM alterations may contribute to adverse drug reactions. These insights highlight GM as a potential target for optimizing TB therapeutics through improved efficacy and reduced toxicity. This section systematically examines three key aspects of GM–TB interactions: (1) ATD‐induced GM dysbiosis, (2) GM‐mediated modulation of ATD pharmacokinetics/pharmacodynamics, and (3) the mechanism of interaction between GM and ATD (Table 2) [18, 29, 49, 61, 62, 63, 64, 65, 66, 67]. Elucidating these mechanisms could enable personalized treatment strategies to enhance clinical outcomes.
TABLE 2.
Effects of anti‐TB treatment on GM.
| Refs | Year | Drugs | Species (Sample size) | Analysis method | Changes in GM | Impact or significance |
|---|---|---|---|---|---|---|
| [61] | 2024 | HRZE | Human (n = 24, including 5 naive TB patients, 6 DS‐TB, 10 DR‐TB‐inj–, 3 DR‐TB‐inj+) | 16S rRNA sequencing (V3–V4 region); Illumina platform; OTUs/ASVs | Reduced α diversity; decrease in Collinsella, Bacillota, and Prevotella; increase in Lactobacillus (Bacillota) | May influence TB progression and treatment outcomes |
| [62] | 2023 | INH | Mouse (n = 7/group, group = 10) | 16S rRNA sequencing (V3–V4 region); Illumina NovaSeq platform; ASVs | Significant increase in α diversity; increase in Muribaculaceae and Bifidobacterium; decrease in Bifidobacteria | Impairment of the immune system, increased risk of liver damage |
| [63] | 2023 | HRZ | Mouse (n = 5–7/group, group = 4) | HPLC/MS‐MS | Rapid and significant changes in GM composition | Affects the pharmacokinetics of certain TB antibiotics |
| [18] | 2021 | INH | Human (n = 99, including 29 naive TB patients, 29 TB2MT, 19 TB6MT, 22 HCs) | 16S rRNA sequencing (V3–V4 region); ITS2; Illumina HiSeq. 2500 platform; ASVs | Reduced diversity of bacteria and fungi, with altered composition; dysbiosis characterized by a decrease in beneficial bacterial and fungal overgrowth; increase in Bacteroides, decrease in Actinomycetota and Pseudomonadota | Increased susceptibility to TB and limitation in INH‐mediated MTB clearance |
| [64] | 2020 | INH | Mouse (n = 4–5/group, group = 6) | 16S rRNA sequencing (V3–V4 region); Illumina MiSeq platform; ASVs | Decrease in beneficial symbiotic bacteria; increase in Bifidobacterium and Enterococcus; decrease in Bacteroides, Campylobacter, and Lactobacillus | Impairment of T cell activation, proliferation, and memory T cell formation, weakening the immune response to INH treatment |
| [65] | 2019 | HRZ | Human (n = 84, including 13 HCs, 10 latent TB, 28 active TB, 13 TB with 1‐week therapy, 10 TB with 2‐week therapy, 10 cured TB) | qRT‐PCR | Significant reduction in α diversity; decrease in Bacillota, increase in Bacteroides | Risk factors for TB progression and prognosis |
| [49] | 2019 | HZ/R | Mouse (n = 5/group, group = 5) | 16S rRNA sequencing (V4 region); Illumina HiSeq. 2500 platform; OTUs | HZ: Increase in Bacteroidota; R: Decrease in Bacillota, increase in Verrucomicrobia and Bacteroidota | Changes in GM metabolites in peripheral circulation may affect alveolar macrophage metabolism, increasing MTB burden, and negatively influencing macrophage immunity against MTB |
| [29] | 2017 | HRZ | Mouse (n = 4–5/group, group = 11) | 16S rRNA sequencing (V3–V4 region); Illumina MiSeq platform; ASVs | Significant reduction in diversity; decrease in Bacillota, increase in Erysipeloclostridium | Affects host resistance to disease and immune function |
| [66] | 2017 | HRZE | Human (n = 120, including 50 uninfected controls, 19 treatment, 25 LTBI controls, 26 cured controls) | 16S rRNA sequencing (V3–V4 region); Illumina MiSeq platform; ASVs; Metagenomic sequencing; Hiseq. 4000; Whole metagenome data | Decrease in Ruminococcus, Eubacterium, Lactobacillus, and Bacteroides; increase in Erysipeloclostridium and Prevotella | Microbiota disturbance and variability in peripheral immunity may affect the efficacy of TB treatment |
| [67] | 2016 | Broad‐spectrum antibiotics | Mouse (n = 4–5/group, group = 5) | Cultivable microbes (plating on different media); qRT‐PCR for specific bacterial species; ASVs | Reduced diversity; increase in Enterococcus; decrease in Bifidobacterium, Lactobacillus, Campylobacter, and Bacteroides | It promotes the growth of MTB in the lungs and facilitates its spread to other organs. |
Abbreviations: DR‐TB‐inj–, drug‐resistant tuberculosis treated without injectable agents; DR‐TB‐inj+, drug‐resistant tuberculosis treated with injectable agents; EMB, ethambutol; HPLC/MS‐MS, high‐performance liquid chromatography/tandem mass spectrometry; HRZE, Isoniazid (INH, H), Rifampicin (RIF, R), Pyrazinamide (PZA, Z), Ethambutol (EMB, E); MTB, Mycobacterium tuberculosis; qRT‐PCR, quantitative reverse transcription polymerase chain reaction; TB2MT, TB patients after 2 months of treatment; TB6MT, TB patients after 6 months of treatment.
Impact of ATD on GM
As the therapeutic mainstay for TB, ATD exerts broad‐spectrum effects extending beyond MTB to disrupt host GM homeostasis. Key mechanisms include: (1) reduced microbial diversity, (2) depletion of beneficial taxa (e.g., SCFA‐producing bacteria), (3) promotion of antibiotic resistance, (4) gut–immune axis dysregulation, and (5) secondary complications [68, 69]. Wipperman et al. demonstrated that standard ATD regimens significantly diminish GM diversity, particularly affecting Roseburia and Faecalibacterium populations, which correlated with immune variability and potential treatment efficacy reduction [66]. Subsequent work by the same group linked ATD‐driven GM changes to heightened inflammatory responses in TB patients, underscoring the clinical relevance of microbial balance [70]. Longitudinal analyses by Namasivayam et al. revealed persistent dysbiosis patterns during TB treatment, characterized by decreased Bacteroidota and increased Pseudomonadota abundance, potentially compromising host immunity and disease resistance [29]. Importantly, Khan et al. identified functional consequences of this dysbiosis, showing that impaired butyrate production weakens anti‐MTB immune responses [49]. These collective findings position GM integrity as a critical determinant of TB treatment success.
Influence of GM on ATD metabolism
The GM‐ATD relationship exhibits bidirectionality, with GM actively participating in drug absorption, distribution, metabolism, and excretion processes [71, 72, 73]. Clinical studies demonstrate that GM metabolic activity can modify drug bioavailability and toxicity profiles. Maji et al. identified specific bacterial taxa involved in rifampicin (RIF) metabolism, directly influencing drug absorption dynamics [28]. Negi et al. further established that GM disruption impairs isoniazid (INH)‐mediated MTB clearance while exacerbating tissue pathology, suggesting microbiome‐dependent treatment responses [64], Certain supportive therapeutic applications can reduce the toxic and side effects of ATD. For example, Li et al. demonstrated that Lactobacillus casei supplementation alleviates ATD‐induced gastrointestinal toxicity in murine models via SCFA regulation [45]. These findings emphasize the need for microbiome‐informed dosing strategies in TB management.
The mechanism of interaction between GM and ATD
The interaction mechanism between GM and ATD is complex and diverse, involving physiological and biochemical processes at multiple levels. Next‐Generation Sequencing (NGS)‐driven pharmacomicrobiomics reveals GM's role in anti‐TB drug efficacy and toxicity. For example, microbial β‐glucuronidase activity reduces INH bioavailability through enzymatic inactivation, whereas RIF‐induced dysbiosis alters bile acid metabolism, increasing the risks of hepatotoxicity. Functional metagenomic analyses identify microbial pathways capable of detoxifying drug metabolites, suggesting potential targets for adjunctive therapies [40, 74, 75]. These findings align with Doestzada et al.'s proposal that GM can alter drug pharmacodynamics through both direct enzymatic modification and indirect immune or metabolic modulation, potentially explaining interindividual variability in ATD efficacy and tolerability [76]. This section highlights the multifaceted nature of GM–ATD interactions and sets the stage for a detailed examination of the specific mechanisms involved (Figure 3).
FIGURE 3.

Bidirectional interactions between anti‐TB drugs (ATD) and gut microbiota (GM), and their impact on GM homeostasis. This figure illustrates: (1) The detrimental effects of ATD on GM compositions and functions, mediated through multiple mechanisms including the induction of intestinal inflammation, disruption of epithelial barrier integrity, metabolic perturbations, and direct bactericidal activity. (2) How GM dysbiosis reciprocally modulates the pharmacodynamics and toxicity of ATD, through mechanisms including altered drug metabolism, impaired immune modulation, reduced drug bioavailability, enhanced hepatotoxicity, and the facilitation of antimicrobial resistance. (3) Emerging therapeutic strategies aimed at restoring GM homeostasis, such as probiotics supplementation and administration of magnesium isoglycyrrhizinate, which may attenuate inflammation, promote barrier repair, and re‐establish microbial balance. Overall, this figure highlights the complex, bidirectional crosstalk between the GM and ATD, and suggests the potential of microbiota‐targeted interventions to optimize antituberculosis therapy. FMT, fecal microbiota transplantation. MgIG, magnesium isoglycyrrhizinate.
Mechanisms of ATD‐induced GM dysbiosis
Recent studies have elucidated the mechanisms by which ATD induces GM dysbiosis. For instance, Wu et al. [77] demonstrated that second‐line anti‐TB drugs used in the treatment of rifampicin‐resistant TB (RR‐TB) significantly alter the structural composition of the intestinal microbiota. Specifically, the relative abundance of beneficial species such as Prevotella copri decreases, while potentially pathogenic species like Escherichia coli and Salmonella enterica increase. This shift disrupts the gut's immune homeostasis and impairs the host's ability to mount an effective immune response against MTB. Functional analysis revealed that the biosyntheses of phenylalanine, tyrosine, and tryptophan are significantly inhibited during treatment, affecting the production of essential metabolites and neurotransmitters crucial for maintaining gut health and immune function.
Pei et al. [78] further explored the GM characteristics in TB patients experiencing liver injury following anti‐TB treatment. The study found that anti‐TB treatment leads to decreased microbial diversity and significant structural changes in the GM. At different time points, distinct differences in microbial composition were observed between patients with and without liver injury. These findings suggest that specific alterations in the GM may contribute to drug‐induced liver injury (DILI) in TB patients, potentially through modulating the host's immune and inflammatory responses.
Mechanisms of GM modulation of ATD pharmacokinetics/pharmacodynamics
New research has shed light on how GM influences ATD metabolism. Gong et al. [79] investigated the protective effect of magnesium isoglycyrrhizinate (MgIG) against anti‐TB DILI in mice. The study showed that MgIG significantly ameliorated HRZE‐induced liver injury by modifying the GM composition. Specifically, MgIG increased the abundance of beneficial bacteria such as Lactobacillus and enhanced the expression of tight junction proteins, improving intestinal barrier function and reducing intestinal permeability. This, in turn, decreased the levels of LPS and inhibited the activation of the LPS/TLRs/NF‐κB signaling pathway, which is associated with inflammation and liver injury. These findings highlight the role of GM in modulating the gut–liver axis and immune responses, thereby influencing the pharmacodynamics of ATD. Pei et al. [78] also showed that different anti‐TB drugs or drug combinations induce distinct GM profiles in mice models of liver injury. Each drug or combination causes dysbiosis characterized by unique changes in bacterial genera, which may affect the metabolism and toxicity of the drugs. These findings underscore the importance of considering specific microbiota signatures when evaluating the effects of different ATD on the host.
In summary, the mechanisms underlying the interaction between GM and ATD are multifaceted. ATD can induce dysbiosis through altering microbial composition and function, while GM can modulate ATD pharmacokinetics/pharmacodynamics by affecting drug metabolism and host immune responses. Understanding these mechanisms is essential for developing microbiome‐targeted interventions to optimize TB treatment efficacy and safety.
GM AND TB IMMUNE RESPONSE
The host immune system serves as a critical defense mechanism against pathogenic invasion, functioning as the body's primary protective framework [80, 81, 82, 83]. Advances in metabolomics have highlighted the bidirectional interactions between GM and immune regulation, revealing GM's capacity to modulate host immune responses both locally and systemically—though precise mechanistic pathways remain incompletely characterized [84, 85]. These intricate GM–immune interactions undeniably influence TB pathogenesis and progression through multiple pathways. In addition, recent mechanistic studies have demonstrated that alterations in specific gut microbial taxa can directly affect the magnitude and quality of the immune response to MTB, thereby modifying disease progression [86].
Emerging evidence demonstrates three principal mechanisms through which GM modulates anti‐MTB immune responses: (1) direct immunocyte regulation, (2) metabolite‐mediated immune modulation, and (3) lung microenvironment alteration via the gut–lung axis. Elucidating these interactions provides critical insights into TB pathogenesis while informing novel preventive and therapeutic strategies. This section systematically examines GM‐immune interplay through four dimensions: innate immunity, adaptive immunity, gut–lung axis dynamics, and immunocyte functional regulation (Figure 4). Collectively, these advances offer transformative perspectives for TB management.
FIGURE 4.

Multimodal regulation of host immunity by the gut microbiota (GM). The GM modulates the host immune system through multiple pathways. First, microbial components are recognized by pattern recognition receptors (e.g., TLRs) expressed on innate immune cells, activating downstream signaling cascades such as the phosphoinositide 3‐kinase (PI3K) pathway. This leads to the inactivation of glycogen synthase kinase 3β (GSK‐3β) and subsequent activation of cyclic adenosine monophosphate (cAMP) response element‐binding protein (CREB)‐dependent transcription of anti‐inflammatory genes, promoting the release of cytokines and other immunomodulatory factors. Second, microbial metabolites, such as SCFAs, bind to G protein‐coupled receptors (GPCRs) to drive the expansion of mucosal regulatory T cells (Tregs), thereby suppressing pro‐inflammatory responses. SCFAs also promote macrophage polarization through the gut–lung axis and enhance the memory function of CD8 + T cells, while stimulating B cell‐mediated secretion of immunoglobulin A (IgA), thereby strengthening mucosal and systemic immunity. In addition, specific bacteria taxa (e.g., Bacteroides, Clostridium, and Prevotella) promote the differentiation of CD4+ T cells into T helper 17 (Th17) and Tregs, contributing to the regulation of adaptive immunity. Moreover, bacterial polysaccharides can regulate T helper cell fate through activating signal transducer and activator of transcription 4 (STAT4) signaling in the presence of IL‐12 to promote Th1 differentiation, or STAT6 signaling under IL‐4 to promote Th2 responses, thus maintaining Th1/Th2 homeostasis.
GM and innate immune response
GM modulates innate immunity through multiple mechanisms, establishing a sophisticated regulatory network. Microbial components such as LPS and peptidoglycan, engage host pattern recognition receptors—including TLRs and NOD‐like receptors—expressed on macrophages, dendritic cells, and intestinal epithelia [87, 88]. Activation of pattern recognition receptors by microbe‐associated molecular patterns initiates downstream signaling cascades that regulate cytokine/chemokine production and inflammatory responses [89, 90]. Brown et al. [91] identified a TLR1/TLR2‐Dectin‐1‐mediated phosphoinositide 3‐kinase pathway activation, resulting in glycogen synthase kinase 3β inactivation and cyclic adenosine monophosphate (cAMP) response element‐binding protein‐dependent anti‐inflammatory gene expression—a critical frontline defense mechanism.
Beyond pathogen recognition, the GM regulates the innate immune response to TB through its metabolites such as SCFAs, including acetate, propionate, and butyrate. Depleted SCFA levels disrupt IL‐10 production and macrophage polarization, while increased LPS from pathobionts activate TLR4 signaling, amplifying pro‐inflammatory cascades. Secondary bile acid metabolism perturbations further contribute to macrophage dysfunction, illustrating the complex interplay between microbial metabolites and host immunity [92, 93].
GM further supports immune regulation through intestinal barrier maintenance [94]. By enhancing mucus production, tightening epithelial junctions, and inducing antimicrobial peptide secretion, GM creates physicochemical barrier against pathogen translocation [37, 95]. Barrier integrity prevents excessive inflammation and systemic immune activation, while GM dysbiosis disrupts this equilibrium—predisposing to inflammatory bowel disease, metabolic disorders, and autoimmunity [96]. These findings position GM as both a central innate immune regulator and potential therapeutic target.
Recent studies have also shown that GM can influence the function of alveolar macrophages, which are crucial for the initial defense against MTB in the lungs. For instance, Khan et al. [49] demonstrated that treatment with certain antituberculosis drugs, such as INH and pyrazinamide (PYZ), can alter the GM and subsequently impair the bactericidal activity of alveolar macrophages. This impairment is characterized by reduced expression of MHC II and decreased production of key anti‐mycobacterial cytokines like TNF‐α and IL‐1β, leading to increased susceptibility to MTB infection.
Furthermore, additional mechanistic insights reveal that GM‐derived metabolites can activate the NLRP3 inflammasome in alveolar macrophages, enhancing IL‐1β secretion, which is pivotal for early MTB control [21]. Moreover, studies have shown that GM‐derived butyrate not only modulates macrophage activity but also regulates gut mucus barrier repair via the macrophage/WNT/ERK signaling pathway, thereby indirectly influencing pulmonary immune defense against MTB [97].
GM and adaptive immune response
Emerging evidence demonstrates that GM critically influences not only innate immunity but also orchestrates adaptive immune responses. The adaptive immune system, mediated by B and T lymphocytes, establishes antigen‐specific immunological memory for long‐term protection [98, 99]. GM profoundly modulates T‐cell differentiation patterns: Bacteroides and Clostridia species induce Treg development, Bordetella pertussis enhances Th1 cell maturation, while segmented filamentous bacteria, Citrobacter rodentium, and Escherichia coli drive Th17 differentiation in murine models [100, 101, 102]. Notably, Loftfield et al. [103] demonstrated in pulmonary studies that Prevotella species activate TLR2 signaling, stimulating antigen‐presenting cells to secrete IL‐1β, IL‐6, and IL‐23, thereby promoting Th17 differentiation from CD4+ T cells.
GM‐derived metabolites significantly regulate adaptive immunity. Butyrate, crucial for combating intracellular pathogens and malignancies, enhances CD8+ T cell metabolic fitness and memory potential [104]. SCFAs promote Tregs differentiation while suppressing pro‐inflammatory cytokine production, thereby maintaining immune homeostasis [105, 106, 107, 108]. Smith et al. [109] demonstrated free fatty acid receptor 2 dependent GM‐SCFA regulation of colonic Treg populations and colitis prevention in murine models. Concurrently, SCFAs modulate B cell metabolism and antibody production, with DA Peterson et al. [110] showing that SCFA‐mediated secretory IgA responses reinforce mucosal immunity against enteric pathogens. Furthermore, GM components like polysaccharide A from Bacteroides fragilis correct systemic T cell deficiencies and Th1/Th2 imbalances, as evidenced by comparative studies in germfree versus colonized animals [111]. These intricate interactions underscore the bidirectional GM‐immune crosstalk. However, GM dysbiosis disrupts this equilibrium, characterized by Treg depletion and pathogenic Th1/Th17 expansion, potentially driving chronic inflammation and autoimmunity [112]. These findings highlight GM's pivotal role in adaptive immune homeostasis and its therapeutic implications.
In the context of TB, GM dysbiosis has been shown to affect the balance of T‐cell subsets. For example, Segal et al. [46, 47] found that increased levels of SCFAs, such as butyrate, in human immunodeficiency virus (HIV) ‐infected individuals on antiretroviral therapy (ART) were associated with higher TB susceptibility. Butyrate inhibited the production of IFN‐γ and IL‐17A, which are crucial for controlling MTB infection, while promoting the induction of FoxP3+ Tregs that can suppress protective immune responses. Similarly, Lachmandas et al. [46, 47] demonstrated that butyrate could reduce pro‐inflammatory cytokine responses to MTB in human peripheral blood mononuclear cells (PBMCs) while increasing IL‐10 production, an effect that was independent of its HDAC inhibitory activity.
Moreover, emerging data indicate that shifts in the GM composition can influence antigen‐presenting cell function, thereby modulating Th1 and Th17 responses critical for MTB clearance. Specifically, experimental models have shown that GM dysbiosis leads to attenuated IFN‐γ production by T cells—a mechanism that may underpin increased TB susceptibility [53].
GM and the gut–lung axis
The gut–lung axis, first conceptualized by Turner‐Warwick (1968) [113], represents a bidirectional immunoregulatory network connecting intestinal and pulmonary mucosal systems. Emerging evidence implicates this axis in respiratory infections, with Dumas et al. [60] proposing GM‐mediated modulation of MTB colonization through metabolite production, immune cell regulation, and pulmonary microenvironment remodeling.
GM influences pulmonary immunity through three principal mechanisms: (1) Circulating microbial metabolites (e.g., SCFAs) regulate alveolar macrophage function [60, 114]; (2) Bacterial components prime systemic immune responses; (3) Immune cell trafficking between mucosal sites. Conversely, pulmonary perturbations reciprocally affect gut homeostasis. Anti‐TB therapy exemplifies this interdependence—Wipperman et al. [70] observed that antimicrobial‐induced microbiome alterations during early TB treatment (≤14 days) may impair inflammatory resolution through systemic immune modulation. These findings elucidate the gut–lung axis's complex role in TB pathogenesis and therapeutic response.
Notably, GM perturbations by specific anti‐TB drugs may trigger cascading effects. Khan et al. [49] demonstrated that INH‐ and PYZ‐induced GM dysbiosis impairs alveolar macrophage functionality—a frontline defense against pulmonary MTB. Crucially, fecal microbiota transplantation (FMT) reverses this impairment, directly validating GM's role in maintaining alveolar macrophage homeostasis via the gut–lung axis. Emerging evidence further expands this paradigm: Gut‐derived microbial signals regulate lung dendritic cell migration and function, enhancing granuloma formation and anti‐MTB immunity [51, 52]. Conversely, high‐fat diet‐induced GM alterations remodel the pulmonary transcriptome via NOS2 upregulation, compromising intracellular antimicrobial defenses and promoting MTB colonization [17]. Collectively, these findings delineate a multidimensional regulatory network through which GM influences TB pathogenesis and therapeutic outcomes via the gut–lung axis.
GM and immune cell dynamics
GM critically regulates immune cell dynamics central to TB pathogenesis, particularly Th1‐polarized responses essential for macrophage activation via IFN‐γ signaling [83]. Mechanistic studies reveal GM's modulation of unconventional T cell subsets: Vorkas et al. [115] demonstrated GM‐mediated regulation of mucosal‐associated invariant T cells, while Constantinides et al. [116] identified γδ T cell functional modulation. Dysbiosis‐induced Th1/Th2 imbalance may compromise anti‐mycobacterial immunity [117].
GM further impacts systemic inflammation through intestinal barrier regulation. Dysbiosis increases intestinal permeability, permitting translocation of microbial products (e.g., LPS) that drives chronic inflammation—a potential accelerator of TB progression [58]. These collective mechanisms position GM as a critical modulator of TB immunopathology and a promising target for immunological interventions.
Recent mechanistic studies have also uncovered that GM can modulate the cytotoxic function of NK cells, enhancing the clearance of MTB‐infected cells. Furthermore, GM‐induced alterations in systemic cytokine profiles may shift the balance between pro‐inflammatory and regulatory immune responses, further influencing TB outcomes [118]. In addition, gut dysbiosis has been associated with increased intestinal permeability and upregulation of NOS2 in lung tissues, which leads to reduced reactive oxygen species production and decreased expression of DEFB1, thereby creating a microenvironment favorable for MTB colonization [119].
GM AS A POTENTIAL TARGET FOR DIAGNOSIS AND TREATMENT OF TB
The growing recognition of GM in human pathophysiology has positioned it as an emerging focus for TB management strategies. Mounting evidence demonstrates profound GM dysbiosis in TB patients, with these microbial alterations not only serving as disease biomarkers but potentially influencing clinical progression and therapeutic responses. This dual diagnostic‐therapeutic potential stems from GM's noninvasive accessibility and capacity to modulate both host immunity and xenobiotic metabolism. Current research explores two principal applications: (1) development of microbial signature‐based diagnostic tools, and (2) microbiome‐targeted interventions to enhance treatment efficacy. This section examines recent advances in GM‐based TB management approaches and their clinical implications (Figure 5).
FIGURE 5.
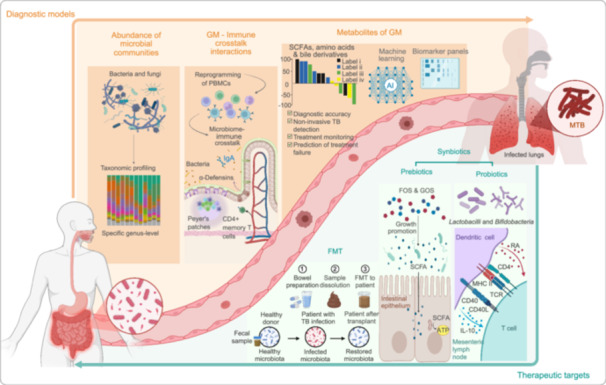
Gut microbiota (GM) as a diagnostic and therapeutic target in tuberculosis (TB). Emerging evidence highlights the GM as a key modulator of TB pathogenesis and a promising target for diagnosis and treatment. At the molecular level, the GM influences TB development through systemic circulation, with alterations in microbial composition, fungal–bacterial interactions, gut‐immune crosstalk, and metabolite profiles offering diagnostic potential. Elucidating these mechanisms is critical for advancing TB therapeutics. Microbiota‐targeted interventions, including probiotics, prebiotics, synbiotics, and FMT can restore GM homeostasis, reduce treatment‐associated complications, and improve gut and immune health.
GM as a biomarker for the diagnosis and treatment monitoring of TB
GM as a diagnostic biomarker for TB
Emerging evidence highlights the capacity of GM alterations to serve as sensitive indicators of host health status, with particular relevance to TB diagnostics. Taxonomic profiling reveals characteristic patterns of reduced microbial diversity and specific genus‐level abundance changes in TB patients. Han et al. identified distinct bacterial‐fungal co‐occurrence networks that improved diagnostic accuracy when incorporated into combinatorial models [24]. Complementary work by Hu et al. demonstrated robust classification performance using bacterial abundance profiles alone, achieving effective discrimination between healthy controls (HCs) and TB patients [27]. Mechanistically, Namasivayam et al. established correlations between GM alterations and functional reprogramming of PBMCs, suggesting microbiome‐immune crosstalk could inform diagnostic development [34].
The diagnostic potential extends beyond taxonomic composition to microbial metabolic output. Metabolomic analyses reveal disease‐associated perturbations in SCFAs, bile acid derivatives, and amino acid profiles. Maji et al. specifically implicated depletion of SCFA‐producing taxa as a potential diagnostic marker, linking microbial metabolism to TB pathophysiology [28]. Compared to conventional diagnostics relying on sputum analysis (smear microscopy, culture) or tuberculin skin testing, GM‐based approaches offer distinct advantages in sample accessibility and analytical throughput. Lu et al. demonstrated this potential through machine learning (ML) integration of microbiome data, achieving diagnostic accuracy comparable to established methods while eliminating sputum collection requirements [120]. Luo et al. [121] developed fecal biomarker panels (e.g., deoxycholate, LysoPC 12:1) and a nomogram integrating age/sex to stratify latent TB infection (LTBI) progression risk. These developments position GM analysis as a promising platform for noninvasive TB detection and treatment monitoring.
Treatment monitoring based on GM metabolic characteristics
The research progress of GM metabolomics provides crucial evidence for treatment monitoring and individualized interventions. Sahu et al. [122] tracked partial GM restoration and metabolic normalization in TB patients during treatment. Luies et al. [123] identified GM‐derived metabolites (3,5‐dihydroxybenzoate, 3‐(4‐hydroxy‐3‐methoxyphenyl)propionate) predictive of treatment failure (AUC = 0.94). These findings lay the foundation for a TB efficacy evaluation system based on GM metabolic characteristics.
GM as a therapeutic target for TB
TB therapies are known to disrupt GM equilibrium, impairing immunity‐associated metabolites like bile acids and SCFAs [34, 66, 124]. Emerging evidence supports GM modulation as a promising adjuvant approach for TB treatment [125, 126]. Accumulating evidence establishes GM's critical involvement in three therapeutic‐relevant processes: immunomodulation, drug biotransformation, and pathogen containment. This functional triad underpins growing interest in microbiome engineering as an adjunct to conventional anti‐TB regimens. Therapeutic strategies broadly fall into two categories: (1) pharmacological modulation of microbial communities, and (2) ecological restoration through microbiota transplantation.
Pharmacological modulation of microbial communities
In the research of TB treatment, pharmacological interventions primarily utilize probiotics, prebiotics, and synbiotics to reshape microbial communities. Diallo et al. conceptualized a “Host‐Microbiota‐Directed Therapy,” demonstrating improved treatment outcomes through microbiome optimization [127]. Experimental validation is provided by Gavrilova et al., who identified anti‐mycobacterial activity in Lactobacillus strains through direct microbial antagonism [128]. Negi et al. further elucidated an immunological mechanism, showing probiotic‐mediated restoration of histocompatibility complex class II expression on lung dendritic cells coupled with Treg cell reduction enhances mycobacterial clearance [53]. Although different studies consistently emphasize the importance of immunomodulation, suggesting a dominant therapeutic pathway, differences in implementation, effect evaluation, and dosing strategies have been observed. These variability issues underscore the need for further standardization of experimental protocols and a deeper exploration of optimal clinical strategies.
Ecological restoration through microbiota transplantation
Microbiota transplantation has emerged as a promising strategy for ecological restoration in TB therapeutics, aiming to rectify treatment‐associated dysbiosis by reconstituting balanced microbial communities. FMT, a therapeutic modality with well‐documented efficacy in Clostridioides difficile infection management, is now being explored for its potential in TB treatment optimization. Pioneering work by Trivedi et al. [129] demonstrated that FMT could effectively mitigate antibiotic‐induced GM dysbiosis during TB therapy, potentially enhancing treatment outcomes through microbiome stabilization. This finding is further supported by Eribo et al. [130], who reported that post‐antibiotic FMT administration not only accelerates microbial community recovery but also reduces treatment complications and may decrease transmission risks in drug‐resistant TB (DR‐TB) through microbiome‐mediated immune modulation.
Critical evaluation of existing evidence reveals that the therapeutic efficacy of FMT is modulated by three principal factors: donor‐recipient compatibility, temporal precision of intervention, and host‐specific biological variables. Rigorous donor screening protocols—particularly those incorporating microbial diversity metrics and pathogen exclusion criteria—have shown direct correlation with clinical success rates. The timing of FMT administration relative to antibiotic exposure emerges as a crucial determinant of microbial engraftment efficiency, with early‐phase interventions demonstrating superior ecological restoration capacity. Furthermore, interindividual variations in immune status, baseline GM architecture, and genetic predisposition significantly influence therapeutic responsiveness.
Integrated perspectives and future directions
Current research substantiates a paradigm shift toward integrated TB management frameworks that synergize conventional antimicrobial therapy with microbiome‐stabilizing interventions. Accumulating evidence from preclinical models and clinical observations suggests that GM modulation could address three persistent challenges in TB care: (1) treatment duration reduction through enhanced chemotherapeutic efficacy mediated by microbial β‐glucuronidase activity [53], (2) side effect mitigation via microbiome‐dependent optimization of drug metabolism pathways [62, 131], and (3) relapse prevention through sustained immunomodulation of tissue‐resident memory T cells [26, 132]. Notably, emerging combinatorial approaches demonstrate amplified therapeutic potential—controlled trials reveal that high‐fiber diets potentiate probiotic efficacy by elevating SCFA production, while polyphenol‐rich nutritional regimens suppress pro‐inflammatory taxa proliferation during anti‐TB treatment [133]. This metabolic‐immune crosstalk is further exemplified by Martineau et al.'s landmark discovery that vitamin D supplementation augments TB treatment responses through GM‐dependent induction of antimicrobial peptides [134].
The next frontier in TB therapeutics lies in rationally integrating pharmacological and ecological restoration strategies. To achieve this, future investigations must systematically decipher the tripartite interaction dynamics between gut ecosystems, pharmacological agents, and host immunity using longitudinal multi‐omics profiling (metagenomic, metabolomic, proteomic) coupled with gnotobiotic animal models. Concurrently, phase III clinical trials employing precision microbiota interventions should establish three critical parameters: (a) patient stratification biomarkers predictive of intervention responsiveness, (b) optimal therapeutic windows relative to antibiotic exposure phases, and (c) standardized efficacy metrics encompassing microbial diversity indices, inflammatory biomarkers, and treatment completion rates. The convergence of these research trajectories promises to advance personalized TB regimens tailored to individual microbial and immunological profiles. Such integrative approaches not only reinforce global TB control through improved treatment adherence and reduced transmission risks but also provide a mechanistic framework for developing next‐generation therapeutics targeting host–microbe–drug interactions.
MULTI‐OMICS AND AI TECHNOLOGIES IN GM‐TB RESEARCH
NGS: Revolutionizing genomic investigations
NGS, as a group of high‐throughput technologies, can rapidly and cost‐effectively analyze complete genomes, transcriptomes, and epigenomes. Unlike traditional Sanger sequencing, which processes individual DNA sequences sequentially, NGS achieves parallel processing of millions of DNA fragments through automated workflows. This breakthrough has transformed genomic research such as medicine and has been critical for pathogen characterization, drug resistance detection, and epidemiological surveillance [135]. Building on the technological developments, NGS has significantly advanced microbial community profiling.
Technologies, developments, and multifaceted applications
The core principle of NGS involves parallel sequencing of DNA or RNA fragments immobilized on solid surfaces such as beads or flow cells (Figure 6A). After a series of operations including adapter ligation and clonal amplification, sequencing clusters are generated. With diverse major detection methods, Illumina technology is widely applied, while Pacific Biosciences (PacBio) and Oxford Nanopore are proficient in long‐read sequencing [136].
FIGURE 6.

Overview of the next‐generation sequencing (NGS) workflow and its applications. (A) Schematic representation of the core stages in the NGS workflow: DNA fragmentation and library preparation, high‐throughput sequencing, and bioinformatics analysis and data interpretation. (B) Illustration of the diverse applications of NGS across genomics and biomedical research.
The commercial development of NGS platforms began with the 454 pyrosequencer [137]. Early platforms differed in several aspects [138]. Subsequently, third‐generation sequencing platforms (such as PacBio and Oxford Nanopore), which can directly sequence single DNA molecules emerged (Table 3) [139, 140, 141, 142, 143, 144, 145, 146, 147, 148, 149, 150]. Emerging methods combine short‐read efficiency with long‐range genomic information.
TABLE 3.
Comparison of the most important sequencing platforms of NGS.
| Method | Properties | Refs |
|---|---|---|
| Illumina sequencing | Short‐read sequencing | [139, 140, 141] |
| The most commonly used NGS platform | ||
| Relies on sequencing‐by‐synthesis technology (fluorescently labeled nucleotides are incorporated into a growing DNA strand during sequencing) | ||
| Identifying the detected resulting signal to determine the incorporated base | ||
| Simultaneously sequencing millions of short reads (typically 100–300 bp in length) | ||
| High accuracy, | ||
| Extensive bioinformatics support | ||
| Scalability | ||
| Wide range of applications, including whole‐genome sequencin, whole‐exome sequencing, targeted sequencing, and RNA‐ sequencin | ||
| Ion torrent sequencing | Semiconductor Sequencing (detects the release of protons when a nucleotide is incorporated into the DNA strand) | [142, 143, 144] |
| Eliminates the need for optical detection | ||
| Uses changes in pH to identify the base being added to the growing strand | ||
| Fast turnaround time | ||
| Lower cost | ||
| Shorter reads (ranging from 50 to 400 bp) | ||
| Commonly employed in targeted sequencing applications, such as gene panels and microbial sequencing, and for research requiring high‐speed results. | ||
| PacBio sequencing | Long‐read sequencing | [145, 146, 147] |
| Known as Single Molecule Real‐Time sequencing | ||
| Reading of much longer DNA fragments compared to Illumina and Ion Torrent platforms (average read lengths range from 10 kb to >100 kb) | ||
| Utilizes zero‐mode waveguide technology to capture real‐time sequencing events | ||
| Generating each read by combining DNA polymerase with nucleotides and detecting the light emitted by the combined bases | ||
| Advantageous for resolving complex genomic regions, structural variants, and repetitive sequences that are difficult to sequence with short‐read technologies | ||
| Used widely for de novo genome assembly, transcriptome analysis, and the study of large, complex genomes | ||
| Oxford nanopore sequencing (Nanopore technology) | Based on the detection of changes in electrical current as DNA strands pass through nanopores embedded in a membrane | [140, 148, 149, 150] |
| Real‐time sequencing method | ||
| Generating ultra‐long reads (with some individual reads extending over several megabases) | ||
| Offering a portable and flexible sequencing solution | ||
| It can be deployed in various environments using devices such as the MinION | ||
| Providing significant advantages in read length and real‐time sequencing, enabling applications such as rapid pathogen detection, environmental monitoring, and metagenomics | ||
| Higher error rate compared to Illumina and PacBio (but advances in software and base‐calling algorithms have steadily improved accuracy and reliability) |
Abbreviations: NGS, next‐generation sequencing; PacBio, Pacific Biosciences.
Different NGS methods are suitable for different experimental purposes. Illumina is the gold standard for large‐scale genomic analyses and PacBio and Oxford Nanopore are applicable to long‐read resolution, Ion Torrent and others have unique advantages in clinical applications. The emerging technologies such as single‐cell RNA‐sequencing (RNA‐Seq), have promoted NGS applications and facilitated the study of genetic diversity and cellular heterogeneity at unprecedented depth [151, 152].
The versatility of NGS covers multiple disciplines, and its core applications involve seven key areas (Figure 6B). In genomic research, whole‐genome sequencing (WGS) serves as the foundation, showing remarkable advantages in clinical applications [153]; targeted sequencing is a cost‐effective alternatives, and exome sequencing can identify pathogenic mutations [154]. NGS‐based RNA‐Seq has revolutionized transcriptome analysis [155, 156]. Epigenetics can analyze DNA methylation with the help of NGS [157, 158]. In microbial ecology, NGS has revolutionized metagenomics research [159, 160]. In oncology, NGS is used to identify genetic alterations, and liquid biopsy enhances its clinical impact [161]. Furthermore, it has been reported that pharmacogenomics optimizes therapeutic strategies through NGS to improve patient outcomes [162].
NGS‐driven microbial community profiling
NGS has fundamentally transformed microbial community analysis by overcoming the limitations of traditional culture‐dependent methods. Conventional approaches fail to detect approximately 80% of environmental microbes due to unculturability, whereas NGS enables comprehensive detection of microbial constituents through direct nucleic acid sequencing, providing insights into community composition, functional potential, and metabolic activity across diverse ecosystems [163].
For microbial profiling, distinct NGS strategies are used based on research goals. 16S rRNA gene sequencing is the gold standard for taxonomic classification at genus or species level. Whole‐genome shotgun sequencing analyzes functional genes and metabolic pathways, and metatranscriptomics captures community gene expression. Platform selection among Illumina, PacBio, and Nanopore technologies depends on read length, throughput, and error tolerance [135].
Deep sequencing enables high‐resolution diversity analysis, revealing dominant and low‐abundance microbial members. This helps identify keystone species, detect pathogens, and monitor population shifts. The depth of sequencing directly correlates with the accuracy of microbial interaction network reconstruction for precise modeling of community dynamics [164].
In clinical microbiology, NGS‐based profiling has elucidated the human microbiome's role in health and disease, such as dysbiosis in gut microbial communities linked to disorders. In environmental applications, NGS clarifies microbial contributions to biogeochemical cycles, and in extreme environment studies, it helps catalog extremophile communities and their adaptations [165].
Applications in GM‐TB research
The GM interplay with MTB infection has emerged as a critical research frontier, particularly through the gut–lung axis concept. NGS, as the preeminent technology, plays a crucial role in GM‐TB research. This bidirectional communication pathway suggests GM modulation of pulmonary immunity, and NGS serves as the primary tool for characterizing microbial shifts during TB progression (Figure 7). High‐resolution sequencing reveals specific dysbiosis patterns in TB patients, including enrichment of pro‐inflammatory taxa and depletion of beneficial SCFA‐producing bacteria [11, 20].
FIGURE 7.
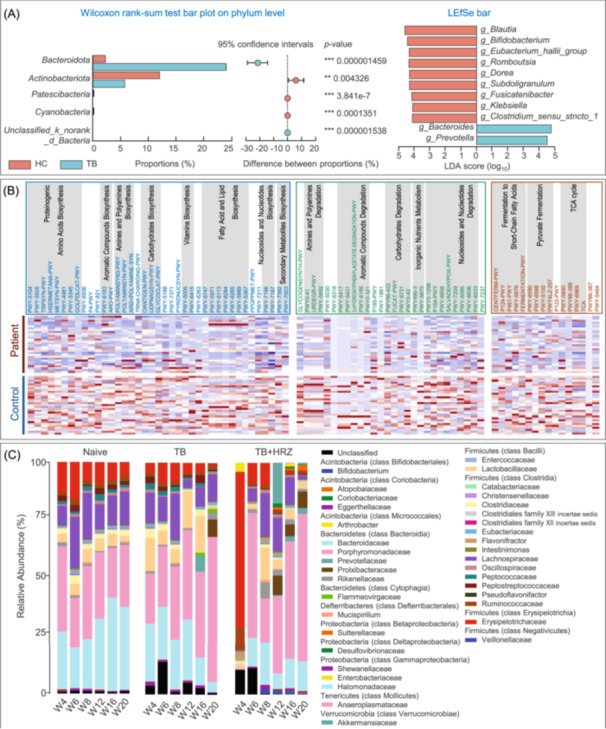
Applications of NGS in gut microbiota‐tuberculosis (TB) research. (A) Microbial composition analysis: Relative abundances of intestinal bacterial phyla in TB patients versus healthy controls (HC) were compared using the Wilcoxon rank‐sum test. Differentially abundant taxa were identified by linear discriminant analysis effect size (LEfSe) with LDA score > 4 and p < 0.05. Adapted with permission from [24], Copyright 2024, BMC. (B) Metabolic pathway analysis: Heatmap shows differential metabolic pathway abundance between TB and HC groups based on NGS‐derived functional profiling. Pathways were ranked according to consensus functional classification; statistical significance was determined using the Wilcoxon rank‐sum test with false discovery rate (FDR) < 0.1. Abundances were standardized as row Z‐scores. Adapted with permission from [27], Copyright 2019, Frontiers. (C) Longitudinal microbiota dynamics during anti‐TB therapy: Temporal changes in the average relative abundance of bacterial families were tracked using stool sample sequencing data across multiple treatment time points and experimental groups. Bacterial families are grouped by phylum and class in the accompanying color key. Time points are indicated along the x‐axis, grouped by treatment conditions. Adapted with permission from [29], Copyright 2017, BMC.
For example, through 16S rRNA sequencing and shotgun metagenomics (both NGS‐based techniques), Pseudomonadota and Bacillota are consistently identified as dominant phyla in TB‐associated dysbiosis. NGS‐derived data also indicate that these microbial shifts correlate with heightened systemic inflammation and impaired granuloma stability, exacerbating lung pathology. Concurrent reductions in Faecalibacterium and Bifidobacterium populations, detected by NGS, diminish SCFA production, weakening gut barrier integrity and regulatory T‐cell differentiation—key factors in TB immune regulation [166, 167, 168].
Molecular mechanisms connecting GM alterations to TB pathology, which are deciphered with the help of NGS, involve metabolite‐mediated immune modulation. Host‐microbiota immune crosstalk studies demonstrate GM's systemic impact on TB pathogenesis. Increased intestinal permeability (“leaky gut”) allows microbial product translocation into circulation, triggering inflammatory pathways that compromise MTB containment. Dendritic cell‐mediated T‐cell priming in mesenteric lymph nodes and subsequent lung homing highlight the gut–lung immunological axis, with dysbiosis impairing this protective mechanism [39, 169].
Bioinformatics advancements, in dealing with the complexity of NGS data in GM‐TB studies, are essential. Tools like QIIME2 and MetaPhlAn2 enable robust taxonomic profiling, while PICRUSt and HUMANN2 predict functional potentials. ML integration identifies microbial biomarkers predictive of treatment response, facilitating personalized therapeutic strategies through multi‐omics data fusion [170, 171].
Emerging microbiome‐targeted therapies, guided by NGS‐revealed microbial profiles, extend beyond conventional probiotics. Postbiotics containing microbial‐derived SCFAs or antimicrobial peptides offer safer alternatives for immunocompromised patients. Engineered probiotics designed for targeted metabolite delivery and bile acid modulation represent a novel therapeutic frontier. For instance, genetically modified Lactobacillus strains engineered to constitutively express butyrate synthesis pathways have shown promise in preclinical TB models for restoring gut barrier function and anti‐inflammatory responses. Similarly, E. coli Nissle 1917 derivatives equipped with bile salt hydrolase genes demonstrate enhanced capacity to mitigate RIF‐induced hepatotoxicity through bile acid detoxification [172].
FMT, an approach under investigation, also benefits from NGS. Early‐phase trials in TB patients, with NGS‐monitored microbial diversity recovery, explore FMT's potential to accelerate microbial diversity recovery post‐antibiotic treatment. Preliminary evidence suggesting reduced inflammatory markers and improved anti‐TB drug tolerance. However, safety protocols require rigorous standardization given the risk of pathogen transmission in immunocompromised hosts, which is also monitored using NGS [173].
The integration of microbiome science into TB management, facilitated by NGS‐derived microbial signatures, is progressing through two parallel pathways: predictive diagnostics and adjunctive therapies. Microbial signature panels derived from NGS data show potential as prognostic biomarkers, identifying patients at high risk of treatment failure or relapse based on baseline dysbiosis patterns. This stratification, enabled by NGS, allows personalized therapeutic regimens combining standard anti‐TB drugs with microbiome‐modulating interventions [174].
Longitudinal NGS monitoring reveals dynamic GM changes throughout anti‐TB therapy, highlighting critical intervention windows. The initial treatment phase, monitored by NGS, induces the most severe dysbiosis, suggesting early supplementation with SCFA‐producing strains or prebiotic fibers may preserve microbial resilience. Posttreatment phases, also tracked by NGS, exhibit incomplete GM recovery, warranting extended microbiota support strategies to prevent reinfection [175].
Future clinical trials, with a focus on NGS‐guided microbiome‐targeted therapy, must address key challenges in microbiome‐targeted therapy: (1) Standardization of intervention protocols across diverse populations, informed by NGS‐based population—specific microbial profiles; (2) Resolution of donor‐recipient compatibility issues in FMT, aided by NGS‐based microbial matching; (3) Development of TB‐specific microbial consortia with defined therapeutic functions, based on NGS‐predicted functional potentials; (4) Integration of host genetic factors influencing microbiome–drug interactions, explored through NGS‐enabled host–microbiome–drug interaction studies [176].
The ultimate goal involves developing microbiome‐informed precision medicine frameworks for TB, relying on NGS‐derived comprehensive microbial and host information. By correlating individual microbial profiles, obtained by NGS, with drug metabolism rates and immune response patterns, clinicians could optimize: (1) Antibiotic dosing schedules to minimize GM disruption, guided by NGS‐monitored drug–microbe interactions; (2) Probiotic/postbiotic selection based on strain‐specific functional deficits, identified by NGS‐based functional profiling; (3) Dietary recommendations tailored to microbial metabolic needs, deduced from NGS‐revealed microbial metabolic pathways [177].
NGS technologies have fundamentally redefined the GM‐TB research paradigm. From characterizing dysbiosis patterns to decoding metabolite–immune interactions, NGS‐driven insights are catalyzing a therapeutic revolution. The next decade will likely witness the clinical implementation of microbiome‐adjuvant therapies, transforming TB management from empirical antibiotic regimens to precision ecology‐based medicine, all centered around the application of NGS in GM‐TB research [178].
Proteomics and the GM
Proteomic techniques for microbiome analysis
Proteomics, a crucial method for characterizing protein profiles in complex biological systems [179], can precisely identify and quantify proteins, detect posttranslational modifications (PTMs), and is useful for GM functional studies [180]. Dysbiosis in GM leads to changes in the gut metaproteome, related to conditions like metabolic syndrome and nutritional disorders [181].
Metaproteomics, which analyzes complete protein complements within environmental microbial communities [182], differs from conventional proteomics as it examines polymicrobial systems and captures organism‐specific PTMs that mediate microbial adaptation through mechanisms such as phosphorylation, acetylation, and oxidation [183]. Current analytical workflows combine advanced mass spectrometry (MS) with bioinformatics, while new synthetic databases improve peptide identification [183, 184].
However, there are challenges in computational requirements, standardization of protein grouping, and database construction. Metaproteomic analysis of neonatal fecal samples shows its potential in clinical diagnostics [184]. In GM analysis, MS‐based shotgun metaproteomics is commonly used with various sample sources (Table 4) [185, 186, 187, 188, 189, 190, 191, 192, 193, 194, 195, 196, 197, 198, 199], and has identified novel therapeutic targets in TB. Future developments should prioritize: (1) Diagnostic protocol optimization for clinical fecal sample processing, (2) Computational infrastructure enhancement for large‐scale data analysis, and (3) Integrative multi‐omics frameworks combining metaproteomic data with metabolomic and transcriptomic datasets to decipher host–microbe crosstalk.
TABLE 4.
Comprehensive overview of proteomic strategies for identifying drug targets in TB treatment.
| Mechanism | Advantage | Disadvantage | Refs |
|---|---|---|---|
| Identifying essential proteins in MTB via shotgun proteomics | It helps uncover critical pathways unique to the bacteria for selective drug targeting | Requires large protein databases; prone to sample complexity issues | [185] |
| Quantitative proteomics using SILAC or TMT | Enables precise comparison of protein expression under drug‐treated versus untreated conditions | Expensive; dependent on high‐quality MS facilities | [186] |
| Surface proteomics for membrane protein identification | Identifies proteins involved in host–pathogen interactions; potential vaccine candidates | Difficult to isolate membrane proteins; low abundance of targets | [187, 188] |
| PTM analysis of bacterial proteins | Reveals functional regulation mechanisms critical for survival or resistance | Complex data analysis, limited by incomplete PTM databases | [189] |
| Host proteomics to study immune responses | Identifies host pathways affected by TB, enabling host‐directed therapy discovery | May yield indirect targets; potential off‐target effects in therapies | [190] |
| Drug‐binding protein identification using affinity‐based approaches | Identifies bacterial proteins that directly interact with existing or experimental drugs | Limited to drugs with known mechanisms; may miss noncovalent interactions | [191] |
| Phosphoproteomics to map bacterial signaling pathways | Identifies kinases and phosphatases critical for TB survival and replication | It requires enrichment techniques and often suffers from low phosphopeptide abundance. | [192] |
| Comparative proteomics across drug‐resistant and susceptible strains | Identifies resistance‐related proteins and mechanisms | Interpretation can be complex due to strain‐specific variations | [193] |
| Proteomics for secretome analysis | Identifies proteins secreted by TB during infection, which are potential vaccine or drug targets | May miss low‐abundance proteins; secretome changes under different conditions | [194] |
| Integrative proteomics with transcriptomics | Correlates protein expression with transcriptional changes for robust target discovery | Proteomic data may not always correlate directly with transcriptomic data | [195] |
| Multi‐drug proteomic profiling | Examines protein changes under treatment with multiple drugs to detect synergistic effects | It may require extensive bioinformatics to interpret complex datasets | [196] |
| Functional proteomics with CRISPR screens | Combines proteomics with CRISPR gene editing to validate essential TB proteins as drug targets | High complexity; limited to labs with CRISPR expertise | [197] |
| Time‐resolved proteomics during TB infection | Tracks protein expression over infection stages, identifying targets for specific disease phases | Requires longitudinal sample collection; high costs | [198] |
| Proteomics‐guided vaccine target discovery | Identifies immunogenic proteins for designing TB vaccines | Proteins identified may lack sufficient antigenicity in human trials | [199] |
Abbreviations: CRISPR, clustered regularly interspaced short palindromic repeats; MS, mass spectrometry; PTMs, posttranslational modifications; SILAC, stable isotope labeling by amino acids in cell culture; TB, tuberculosis; TMT, tandem mass tags.
Proteomic techniques in TB research
Shotgun proteomics
Proteomic techniques play crucial roles in TB research. Shotgun proteomics, using MS‐based workflows, enzymatically digests protein samples, separates peptides via multidimensional liquid chromatography (LC), conducts tandem MS analysis, and matches with databases for protein identification [200]. It can analyze complex peptide mixtures and is used for both qualitative and quantitative analyses.
In MTB research, shotgun proteomics has led to important discoveries. Nikitushkin et al. [201] performed LC‐MS proteomic profiling of latent MTB cells, identifying 1379 conserved proteins with 468 exhibiting significant quantitative shifts. Their work revealed metabolic adaptations involving methyl citrate cycle activation, glyoxylate shunt utilization, and pH‐dependent PhoP regulation. It also identified dormancy‐associated biotransformation enzymes critical for prodrug activation, which provides key insights for developing diagnostics and therapies against latent TB. Concurrently, Tucci et al. [202] analyzed MTB culture filtrates via LC‐MS/MS, detecting 1314 proteins including 6‐kDa eary secretory antigenic target secretion system X (ESX) secretion system components and pro‐glu/pro‐pro‐glu family (PE/PPE) virulence factors. Their identification of 46 O‐glycosylated proteins (33 novel) advanced understanding of MTB's antigenic landscape and survival strategies.
Clinical studies further validate shotgun proteomics' utility. Mateos et al. [203] identified distinct proteomic signatures in TB patients' biofluids, contrasting upregulated inflammatory mediators with enhanced innate immunity markers in uninfected contacts. From a therapeutic perspective, Villela et al. [204] confirmed guanosine monophosphate synthetase as essential through targeted validation, demonstrating that guaA deletion induces MTB lethality reversible via Mycobacterium smegmatis homolog complementation—establishing guanosine monophosphate synthetase inhibition as a viable therapeutic strategy.
Targeted proteomics
Targeted proteomics has emerged as a gold standard for precise protein quantification. It focuses on predefined protein subsets, different from untargeted methods that indiscriminately survey thousands of proteins [205]. There are three main methodologies in this field: Selected/multiple reaction monitoring (SRM/MRM), parallel reaction monitoring (PRM), and data‐independent acquisition (such as SWATH‐MS). SRM/MRM, implemented on triple quadrupole instruments, achieves exceptional selectivity via dual mass filtering and extended dwell times, enhancing sensitivity 10–100 × over full‐scan methods and matching immunoassay precision [206, 207]. PRM, while retaining SRM's precursor isolation, uses high‐resolution Orbitrap/time‐of‐flight (TOF) analyzers to capture full MS/MS spectra for better data analysis [208]. Data‐independent acquisition systematically partitions precursor windows across chromatographic elution profiles to generate comprehensive MS/MS maps [209].
Clinical applications demonstrate targeted proteomics' diagnostic potential. Yao et al. [210] identified a 9‐protein plasma signature distinguishing active TB from controls via analysis of 92 immune proteins, achieving 93.1% specificity for severe cases through ML. Robak et al. [211] developed a pleural effusion classification assay with 89% accuracy, differentiating TB‐associated pleural effusion from malignant/infectious etiologies. For drug resistance detection, Chen et al. [212] established a diagnostic panel (sCD14, PGLYRP2, FGA) demonstrating 92% sensitivity and 88% specificity for multidrug‐resistant TB (MDR‐TB) identification.
Single‐cell proteomics (SCP)
SCP has revolutionized cellular heterogeneity studies by resolving protein‐level variations that are not obscured in bulk analyses. Although single‐cell transcriptomics are popular in cellular resolution research, recent SCP advancements now enable functional proteome characterization at ultralow protein abundances (10⁻¹⁸–10⁻¹⁵ g/cell) [213]. Unlike bulk proteomics, averaging thousands of cells, SCP preserves cellular individuality. It can uncover rare pathogenic subpopulations, stochastic protein expression patterns, and dynamic PTMs in living systems [214]. This unique capability allows real‐time monitoring of protein dynamics in viable cells, contrasting with static snapshots from lysed cell aggregates (Table 5) [203, 210, 214, 215, 216, 217, 218, 219, 220, 221].
TABLE 5.
Summarized proteomics techniques in microbiome and TB research.
| Sr. no | Technique | Limitations/challenges | Potential solutions | Application in TB research | Mechanism of action | Refs |
|---|---|---|---|---|---|---|
| 1 | Shotgun metaproteomics |
|
|
Profiling MTB proteins for biomarker discovery and therapeutic targets | Enzymatic digestion of proteins into peptides followed by LC‐MS/MS for identification and quantification | [203, 215, 216] |
| 2 | Targeted proteomics |
|
|
Quantitative measurement of MTB proteins for biomarker validation and drug target assessment | SRM/PRM techniques for selective peptide detection and quantification with high sensitivity | [210, 217] |
| 3 | SRM and PRM |
|
|
Quantifying specific MTB proteins for validating biomarkers and drug targets | SRM/PRM in MS for selective peptide detection with high specificity | [218] |
| 4 | SCP |
|
|
Investigating protein expression at the single‐cell level to understand host immune responses to MTB | MS for analyzing proteins from individual cells, examining pathogen–host interactions | [219, 220] |
| 5 | Top‐down proteomics |
|
|
Characterizing intact MTB proteins, including posttranslational modifications | Direct analysis of whole proteins without prior digestion, identifying isoforms and modifications | [221] |
Abbreviations: AI, artificial intelligence; LC‐MS, liquid chromatography‐mass spectrometry; PRM, parallel reaction monitoring; SCP, single‐cell proteomics; SRM/MRM, selected/multiple reaction monitoring; TB, tuberculosis.
In TB research, SCP has provided important insights. Chung et al. [222] revealed MTB remodels bone marrow mesenchymal stem cell proteomes to evade immune detection by downregulating antigen presentation machinery. Kaur et al. [223] demonstrated virulent MTB strains manipulate host differentiation pathways (Notch/Wnt), mitochondrial metabolism (oxidative phosphorylation suppression), and RNA splicing to establish persistent infection niches. At the host‐pathogen interface, Devasundaram et al. [224] identified divergent T‐cell responses to early secretory antigen‐6 (ESAT‐6) and Lpd antigens between latent and active TB patients via single‐cell cytokine profiling, suggesting pathogen‐directed immune modulation. These findings align with MTB's hypoxia adaptation mechanisms, where Forrellad et al. [225] observed upregulated DosR regulon proteins promoting dormancy in low‐oxygen bone marrow microenvironments.
Host–microbe interactions and proteomics in TB
MTB is a formidable pathogen due to its sophisticated co‐evolutionary adaptations [225]. The interplay between MTB and host immunity involves multilayered interactions spanning immune signaling, metabolic reprogramming, and cellular stress regulation [226], affecting host‐microbiome equilibrium and extracellular communication [227].
MTB actively subverts host defenses through molecular effectors such as Hip1 and ESAT‐6, disrupting T‐cell responses and creating an environment with restricted MTB spread and chronic inflammation [227]. Microbiome‐derived metabolites influence the host‐MTB metabolic crosstalk. For example, indole‐3‐propionic acid can inhibit MTB growth, showing its dual role in TB control [130].
Antibiotic therapies (e.g., INH, pyrazinamide [PZA]) induce GM dysbiosis in TB models, reducing macrophage microbicidal activity [130]. Proteomic techniques help study these interactions. Metaproteomics can analyze protein complements in the host‐MTB microbial communities, identifying proteins and PTMs for understanding molecular‐level interactions. Integrating proteomic data with other omics, as in future metaproteomics development, aids in understanding host–microbe crosstalk. Figure 8 synthesizes these integrated host mechanisms governing TB pathogenesis and resolution, guiding interpretation of proteomic data.
FIGURE 8.

An overview of the host–microbe interactions in TB progression. The bidirectional influence of the lung–GM axis on host immunity and MTB survival. On the left (green), a balanced microbiome supports effective immune surveillance through enhanced immune signaling, a metabolically favorable state (characterized by elevated glycolysis and reduced oxidative phosphorylation), maintenance of stable immune epigenetic landscapes, and tightly regulated cytokines production, promoting MTB clearance. In contrast, the right side (pink) depicts a dysbiotic microbiome that contributes to impaired host defense through disrupted immune signaling (e.g., reduced cell–cell communication), metabolic dysregulation, and skewed pro‐/anti‐inflammatory responses, thus supporting MTB persistence and immune evasion. GM, gut microbiota; TB, tuberculosis.
Potential for identifying novel therapeutic targets
Current anti‐TB regimens aim to achieve three primary objectives: complete MTB eradication, symptomatic relief, and prevention of disease transmission. The World Health Organization (WHO)‐endorsed directly observed treatment, short‐course strategy for drug‐sensitive TB combines four first‐line drugs during the intensive phase (INH, RIF, ethambutol [EMB], PZA) followed by a 4‐month continuation phase with INH and RIF. This standardized approach has contributed to the successful treatment of 66 million TB cases since 2000, as reported in the 2023 WHO guidelines [228, 229]. However, the management of MDR‐TB and extensively drug‐resistant TB (XDR‐TB) remains clinically challenging, requiring prolonged regimens (up to 2 years) with toxic second‐/third‐line agents that yield suboptimal outcomes.
The emergence of drug resistance stems from three principal mechanisms: (1) mutations in drug‐targeted genes or activating enzymes, (2) compensatory evolutionary adaptations, and (3) overexpression of efflux pump systems. These resistance pathways, coupled with the limited diversity of existing anti‐TB drugs, underscore the critical need for novel therapeutic strategies. Priority areas include developing agents capable of circumventing resistance mechanisms, shortening MDR‐TB treatment durations, and enhancing treatment tolerability.
Proteomics‐driven approaches are accelerating the discovery of next‐generation targets through two complementary strategies: (1) systematic identification of MTB vulnerabilities via essential protein mapping, and (2) characterization of host‐microbiome‐derived proteins with therapeutic potential. Table 6 summarizes proteomic methodologies for TB drug target identification [230, 231, 232, 233, 234, 235, 236, 237, 238, 239, 240, 241, 242, 243, 244, 245, 246, 247, 248, 249, 250], while Table 7 catalogs microbiome‐associated proteins demonstrating anti‐mycobacterial activity [251, 252, 253, 254, 255, 256, 257, 258, 259, 260]. These emerging resources provide a framework for rational drug design aimed at overcoming current therapeutic limitations in TB management.
TABLE 6.
Discoveries using proteomics to identify drug targets in TB treatment.
| Sr. no | Discovery/protein target | Role in TB pathogenesis | Methodology used | Refs |
|---|---|---|---|---|
| 1 | DnaK chaperone | Assists in protein folding under stress conditions, crucial for bacterial survival within host macrophages. | LC‐MS/MS‐based proteomics | [230] |
| 2 | Isocitrate lyase | Essential for MTB's glyoxylate shunt, enabling survival in nutrient‐limited conditions. | 2D gel electrophoresis with MS analysis | [231] |
| 3 | ESX secretion system proteins | Involved in virulence and immune evasion by facilitating the secretion of effector proteins like ESAT‐6. | Label‐free quantitative proteomics | [232] |
| 4 | Mycothiol biosynthesis enzymes | Protects MTB from oxidative stress and maintains redox balance. | Targeted proteomics (MRM) | [233] |
| 5 | ATP synthase subunits | The key enzyme for energy production in MTB that inhibition can lead to bacterial death. | Top‐down proteomics | [234] |
| 6 | Phosphopantetheinyl transferase | Involved in lipid metabolism and mycolic acid synthesis, crucial for MTB cell wall integrity. | Shotgun proteomics | [235] |
| 7 | Clp protease complex | Degrades damaged or misfolded proteins, essential for bacterial stress response and survival. | LC‐MS/MS and bioinformatics | [236] |
| 8 | LprG‐Rv1410c lipid transport system | Facilitates lipid transfer for immune modulation, contributing to MTB's persistence in host tissues. | Proteomics‐integrated lipidomics | [237] |
| 9 | PknG kinase | Regulates host–pathogen interactions and inhibits phagosome‐lysosome fusion. | Phosphoproteomics | [238] |
| 10 | DosR regulon proteins | Helps MTB adapt to hypoxic conditions during latent infection, making them potential targets for latency control. | Differential proteomics | [239] |
| 11 | Oxidative stress response proteins | Include peroxiredoxins and catalases, which neutralize host‐induced oxidative damage to MTB. | Comparative proteomics | [240] |
| 12 | PE/PPE family proteins | Modulate host immune responses and contribute to antigenic variation and immune evasion. | Structural proteomics | [241] |
| 13 | RpoB | Mutation in RpoB leads to RIF resistance, making it a critical target for new drug development. | Proteomic profiling of resistant strains | [242] |
| 14 | Trehalose dimycolate synthesis enzymes | Contribute to MTB's cell wall formation and immune evasion. | Functional proteomics | [243] |
| 15 | KasA/KasB enzymes | It is key in mycolic acid synthesis and is essential for MTB cell wall structure. | Label‐free quantitation proteomics | [244] |
| 16 | EmbCAB complex | Involved in arabinogalactan biosynthesis, a crucial component of the mycobacterial cell wall. | Isobaric tagging proteomics | [245] |
| 17 | MmpL transport proteins | Participate in the transport of lipids necessary for MTB virulence and survival. | Shotgun proteomics | [246] |
| 18 | SigH sigma factor | Regulates the expression of stress response genes during host immune attack. | Differential proteomics | [247, 248] |
| 19 | EspE proteins | Facilitate host immune modulation, helping MTB evade detection. | Label‐free proteomics | [249] |
Abbreviations: ESAT‐6, early secreted antigenic target‐6; PE/PPE, pro‐glu/pro‐pro‐glu family; RIF, Rifampicin; TB, tuberculosis.
TABLE 7.
Development of microbiome‐based therapeutic proteins in TB treatment.
| Sr. no | Therapeutic protein | Source (microbiome or engineered microbe) | Mechanism of action | Current status | Refs |
|---|---|---|---|---|---|
| 1 | Recombinant Mycobacterium proteins (e.g., Ag85 complex) | Engineered Mycobacterium smegmatis | Boosts host immunity by acting as a vaccine adjuvant and enhancing T‐cell responses. | Clinical trials as vaccine adjuvants in TB therapy | [251] |
| 2 | EsxA protein secretion inhibitors | Bacterial microbiome‐derived inhibitors | Blocks the EsxA protein secretion system to reduce virulence and immune evasion of MTB. | Early‐stage drug discovery pipeline | [252] |
| 3 | Resuscitation‐promoting factors | Dormant MTB and environmental microbiota | Revives dormant MTB to render it susceptible to conventional TB drugs. | Explored in conjunction with standard TB therapy to target latent infections | [253] |
| 4 | Human microbiota‐engineered peptides (e.g., LL‐37 mimetics) | Engineered gut commensals | Mimics natural antimicrobial peptides like LL‐37 to enhance bacterial clearance. | Preclinical studies in mouse TB models | [254] |
| 5 | Mycobacteriophage‐derived lysins | Engineered bacteriophages targeting MTB | Degrades MTB cell walls specifically, reducing bacterial load while sparing host microbiota. | Phase I clinical trials for safety and efficacy | [255] |
| 6 | Butyrate‐producing microbiota | GM (Clostridium species) | Enhances host immunity and regulates inflammation by promoting butyrate synthesis. | Potential adjunct in TB immunotherapy | [28] |
| 7 | Probiotic‐derived enzymes (e.g., superoxide dismutase) | Probiotic strains (e.g., Lactobacillus reuteri) | Protects host tissues from oxidative stress induced by MTB infection. | Experimental models showing reduced TB‐induced inflammation | [256] |
| 8 | Heat Shock Protein 65 | Recombinant Lactococcus lactis | Boosts host T‐cell‐mediated immunity against MTB. | Preclinical vaccine candidate showing promise | [257] |
| 9 | Microbiota‐SCFAs (e.g., acetate, butyrate) | GM (e.g., Bacteroides) | Modulate immune responses by enhancing macrophage antimicrobial activity. | Explored to enhance the efficacy of TB vaccines | [258] |
| 10 | Siderophores (iron‐binding proteins) | Probiotic microbiota | Compete with MTB for iron, depriving the bacteria of a vital nutrient. | Investigated as part of combination therapies | [259] |
Abbreviations: LL‐37, cathelicidin‐related antimicrobial peptide; TB, tuberculosis.
Metabolomics approaches
Metabolomics, defined as the comprehensive analysis of metabolites (small molecules <50 Da) within a biological system, is a critical component of systems biology. It can show strong correlations with phenotypic changes, helping to understand physiological balance and how internal or external factors affect metabolic networks [261]. In GM research, it's useful for studying microbial activity and composition by quantifying GM‐related metabolites [262]. For instance, Folz et al. found that dietary and microbially derived metabolites contribute to metabolic differences among 15 healthy people [263]. It can also identify metabolic pathway alterations and track adaptive responses of the microbiota to environmental factors, including disease states [261, 264].
Techniques used in metabolomics
Metabolomic studies predominantly employ high‐throughput analytical platforms such as MS and nuclear magnetic resonance (NMR). MS‐based methods, including quadrupole MS and time‐of‐flight MS (TOFMS), are widely adopted due to their superior sensitivity, broad dynamic range, and capacity for multiplex metabolite detection [261, 262, 264]. Gas chromatography (GC)‐MS is good for analyzing primary metabolites, while LC‐MS is better for nonvolatile polar metabolites [261, 262, 264]. Capillary electrophoresis (CE) can separate charged small molecules efficiently [265]. Fernández‐García et al. [266] integrated GC‐quadrupole time‐of‐flight (QTOF)‐MS (volatile/acidic metabolites), LC‐QTOF‐MS (lipids), and CE‐TOFMS (positively charged metabolites) to characterize MTB‐induced metabolic reprogramming in murine lung tissue, uncovering novel host–pathogen interaction pathways.
Emerging MS imaging (MSI) techniques, such as matrix‐assisted laser desorption/ionization (MALDI) and desorption electrospray ionization, can directly show metabolite distribution in tissue sections [267, 268]. Prideaux et al. [269] applied MALDI‐MSI to study distributions of antitubercular drugs in infected lungs, and others correlated mycobacterial lipid marker localization with therapeutic efficacy [270].
NMR spectroscopy can identify metabolites with less sample preparation, although its sensitivity is lower. It is effective at detecting polar metabolites and can complement LC‐MS analyses. Andreas et al. [271] synergized NMR and LC‐MS to identify pediatric TB serum biomarkers, achieving high‐sensitivity detection of both lipophilic and hydrophilic metabolites.
The choice between untargeted and targeted metabolomics depends on experimental objectives. Untargeted approaches utilize high‐sensitivity instruments (e.g., LC‐TOFMS, GC × GC‐TOFMS) for global metabolite profiling [261]. For example, a GC × GC‐TOFMS study identified HIV/TB coinfection‐specific urinary metabolic signatures [272]. Targeted metabolomics, often employing LC‐MS or NMR, focuses on predefined pathways or metabolites. Building on prior untargeted findings [273], Isaiah et al. [274] used LC‐MS/MS to quantify GM‐associated organic acids in pediatric tuberculous meningitis (TBM) cases, establishing TBM‐specific metabolic dysregulation profiles.
Data preprocessing varies for MS and NMR. MS data require peak deconvolution, alignment, and noise reduction [275, 276], while NMR data undergo Fourier transformation, spectral correction, and dimension reduction via binning [275]. Normalization and statistical analyses help find differential metabolites [275, 276]. Figure 9 summarizes a standardized metabolomics workflow. Building on these advanced analytical techniques, recent studies have applied metabolomics to unravel the complex interactions between the GM and TB.
FIGURE 9.

Overview of the general metabolomics workflow. The metabolomics pipeline comprises several key steps, beginning with sample collection and preparation, followed by analytical profiling using high‐resolution platforms such as chromatography, mass spectrometry (MS), nuclear magnetic resonance (NMR) spectroscopy. Data acquisition includes pre‐processing, clean‐up, and statistical analysis. The final step involves the identification and interpretation of metabolic signatures to reveal underlying biological processes and disease mechanisms. Some of the illustrative elements were created using BioRender (https://BioRender.com).
Metabolome‐profiling in GM–TB interactions
MTB establishes its infectious niche by remodeling host metabolic networks, primarily disrupting lipid, carbohydrate, and amino acid homeostasis. Wang et al. [25] employed untargeted GC‐MS and 16S rRNA gene sequencing to demonstrate that active TB significantly depletes SCFAs and reshapes the fecal metabolome compared to HCs. SCFAs play dual roles in maintaining intestinal barrier integrity and modulating immune responses, and their reduction may exacerbate inflammatory cascades [25].
Beyond energy metabolism, amino acid dysregulation emerges as a critical feature of TB pathogenesis. TB patients show reduced serum levels of histidine and glutamine [277, 278], while perturbations in tryptophan, arginine, and gamma‐aminobutyrate (GABA) pathways directly influence immune signaling and host defense mechanisms [279]. Albors‐Vaquer et al. [280] further identified distinct serum metabolic signatures that differentiate active TB patients from asymptomatic household contacts and HCs, highlighting amino acid reprogramming as a biomarker of infection status.
Metabolomics provides unique insights into host‐microbiome crosstalk during TB progression. Mason et al. [273] utilized GC‐MS to identify cerebrospinal fluid biomarkers for pediatric TBM, while Yang et al. [281] integrated fecal amplicon sequencing with LC‐MS/MS metabolomics to distinguish TB, LTBI, and HCs. Notably, metabolomic approaches demonstrate exceptional diagnostic specificity: Chen et al. identified phenylalanylphenylalanine as a serum biomarker differentiating TB from lung cancer using ultra performance liquid chromatography (UPLC)‐QTOF/MS [282], and urinary metabolomic profiles effectively discriminate TB from non‐tuberculous mycobacterial infections [283].
Longitudinal metabolomic profiling can predict TB progression. Weiner et al. [284] identified trans‐African serum biosignatures predicting TB progression in household contacts. Luo et al. [121] developed fecal biomarker panels to stratify LTBI progression risk.
Metabolomics holds translational potential for treatment optimization. Sahu et al. [122] tracked partial GM restoration and metabolic normalization in TB patients during treatment, and Jeyanathan et al. [285] studied BCG vaccination effects. Lipidomic profiling by UPLC‐QTOF MS found potential treatment response biomarkers [277, 286]. Opperman et al. and Luies et al. identified GM‐derived metabolites related to treatment failure [123, 287].
Despite these advances, challenges persist in generalizing findings due to interindividual metabolic variability influenced by GM composition, diet, and environmental factors. Large‐scale validation across diverse cohorts remains essential before clinical translation [261]. Nevertheless, metabolomics—particularly when integrated with multi‐omics and ML—offers unprecedented insights into GM‐TB interplay, advancing diagnostic, prognostic, and therapeutic strategies.
AI technologies
The 2024 Nobel Prize in Physics recognized Geoffrey Hinton and John Hopfield for pioneering neural network architectures that emulate neurobiological processes, while the Chemistry Prize honored David Baker, Demis Hassabis, and John Jumper for their AI‐driven breakthroughs in protein structure prediction. These accolades underscore AI's transformative role in decoding complex biological systems, including genomic and microbiome datasets, to advance personalized diagnostics and therapeutic strategies [288, 289].
AI in analyzing and interpreting complex GM data
GM research generates high‐dimensional, heterogeneous datasets that are hard to analyze with conventional analytical methods. Microbiome datasets from sequencing need preprocessing due to class imbalance and sparsity before applying AI for pattern recognition (Tables 8 and 9) [291, 292, 293, 294, 295, 296, 297, 298, 299, 300, 301, 302, 303].
Table 8.
Open‐source resources for carrying out GM data analysis.
| Category | Resource/database | Description | Link | Refs |
|---|---|---|---|---|
| Microbiome databases | EasyMetagenome | A user‐friendly and flexible pipeline for shotgun metagenomic analysis | https://github.com/YongxinLiu/EasyMetagenome | [290] |
| HMP | Comprehensive datasets of microbial communities from various human body sites | https://hmpdacc.org/ | [291] | |
| Gut feeling knowledge base | Database on GM–host interactions, including metabolites and disease associations | https://hivelab.biochemistry.gwu.edu/gfkbbaseline | [292] | |
| IMG | Comprehensive resources for microbial genome and metagenome analysis | https://img.jgi.doe.gov/ | [293] | |
| Metabolomics | HMDB | Database of metabolites, including those related to the GM | https://hmdb.ca/ | [294] |
| METLIN | Repository of metabolomic and lipidomic data, useful for gut microbial metabolite analysis | https://metlin.scripps.edu/landing_page.php?pgcontent=mainPage | [295] | |
| Microbial interactions | KEGG | Database linking microbiome functional pathways to host health and disease | https://www.genome.jp/kegg/ | [296] |
| BioCyc | Collection of pathway/genome databases for microbial and host interaction | https://biocyc.org/ | [297] | |
| STRING | Database for protein–protein interactions, including microbial proteins | https://string-db.org/ | [298] | |
| Clinical datasets | EBI metagenomics | A platform for analyzing and archiving metagenomic data, including GM | https://www.ebi.ac.uk/metagenomics | [299] |
| GMrepo | Comprehensive database of human GM‐related studies and datasets | https://gmrepo.humangut.info/home | [300] | |
| Taxonomic databases | Greengenes | Database for 16S rRNA gene sequences to study microbial taxonomy | https://rnacentral.org/expert-database/greengenes | [301] |
| SILVA ribosomal RNA gene database project | High‐quality ribosomal RNA sequences database for microbial taxonomy analysis | https://www.arb-silva.de/ | [302] | |
| RDP | Database for analyzing and comparing rRNA sequences | https://www.glbrc.org/data-and-tools/glbrc-data-sets/ribosomal-database-project | [303] | |
| Disease‐specific resources | GutMDisorder | Database linking the GM to metabolic disorders | http://bio-computing.hrbmu.edu.cn/gutMDisorder/ | [304] |
Abbreviations: EBI, European Bioinformatics Institute; GM, gut microbiota; GMrepo, Human Gut Microbiota Research Database; GutMDisorder, Gut Microbiota‐Metabolic Disorder Database; HMP, human microbiome project; SILVA, Small subunit rRNA gene database; STRING, Search Tool for the Retrieval of Interacting Genes/Proteins.
TABLE 9.
Summary of implementation for AI/ML with their motivations.
| Motive | Use case | AI/ML model utilized | Refs |
|---|---|---|---|
| GM clustering | Clustering enterotype microbiome for pattern identification | K‐Means clustering, PCA | [305] |
| Predictive modeling | Colorectal cancer detection via identifying specific microbes involved in dysbiosis | RF, XGboost, SVM | [306] |
| Diagnostic application | Identifying the microbiome as a biomarker for anti‐TB drug‐induced inflammation | RF | [70] |
| Identification of microbiome pattern linked to ATB‐DILI | RF, SVM | [307] | |
| Automated diagnosis of TB via analyzing chest X‐ray scans | RF | [308] | |
| Treatment outcome | Predictive modeling to address loss to follow‐up in TB management | XGboost | [309] |
| ML models to predict treatment unsuccess in PTB patient | Decision trees | [310] | |
| Personalized treatment | Identifying new TB therapy combinations based on patients with MDR and nonresponsive patients. | Multi‐modal AI agent | [311] |
| Early detection systems to detect adverse prognosis in patients receiving anti‐TB therapy | XGBoost, RF, MLP, light GBM, logistic regression, and SVM | [312] |
Abbreviations: ATB‐DILI, antituberculosis drug‐induced liver injury; GBM, gradient boosting machine; GM, gut microbiota; ML, machine learning; MDR‐TB, multi‐drug‐resistant tuberculosis; MLP, multi‐layer perceptron; PCA, principal component analysis; RF, random forest; SVM, support vector machine.
Types of AI techniques used for GM data analysis
ML has two main types: supervised learning and unsupervised learning. Supervised learning employs labeled data to train models for classification or regression, like Support Vector Machines (SVM), random forest (RF), and Naïve Bayes, which help find microbial biomarkers and disease diagnostics. For instance, RF identified Flavonifractor plautii as a colorectal cancer (CRC)‐specific GM biomarker [313]. Unsupervised learning identifies intrinsic patterns in unlabeled data through clustering (K‐means, DBSCAN), association rule mining, or dimensionality reduction (principal component analysis [PCA], t‐SNE, UMAP). Knights et al. [305] applied K‐means clustering to define GM enterotypes, and Shi et al. [314] demonstrated that combining Bray–Curtis and UniFrac metrics optimizes clustering robustness across diverse datasets.
Deep learning (DL) can handle high‐dimensional data (e.g., genomics, metagenomics) through hierarchical feature extraction, although it suffers from interpretability issues, particularly in clinical applications. Novielli et al. [306] integrated SHapley Additive exPlanations (SHAP) values into CRC‐GM analyses to explain DL models [306, 315].
Resources for databases for GM analysis
Bioinformaticians investigating GM–host interactions can leverage a suite of specialized tools and databases tailored for multi‐omics integration (Table 8) [290, 291, 292, 293, 294, 295, 296, 297, 298, 299, 300, 301, 302, 303, 304]. Foundational frameworks such as the Human Microbiome Project (HMP) [291] provide standardized references for microbial community profiling. For streamlined data processing, platforms like EasyMetagenome offer user‐friendly pipelines for metagenomic analysis [290], while functional annotation is supported by curated databases including the Gut Feeling Knowledge Base and the Integrated Microbial Genomes (IMG) system [292, 293].
Metabolomic integration is facilitated by human metabolome database and METLIN, which host comprehensive metabolite annotations and spectral libraries [294, 295]. To decipher host‐microbe crosstalk, pathway‐centric platforms like KEGG [296], BioCyc [297], and STRING [298] enable systematic analysis of metabolic interactions and protein networks.
Clinical‐translational resources further bridge mechanistic insights to patient‐oriented applications. The European Bioinformatics Institute Metagenomics platform [299] and GMrepo [300] provide clinical metagenomic datasets, whereas taxonomic classification relies on reference databases such as Greengenes [301], SILVA [302], and the Ribosomal Database Project [303]. For disease‐specific investigations, GutMDisorder curates experimentally validated GM‐disease associations [304]. These resources, combined with advanced ML algorithms, enhance our ability to uncover microbiome signatures and translate them into actionable insights for TB management.
ML algorithms for pattern recognition in GM‐TB studies
ML has emerged as a pivotal tool for uncovering microbiome signatures in TB pathogenesis and treatment outcomes (Figure 10). Wipperman et al. [70] used RF regression to analyze multi‐modal data and found biomarkers for inflammatory resolution during anti‐TB therapy. Their model revealed inverse correlations between Bacilli/Pseudomonadota abundance and pro‐inflammatory cytokines (TNF‐α, IL‐6), demonstrating ML's capacity to decode microbiome–immune interactions. Furthermore, Wang et al. [307] addressed anti‐TB DILI (ATB‐DILI) through SVM and RF analysis of urine metabolomics from 69 patients. They identified Negativicoccus and Actinotignum as predictive taxa, achieving high diagnostic accuracy for ATB‐DILI risk stratification. Extending these principles to veterinary medicine, Lee et al. [316] developed an ML model with 96% accuracy for detecting Mycobacterium avium infections in cattle, pinpointing Clostridium as a key diagnostic marker. These studies collectively highlight ML's versatility in translating GM patterns into clinically actionable insights across human and animal TB research.
FIGURE 10.

Applications of artificial intelligence (AI)/machine learning (ML) in microbiome research: from data collection to implementation. The diagram outlines the workflow for AI/ML integration in microbiome studies. The process begins with sample collection and processing, followed by multi‐omics profiling approaches (e.g., DNA, RNA, metabolite, and protein‐based analyses) to generate high‐dimensional datasets (e.g., taxonomic composition, alpha and beta diversity, functional profiling). Unsupervised learning methods, such as clustering and dimensionality reduction, are used to explore microbial community structure and diversity. Supervised learning algorithms (e.g., classification and regression) enable prediction of clinically relevant outcomes, such as disease diagnosis and treatment response. Deep learning (DL) methods, using architectures such as neural networks and Long Short‐Term Memory (LSTM) networks, further enhance the ability to model complex, nonlinear relationships within microbiome data.
Predictive modeling for TB treatment response
Development of predictive models for treatment outcomes
Predictive modeling has emerged as a cornerstone in precision TB management, integrating multi‐dimensional patient data—including microbiome profiles, genetic markers, and immunological parameters—to forecast therapeutic trajectories. For treatment failure prediction, Peetluk et al. [310] developed a multivariate model identifying clinical and demographic predictors of unsuccessful pulmonary TB (PTB) outcomes. Addressing patient adherence, Chen et al. [309] employed XGBoost algorithms to predict loss to follow‐up risks in TB cohorts, enhancing intervention strategies.
Imaging‐driven prognostication further extends predictive capabilities. Lv et al. [308] demonstrated the utility of computed tomography (CT)‐derived radiomic features in forecasting sputum culture conversion timelines for MDR‐TB patients. Supervised learning applications have also advanced outcome stratification: Host et al. [317] applied supervised ML (RF, SVM, gradient‐boosted trees) to predict treatment success in DR‐TB, while Fayaz et al. [318] optimized decision tree algorithms for outcome prediction in PTB cohorts (AUC: 0.90, accuracy: 92.7%).
AI‐driven personalization of TB treatment strategies
Advances in microbiome analysis offer novel opportunities for precision medicine via host‐directed microbiota modulation. Probiotics, prebiotics, and microbiota transplantation could restore GM balance, serving as adjunct therapies tailored to individual GM profiles [127].
Multimodal AI systems that integrate clinical, imaging, and genetic data are enhancing predictive accuracy. Sambarey et al. [311] developed a model using multi‐domain data (radiological, microbiological, demographic) to predict MDR‐TB outcomes and find optimized drug combinations. Ford et al. [319] simplified complex antibiotic regimen design by training ML models on in vitro drug–pair interactions, enabling prediction of >500 higher‐order combinations to surpass current regimen efficacy. Liao et al. [312] created an AI tool for early detection of adverse prognoses (e.g., acute hepatitis, respiratory failure) in 2248 TB patients, demonstrating high AUCs for critical outcomes. Such models enable timely regimen adjustments, improving survival in high‐risk populations.
Predictive modeling for enhanced TB diagnostics
Traditional TB diagnostics (sputum smear microscopy, culture, and polymerase chain reaction) face limitations in sensitivity, speed, and resource demands. AI‐driven solutions are emerging to address these gaps (Table 9). Xiong et al. [320] developed TB‐AI, a CNN model detecting acid‐fast bacilli in microscopy images with 97.94% sensitivity and 83.65% specificity, reducing diagnostic delays and human error. Sharma et al. [321] applied AI to preprocessed chest X‐rays (histogram filtering, CLAHE, median filtering), achieving 98% accuracy with decision trees for pulmonary/extrapulmonary TB detection.
Cough audio analysis represents a promising low‐resource diagnostic frontier. Models like HeAR [322], Swaasa AI [323], and TBscreen [324] leverage self‐supervised learning on datasets from high‐burden regions. However, challenges remain in deploying complex models (e.g., Google's HeAR, trained on 313 million audio clips) on portable devices, which necessitates optimization via distillation or quantization [325].
Challenges and ethical considerations in AI‐driven TB research
The integration of AI into TB research and clinical practice offers transformative potential for improving diagnostics and treatment, yet it introduces critical technical and ethical challenges that demand urgent resolution [326]. A primary concern lies in data limitations and algorithmic bias, as current datasets often inadequately represent diverse global populations, compromising the reliability and generalizability of AI models—a challenge compounded by the “black box” nature of many systems, which undermines clinical trust and underscores the need for explainable AI (XAI) frameworks [327, 328, 329]. Equally pressing are ethical dilemmas surrounding data privacy, equity, and accountability, particularly given the risk of exacerbating global health disparities if AI tools are disproportionately trained on data from high‐income populations, leaving low‐resource, high‐TB‐burden regions underserved [330]. To address these interconnected issues, multidisciplinary collaboration is essential: policymakers must establish robust standards for data privacy and algorithmic auditing, researchers should prioritize creating open‐access, demographically diverse datasets while validating models across heterogeneous cohorts [331], and healthcare systems need to invest in clinician education to align AI capabilities with practical workflows. Only through such coordinated efforts—balancing technological innovation with ethical governance and infrastructural adaptation—can stakeholders overcome these barriers to fully harness AI's potential in reducing the global TB burden.
Microbiome‐TB bioinformatics
Recent advances in sequencing technologies and bioinformatics methodologies [332, 333] have revolutionized microbiome research. However, they bring challenges in managing the scale and complexity of generated data. Microbiome bioinformatics is key to processing and analyzing these datasets in the context of GM‐TB dynamics [22, 334, 335]. This section explores key aspects of microbiome bioinformatics, including data management pipelines, multi‐omics integration, and specialized tools for GM‐TB research.
Data management and analysis pipelines
Effective microbiome‐TB research hinges on standardized workflows for managing heterogeneous datasets. Bioinformatics pipelines address challenges in data storage, computational processing, and analytical reproducibility, enabling researchers to gain insights from complex microbial communities.
Challenges in managing large‐scale microbiome data
High‐throughput sequencing generates voluminous and heterogeneous datasets, posing significant logistical and analytical hurdles. Storing and retrieving this data is difficult, and the variety of data types (like 16S rRNA profiling, shotgun metagenomics, metabolomics, and clinical metadata) makes integration complex due to format and analytical differences [290, 336, 337, 338, 339, 340].
Standardization remains a persistent challenge. Variability in experimental protocols, sequencing platforms, and preprocessing methods introduces batch effects that obscure biological signals [340]. Conventional normalization tools like Combat [341] and Limma [342] often fail to address the sparsity and high dimensionality of microbiome datasets. The absence of universal data standards and metadata reporting guidelines exacerbates these issues, hindering cross‐study comparisons and reproducibility [293].
Computational demands are high for microbiome data analysis, requiring high‐performance computing resources and specialized pipelines for quality control, taxonomic classification, functional annotation, and statistical modeling [343, 344]. Ethical considerations in data sharing, involving sensitive clinical information add to the complexity.
Development of bioinformatics tools for data analysis
Specialized tools address distinct stages of microbiome analysis (Table 10) [300, 323, 345, 346, 347, 348, 349, 350, 351, 352, 353, 354, 355, 356, 357, 358, 359, 360, 361, 362]. Quality control begins with FastQC [363], which evaluates raw sequencing quality, followed by Trimmomatic [364] for adapter removal and read trimming. DADA2 [323] and EasyAmplicon [347] enhance accuracy by resolving amplicon sequence variants, and distinguishing biological sequences from technical artifacts.
TABLE 10.
Bioinformatics tools for data analysis.
| Tool name | Function/application area | Key features | Refs |
|---|---|---|---|
| FastQC | Quality control and preprocessing | Provides comprehensive quality assessments of raw sequencing data, identifying issues like low base quality and adapter contamination. | [345] |
| Trimmomatic | Quality control and preprocessing | To enhance data reliability, perform trimming of low‐quality bases and remove adapter sequences. | [346] |
| DADA2 | Quality control and preprocessing | Denoises sequencing data by distinguishing true biological sequences from sequencing artifacts. | [323] |
| EasyAmplicon | Amplicon data analysis | An easy‐to‐use, open‐source, reproducible, and community ‐based pipeline. | [347] |
| QIIME2 | Taxonomic classification | Versatile platform for end‐to‐end microbiome analysis, integrating plugins for sequence processing, taxonomic assignment, and diversity analysis. | [348] |
| MetaPhlAn 4 | Taxonomic classification | Profiles microbial communities using clade‐specific marker genes for rapid and precise taxonomic assignment. | [349] |
| Kraken2 | Taxonomic classification | Employs ultra‐fast k‐mer‐based sequence classification for high‐resolution microbial profiling. | [350] |
| HUMAnN | Functional profiling | Reconstructs metabolic pathways and quantifies their abundance within microbial communities. | [351] |
| PICRUSt | Functional profiling | Predicts functional gene content based on phylogenetic placement of taxa, inferring metabolic potential from taxonomic profiles. | [352] |
| phyloseq(R package) | Statistical analysis and visualization | Comprehensive framework for importing, analyzing, and visualizing microbiome census data. | [353] |
| vegan (R package) | Statistical analysis and visualization | Advanced ecological analysis tools for examining diversity and community structure. | [354] |
| MicrobiomeAnalyst | Statistical analysis and visualization | Web‐based platform offers a user‐friendly interface for statistical testing, diversity analysis, and data visualization. | [355] |
| MetaWARP | High‐throughput data processing | Accelerates workflows for large‐scale metagenomic data analysis. | [356] |
| UPARSE | OTU clustering | Improves taxonomic assignment and reduces error rates in microbiome data analysis. | [357] |
| Mothur | 16S rRNA sequence processing | Provides sequence processing, alignment, and analysis tools in microbial ecology studies. | [358] |
| Parallel‐META3 | Functional annotation | High‐throughput tool for large‐scale metagenomic data set analysis | [359] |
| MTBseq | WGS analysis | Integrates various analytical steps into a user‐friendly framework for TB research | [360] |
| VSEARCH | OTU clustering | Improves taxonomic assignment and reduces error rates in microbiome data analysis. | [361] |
| GMrepo v2 | Microbiome database | Facilitates storage and querying of microbiome datasets for comparative analysis. | [300] |
| USEARCH12 | OTU picking and clustering | Speeds up OTU assignment and enhances data pre‐processing. | [362] |
Abbreviation: WGS, whole‐genome sequencing.
Taxonomic profiling leverages platforms like QIIME2 [365], which integrates plugins for end‐to‐end analysis, and MetaPhlAn4 [349], which uses clade‐specific marker genes for rapid classification. Kraken2 [366] employs k‐mer matching for high‐resolution taxonomic assignments. TB‐specific tools such as MTBseq [360] enable WGS of MTB, while MUBII‐TB‐DB [367] tracks drug resistance mutations critical for TB control.
Functional insights are derived via HUMAnN [368], which reconstructs metabolic pathways from metagenomic data, and PICRUSt [369], which infers gene content from taxonomic profiles. Downstream analysis employs R packages (phyloseq, vegan) and MicrobiomeAnalyst [370], a web platform for statistical testing, differential abundance analysis, and visualization. These tools collectively enable robust characterization of microbial communities and their functional roles in TB.
Integration of multi‐omics data
TB pathogenesis involves intricate interactions between host genetics, immune responses, and GM dynamics. Multi‐omics integration—combining genomics, transcriptomics, proteomics, and metabolomics—provides a systems‐level understanding of these interactions, identifying biomarkers and mechanistic pathways (Table 11) [371, 372, 373, 374, 375, 376, 377, 378, 379, 380, 381, 382, 383, 384, 385, 386].
TABLE 11.
Overview of multi‐omic data preprocessing and integration.
| Name | Function/application area | Key features | Refs |
|---|---|---|---|
| MultiQC | Data aggregation and reporting | Aggregates results from various bioinformatics analyses into a single report, facilitating comprehensive multi‐omics data visualization. | [371] |
| MetaboAnalyst | Multi‐omics data integration and analysis | It provides tools for metabolomic data analysis, including normalization, statistical testing, and pathway analysis, and it is integrated with other omics data. | [372] |
| iCluster | Integrated clustering of multi‐omics data | Enables simultaneous clustering of multiple omics datasets, identifying shared patterns and associations across different molecular layers. | [373] |
| MixOmics | Multi‐omics data integration and visualization | Offers a suite of statistical methods for integrating and visualizing relationships between different omics datasets, including PLS‐DA and correlation analysis. | [374] |
| MOFA | Dimensionality reduction for multi‐omics data | Implements factor analysis to identify latent factors that capture shared and unique variations across multiple omics layers, enhancing data interpretation. | [375] |
| OmicsNet | Network‐based integration of multi‐omics data | Constructs interaction networks that integrate genomic, transcriptomic, and proteomic data, highlighting key nodes and pathways involved in biological processes. | [376] |
| Cytoscape | Network visualization and analysis | Facilitates the visualization and analysis of complex interaction networks, integrating multi‐omics data to identify functional relationships and network hubs. | [377] |
| Galaxy | Integrative omics pipelines | An open‐source platform that supports the construction of custom workflows for integrating and analyzing multi‐omics data using a wide range of bioinformatics tools. | [378] |
| Qiita | Microbiome data management | Facilitates organization, sharing, and analysis of large‐scale microbiome datasets. | [379] |
| MG‐RAST | Metagenomic data integration | Provides a comprehensive pipeline for analyzing and integrating metagenomic data, including functional annotation and taxonomic classification across multiple datasets. | [380] |
| MetaPhlAn | Microbial community profiling | Provides taxonomic profiles, aiding in the characterization of microbial communities. | [381] |
| NetMoss | Network module structure shift analysis | Using integrated multi‐omics data, evaluating shifts in network modules across different states, and identifying key bacteria and interactions associated with disease transitions. | [382] |
| WGCNA | Network construction and module detection | Constructs weighted gene co‐expression networks, facilitating the identification of modules and hub genes across multi‐omics datasets. | [383] |
| IMP | Multi‐omics integration | Facilitates comprehensive analysis of microbiome‐host interactions. | [384] |
| EBI‐metagenomics | Metagenomics analysis | Provides pipelines for functional and taxonomic annotation of metagenomics data | [299] |
| Enrichr | Gene set enrichment analysis | Provides easy‐to‐use pathways and gene‐set enrichment analysis, enabling functional annotation and biological interpretation. | [385] |
| OmicsIntegrator | Network‐based multi‐omics integration | Maps protein data onto interactome networks and identifies high‐confidence subnetworks relevant to the data. Uses the Prize‐Collecting Steiner Forest algorithm to discover cellular pathways and protein interactions | [386] |
Abbreviation: PLS‐DA, partial least squares‐discriminant analysis.
Combining genomics, transcriptomics, proteomics, and metabolomics data
The systematic integration of multi‐omics data (spanning genomics, transcriptomics, proteomics, and metabolomics) is critical for elucidating the complex biological networks governing host–microbe interactions in TB and GM dynamics [387, 388, 389]. This holistic approach begins with rigorous data alignment across omics layers to ensure biological relevance and analytical coherence, followed by normalization techniques (e.g., z‐score or quantile normalization) to mitigate technical variability and preserve biologically meaningful signals. To further streamline complexity, dimensionality reduction methods such as PCA [390], t‐SNE [391], and gene panel selection algorithms [392, 393] enhance the identification of cross‐omics correlations critical to GM‐TB interplay, ultimately enabling the construction of comprehensive molecular maps that reveal metabolic pathways and functional relationships between GM and MTB (Figure 8). Genomic studies, leveraging high‐throughput sequencing, uncover the genetic basis of host–pathogen interactions, including reduced microbial diversity and antibiotic resistance determinants in TB patients [394, 395], while transcriptomics captures dynamic gene expression patterns such as upregulated pro‐inflammatory cytokines (e.g., IL‐6, TNF‐α) and microbiota‐derived genes in TB pathogenesis [396, 397]. Complementing these layers, proteomics characterizes protein networks and posttranslational modifications linked to immune signaling and MTB survival [189]. Metabolomics further enhances this systems‐level understanding by profiling host and pathogen metabolic changes, revealing altered metabolism and immune‐related metabolite signatures that could reflect both TB severity and treatment response [122, 261, 262, 263, 264]. Finally, computational integration tools like MiBiOmics, DIABLO, and IMP correlate microbial metabolites with host gene expression and protein activity, thereby unraveling pathogenic pathways and identifying potential intervention strategies (Figure 11) [398].
FIGURE 11.

Integrative multi‐omics framework for investigating gut microbiota‐tuberculosis (GM–TB) interactions. This schematic illustrates a comprehensive strategy for multi‐omics integration in GM‐TB research, combining metabolomics, proteomics, transcriptomics, and genomics approaches. The framework incorporates key study parameters, including sample size, phenotypic data, disease models, and clinical or experimental disease characteristics, to facilitate a systems‐level understanding of TB pathogenesis. Central themes include genomic instability and mutations, immune modulation, microbial dysbiosis, and the mechanistic interplay between the lung and gut microbiota.
Multi‐omics approaches to deciphering GM–TB interactions
Multi‐omics methodologies are indispensable for unraveling the complex interplay between GM and MTB pathogenesis [23], enabling systematic dissection of host–microbe interactions that orchestrate immune responses to TB. For instance, correlating host transcriptomic profiles with microbial genomic data identifies specific taxa that modulate immune‐related gene expression, with SCFAs emerging as pivotal mediators in this crosstalk [26, 399, 400]. Notably, GM dysbiosis in TB patients disrupts SCFA synthesis, impairing cytokine pathways critical for MTB containment [401, 402, 403], while metagenomic–metabolomic integration uncovers microbial metabolic pathways driving host–microbe biochemical exchanges, thereby highlighting therapeutic targets [404, 405, 406].
Beyond mechanistic insights, multi‐omics integration enables biomarker discovery: proteomic studies detect microbial enzymes (e.g., SCFA‐producing butyrate kinase) and host proteins linked to granuloma formation, whereas transcriptomic analyses reveal immune‐regulatory genes (e.g., IL‐10, TGF‐β) associated with inflammation resolution [405, 406, 407], collectively informing diagnostic tools and personalized therapies. To synthesize these multidimensional data, computational frameworks like network analysis map interactions between microbial taxa and host immune pathways, identifying hub nodes (e.g., SCFA‐regulated cytokine networks) governing MTB containment [408], while predictive models simulate how perturbations (e.g., antibiotics, diet) alter GM‐TB dynamics, guiding hypothesis‐driven interventions. Crucially, the gut–lung axis serves as a pivotal interface in TB pathogenesis, where multi‐omics elucidates how GM dysbiosis impairs lung immunity via SCFAs and gut‐derived metabolites [32, 40, 409, 410, 411]. Supporting this, metabolomic studies demonstrate that gut‐derived metabolites reshape the lung microenvironment by modulating immune cell recruitment and cytokine balance, positioning GM‐targeted restoration of SCFA production as a promising strategy to enhance anti‐TB immunity.
Bioinformatics tools for GM‐TB research
Bioinformatics plays a pivotal role in addressing the analytical challenges posed by complex datasets in GM‐TB research. Table 12 summarizes essential bioinformatics resources critical to GM‐TB studies, emphasizing their functionalities in managing, analyzing, and interpreting microbiome data [296, 367, 412, 413, 414, 415, 416, 417, 418, 419, 420, 421].
TABLE 12.
Resources, databases, and platforms for GM‐TB research.
| Name | Function/application area | Key features | Refs |
|---|---|---|---|
| HMP | Microbiome database | Comprehensive data on human microbiota across various body sites serve as a comparative study reference. | [412] |
| IMG/M | Microbiome database | Repository for metagenomic datasets, enabling access and analysis of microbiome data relevant to TB studies. | [413] |
| TB Database | TB‐Specific database | Curated genomic, transcriptomic, and proteomic data specific to MTB and related strains. | [414] |
| Mycobrowser | TB‐Specific database | The platform for exploring Mycobacterium species' genomic and functional annotations facilitates host‐microbe studies. | [415] |
| OmicsDI | Multi‐omics integration platform | Integrates various omics datasets, allowing comprehensive analyses involving genomics, transcriptomics, proteomics, etc. | [416] |
| MetaboAnalyst | Multi‐omics integration platform | Supports metabolomics data analysis and integrates with other omics data types for holistic metabolic interaction studies. | [417] |
| KEGG | Functional annotation tool | Provides pathway maps and functional annotations to understand metabolic and signaling pathways in GM–TB interactions. | [296] |
|
SEED SILAC or TMT |
Functional annotation tool | Framework for annotating and analyzing genomic and metagenomic data, exploring functional capabilities of microbial communities. | [418] |
| MUBII‐TB‐DB | Antibiotic resistance | Documents mutations associated with resistance in MTB. | [367] |
| COMBAT‐TB Workbench | Sequence analysis | Provides tools and workflows for variant discovery and phylogenetic analysis for TB research | [419] |
| GTEx Project | Tissue‐specific expression database | Comprehensive resource for studying tissue‐specific gene expression and linking it to microbiota–TB interactions. | [420] |
| ConnectivityMap | Gene expression data repository and analysis platform | Houses a perturbation‐driven gene expression data set for discovering biological connections and analyzing gene expression signatures | [421] |
Abbreviations: GM‐TB, gut microbiota‐tuberculosis; KEGG, kyoto encyclopedia of genes and genomes.
Specialized bioinformatics resources and databases
Specialized databases and analytical platforms are indispensable for advancing GM‐TB research. Microbiome repositories such as the HMP [291] and IMG (IMG/M) [293, 413] provide foundational datasets for comparative studies. HMP catalogs microbiota across human body niches, serving as a reference for GM‐TB investigations, while IMG/M offers metagenomic datasets with curated annotations for contextualizing microbial roles in TB pathogenesis. TB‐focused databases like the TB Database [414, 422] and Mycobrowser [415] centralize genomic, transcriptomic, and proteomic data on MTB strains, enabling comparative analyses of virulence and host adaptation.
Platforms such as Qiita [379] and EBI Metagenomics streamline meta‐analyses of multi‐omics datasets, supporting taxonomic and functional profiling of TB‐associated GM. Tools like PICRUSt predict microbial functional potential from metagenomic data, linking metabolic pathways to host immunity and TB progression. Proteomic interactome resources, including SInCRe [423], map host–pathogen protein interactions, elucidating molecular mechanisms of MTB persistence.
Multi‐omics integration platforms (e.g., OmicsDI [424], MetaboAnalyst [417, 425]) synthesize genomics, transcriptomics, and metabolomics data, revealing metabolic exchanges between GM and host systems. Emerging tools like MOSCA 2.0 [426] and gNOMO [427] enhance analytical precision through spectral clustering and biomarker selection algorithms. Functional annotation resources such as KEGG [296] and SEED [418] decode microbial pathways, while STRING [298] maps protein interaction networks critical to host‐MTB cross‐talk.
Databases like PATRICK [428] and GMTV [429] integrate multi‐omics data with clinical metadata, tracing genomic variations in MTB virulence and DR. Advanced tools such as MiBiOmics [430] and MultiDGD [431] employ DL to model transcriptome–metabolome interactions, identifying dysbiosis‐driven TB regulatory mechanisms. Platforms focused on vaccine development (e.g., MtbVeb [432], TBDReaM [433]) catalog immunogenic targets and DR mutations, bridging computational insights with therapeutic design. The addition of platforms such as MAGqual [434] and MetaWRAP [356] facilitates efficient data processing and analysis, increasing the accuracy and comprehensiveness of GM‐TB research.
Training and collaborative support for researchers
The effective application of bioinformatics tools in microbiome research hinges on comprehensive training programs and robust support systems tailored to researchers' diverse expertise levels. Proficiency in managing large‐scale omics data and addressing challenges associated with non‐standardized metadata requires specialized education for both bioinformaticians and experimental biologists [435, 436]. Institutions such as EBI [437, 438] and online learning platforms like Coursera and edX offer structured courses covering essential bioinformatics concepts, genomic analysis techniques, and multi‐omics integration strategies specifically adapted for microbiome studies. These programs are complemented by practical training through webinars and workshops that provide hands‐on experience with widely used analytical platforms including QIIME2 [365], PICRUSt [352], MiBiOmics [430], and MetaboAnalyst. Such interactive sessions enable researchers to master data processing workflows and optimize tool application for their specific datasets.
To facilitate independent analysis, major resources listed in Tables 10, 11, 12 incorporate detailed documentation, video tutorials, and active user communities that demystify complex analytical procedures. The open‐source paradigm further enhances accessibility through freely modifiable codebases and collaborative development frameworks [439]. Platforms like GitHub serve as critical repositories for sharing analytical workflows and experimental datasets, while international consortia such as the Global Microbiome Consortium and TBPortals Network foster interdisciplinary collaboration between microbiologists, computational biologists, and clinicians [440]. Specialized forums including BioStar, SEQanswers [441], and the Stack Overflow OMICS Forum create vital spaces for troubleshooting, knowledge exchange, and dissemination of best practices. Emerging platforms like KBase [442] extend these capabilities by enabling collaborative workflow development and transparent method sharing.
While these educational and collaborative resources form a critical foundation, equitable access to computational infrastructure remains paramount. Cloud‐based solutions such as Amazon Web Services and Google Cloud provide essential scalable environments for processing large microbiome datasets, ensuring researchers without institutional high‐performance computing resources can still conduct sophisticated analyses. This integrated ecosystem of training, open‐source tools, collaborative platforms, and cloud infrastructure collectively drives innovation in GM‐TB research by lowering technical barriers and accelerating methodological advancements.
CURRENT CHALLENGES AND FUTURE DIRECTIONS IN GM‐TB RESEARCH
While substantial advancements have been made in elucidating the interplay between GM and TB, this field remains nascent, confronting multifaceted challenges that hinder translational progress. Addressing these limitations is critical for refining research methodologies, interpreting findings accurately, and guiding clinical applications. This section systematically examines key obstacles in current research and proposes actionable strategies to advance the field.
Key challenges and limitations
Current GM‐TB research faces six major challenges (Figure 12). First, observational studies dominate the field, limiting causal inference due to potential confounding factors [66, 443]. While these studies identify associations between GM dysbiosis and TB outcomes, they cannot establish causality without controlled experimental designs. Second, small sample sizes in existing studies reduce statistical power and compromise the generalizability of findings, particularly when analyzing high‐dimensional omics data [444]. Third, population‐specific variations in GM composition—driven by genetic, geographic, dietary, and socioeconomic factors—restrict the external validity of regionally constrained studies [445, 446]. Fourth, technical limitations persist in microbiome characterization, including inconsistent sequencing protocols, bioinformatics pipelines, and data normalization methods, which hinder cross‐study comparisons [447]. Fifth, translational gaps exist between animal models and human pathophysiology [448], as murine models fail to fully replicate human TB progression or GM–host interactions. Finally, integrating multi‐omics datasets (metagenomics, metabolomics, transcriptomics) remains technically challenging, requiring advanced computational frameworks to disentangle complex GM–TB relationships.
FIGURE 12.

Challenges and Limitations in gut microbiota‐tuberculosis (GM‐TB) Studies. Current GM‐TB research faces several key challenges and limitations. Observational study designs limit the ability to infer causality, in contrast to the more rigorous evidence provided by randomized controlled trials (RCTs). Small sample sizes weaken statistical power and restrict the generalizability of findings. Population heterogeneity, including genetic background, environmental exposures, and lifestyle factors, is often underrepresented. Technical limitation in microbiome analysis, such as analytical complexity, inconsistence in computational methods, and the lack of standardization, further complicate data interpretation. Translational gaps between preclinical models and human studies remain a significant barrier. Lastly, the integration of multi‐omics data, including metabolomics and proteomics, poses analytical challenges due to differences in data structure, scale, and dimensionality.
Strategic priorities for future research
To address these challenges, seven research priorities emerge (Figure 13). First, mechanistic studies should investigate GM‐mediated modulation of TB vaccine responses, including how specific microbial taxa or metabolites influence antigen presentation and adaptive immunity [449]. Second, longitudinal cohort studies are needed to evaluate GM's prophylactic potential, such as identifying protective microbial signatures or testing FMT in high‐risk populations [450]. Third, biomarker discovery efforts must prioritize validating GM‐derived diagnostic and prognostic signatures across diverse populations using standardized multi‐omics platforms [451]. Fourth, clinical trials should assess whether GM profiling can predict treatment outcomes, optimize anti‐TB drug regimens, or reduce adverse effects. Fifth, the underexplored link between GM dynamics and TB recurrence warrants investigation, particularly in posttreatment microbiological and immunological monitoring. Sixth, methodological advancements are required to harmonize multi‐omics data integration, including ML frameworks to resolve spatial–temporal interactions between GM, host immunity, and MTB. Lastly, international consortia should establish standardized protocols for GM analysis, encompassing sample collection, sequencing, and computational workflows to enhance reproducibility and cross‐study validation.
FIGURE 13.

Future directions and research priorities in gut microbiota‐tuberculosis (GM‐TB) studies. Future research directions in GM‐TB studies encompass elucidating the role of GM in modulating vaccine efficacy and shaping host immune responses, along with exploring the therapeutic potential of probiotics, prebiotics, and FMT in TB prevention. Efforts are also directed towards developing microbiome‐based diagnostic tools by identifying and validating microbial biomarkers and creating rapid, noninvasive diagnostic platforms. Treatment optimization is a growing focus, emphasizing the need to consider GM dynamics to support personalized therapeutic approaches, including tailored antimicrobial regimens. Understanding the contribution of the GM to TB recurrence and latent tuberculosis infection (LTBI) is also critical, with research aimed to identifying predictive markers and recurrence risk factors. Advanced multi‐omics data integration is being facilitated through the development of sophisticated algorithms and bioinformatics tools. Lastly, standardization of microbiome analysis workflows, spanning sample processing and computational analysis, is crucial to ensure reproducibility, comparability, and translational relevance across studies.
By systematically addressing these challenges and priorities, researchers can bridge critical knowledge gaps and accelerate the development of microbiome‐targeted interventions for TB prevention, diagnosis, and therapy.
CONCLUSION
This review comprehensively explores the complex relationship between GM and TB, revealing significant findings and potential applications in this emerging field. Based on a systematic analysis of existing literature, we have drawn the following key conclusions:
Firstly, the GM composition in TB patients significantly differs from that in healthy individuals, characterized by a marked reduction in microbial diversity, changes in the abundance of specific genera, and imbalances in functional microbial communities. These changes reflect the impact of TB on the host's GM and may play a crucial role in the disease's onset, progression, and prognosis.
Secondly, the GM participates in TB's pathological processes through various mechanisms, including regulating host immune responses, influencing the growth and metabolism of MTB, and participating in drug metabolism. Notably, the regulatory role of the GM on the immune system may be a key link between GM and TB, providing new insights into the immune pathogenesis of TB.
Thirdly, the GM shows great potential in diagnosing, treating, and preventing TB. Microbiome‐based diagnostic methods may enhance the accuracy and timeliness of TB diagnosis. Strategies to improve TB treatment by modulating the GM and reducing adverse drug reactions are being explored. Additionally, probiotic supplementation or dietary interventions to prevent TB show promising potential.
Fourthly, applying new technologies in GM‐TB research opens new avenues for understanding and managing the disease. Advanced molecular techniques, such as NGS and metagenomics, enable more detailed and precise characterization of GM. ML and AI are also being applied to identify predictive biomarkers and develop more sophisticated diagnostic and prognostic models. These technological advances are crucial for translating basic research findings into clinical applications and improving patient outcomes.
However, current research faces several limitations, such as the constraints of observational studies, insufficient sample sizes, and population or regional differences. These limitations caution us to carefully interpret and apply research findings, and more high‐quality studies are needed to validate these results. Looking ahead, many areas in the GM‐TB research field warrant further exploration. These include studying the relationship between GM and TB vaccine efficacy, developing microbiome‐based diagnostic and prognostic tools, exploring the application of GM modulation in TB prevention and treatment, and leveraging new technologies to deepen our understanding of the GM–TB interaction. Particularly, large‐scale, long‐term prospective studies will help us better understand the role of the GM in TB's natural history.
In conclusion, research on GM and TB reveals profound connections Between the human microbiome and this ancient disease. As research deepens and technologies advance, we have reason to believe that new strategies based on GM will provide novel tools and insights for TB control, ultimately contributing to more effective management of this global public health issue.
AUTHOR CONTRIBUTIONS
Yanhua Liu: Writing—original draft; visualization; methodology; investigation. Ling Yang: Investigation; writing—original draft; visualization; methodology. Maryam Meskini: Investigation; writing—original draft; methodology. Anjana Goel: Investigation; writing—original draft; visualization. Monique Opperman: Investigation; writing—original draft; methodology. Sagar Singh Shyamal: Investigation; writing—original draft; methodology. Ajay Manaithiya: Investigation; writing—original draft; methodology. Meng Xiao: Investigation; writing—original draft; methodology. Ruizi Ni: Investigation; methodology. Yajing An: Investigation; methodology. Mingming Zhang: Methodology; investigation. Yuan Tian: Investigation; methodology. Shuang Zhou: Investigation; methodology. Zhaoyang Ye: Investigation; methodology. Li Zhuang: Investigation; methodology. Linsheng Li: Investigation; methodology. Istuti Saraswat: Investigation; methodology. Ankita Kar: Investigation; methodology. Syed Luqman Ali: Investigation; methodology. Shakir Ullah: Investigation; methodology. Syed Yasir Ali: Investigation; methodology. Shradha Kaushik: Investigation; methodology. Tianmu Tian: Investigation; methodology. Mingyang Jiao: Investigation; methodology. Shujun Wang: Investigation; methodology. Giulia Ghisleni: Investigation; methodology. Alice Armanni: Investigation; methodology. Sara Fumagalli: Investigation; methodology. WenYu Wang: Investigation; methodology. Chao Cao: Investigation; methodology. Maria Carpena: Investigation; methodology; visualization. Miguel A. Prieto: Conceptualization; writing—original draft; writing—review and editing; resources; visualization. Antonia Bruno: Conceptualization; writing—review and editing; writing—original draft; resources. Chanyuan Jin: Conceptualization; writing—review and editing; writing—original draft; resources. Hanqing Hu: Conceptualization; writing—review and editing; writing—original draft; resources. Yuhang Zhang: Conceptualization; writing—review and editing; writing—original draft; resources. Ilse du Preez: Conceptualization; writing—review and editing; visualization; resources. Ashok Aspatwar: Conceptualization; writing—review and editing; resources; writing—original draft. Lingxia Zhang: Conceptualization; writing—review and editing; methodology; investigation; writing—original draft. Wenping Gong: Conceptualization; writing—review and editing; supervision; visualization; project administration; investigation; writing—original draft; resources.
CONFLICT OF INTEREST STATEMENT
The authors have declared no competing interests.
ETHICS STATEMENT
No animals or humans were involved in this study.
ACKNOWLEDGMENTS
We thank all the reviewers for their valuable comments and suggestions on our manuscript. We also extend our gratitude to the editors for their outstanding contributions to the publication of this review.
Liu, Yanhua , Yang Ling, Meskini Maryam, Goel Anjana, Opperman Monique, Shyamal Sagar Singh, Manaithiya Ajay, et al. 2025. “Gut Microbiota and Tuberculosis.” iMeta 4, e70054. 10.1002/imt2.70054
Yanhua Liu, Ling Yang, Maryam Meskini, Anjana Goel, Monique Opperman, Sagar Singh Shyamal, Ajay Manaithiya, and Meng Xiao contributed equally to this study.
Contributor Information
Miguel A. Prieto, Email: mprieto@uvigo.es.
Antonia Bruno, Email: antonia.bruno@unimib.it.
Chanyuan Jin, Email: jinchanyuanjcy@bjmu.ed.cn.
Hanqing Hu, Email: hanqinghu@bistu.edu.cn.
Yuhang Zhang, Email: yuhang@pkufh.cn.
Ilse du Preez, Email: ilse.dupreez@nwu.ac.za.
Ashok Aspatwar, Email: ashok.aspatwar@tuni.fi.
Lingxia Zhang, Email: 1707025046@stu.sqxy.edu.cn.
Wenping Gong, Email: gwp891015@whu.edu.cn.
DATA AVAILABILITY STATEMENT
No new data were generated or analyzed in this review. Supplementary materials (graphical abstract, slides, videos, Chinese translated version, and update materials) may be found in the online DOI or iMeta Science http://www.imeta.science/.
REFERENCES
- 1. Zumla, Alimuddin , Raviglione Mario, Hafner Richard, and Fordham von Reyn C.. 2013. “Tuberculosis.” New England Journal of Medicine 368: 745–755. 10.1056/NEJMra1200894 [DOI] [PubMed] [Google Scholar]
- 2. Natarajan, Arvind , Beena P. M., Devnikar Anushka V., and Mali Sagar. 2020. “A Systemic Review on Tuberculosis.” Indian Journal of Tuberculosis 67: 295–311. 10.1016/j.ijtb.2020.02.005 [DOI] [PubMed] [Google Scholar]
- 3.World Health Organization. 2024. “Global Tuberculosis Report 2024.” Geneva: World Health Organization: 1−68. https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2024
- 4. Lin, Zixun , Sun Liqin, Wang Cheng, Wang Fuxiang, Wang Jun, Li Qian, and Lu Hongzhou. 2023. “Bottlenecks and Recent Advancements in Detecting Mycobacterium Tuberculosis in Patients With HIV.” iLABMED 1: 44–57. 10.1002/ila2.11 [DOI] [Google Scholar]
- 5. Chen, Zhi , Wang Tao, Du Jingli, Sun Lin, Wang Guirong, Ni Ruizi, An Yajing, et al. 2025. “Decoding the WHO Global Tuberculosis Report 2024: A Critical Analysis of Global and Chinese Key Data.” Zoonoses 5(1): 1. 10.15212/zoonoses-2024-0061 [DOI] [Google Scholar]
- 6. Wang, Juanjuan , Zhu Ningning, Su Xiaomin, Gao Yunhuan, and Yang Rongcun. 2023. “Gut‐Microbiota‐Derived Metabolites Maintain Gut and Systemic Immune Homeostasis.” Cells 12: 793. 10.3390/cells12050793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ma, Pei‐Jun , Wang Meng‐Meng, and Wang Yun. 2022. “Gut Microbiota: A New Insight Into Lung Diseases.” Biomedicine & Pharmacotherapy 155: 113810. 10.1016/j.biopha.2022.113810 [DOI] [PubMed] [Google Scholar]
- 8. Zhao, Min'an , Chu Jiayi, Feng Shiyao, Guo Chuanhao, Xue Baigong, He Kan, and Li Lisha. 2023. “Immunological Mechanisms of Inflammatory Diseases Caused by Gut Microbiota Dysbiosis: A Review.” Biomedicine & Pharmacotherapy 164: 114985. 10.1016/j.biopha.2023.114985 [DOI] [PubMed] [Google Scholar]
- 9. Sittipo, Panida , Lobionda Stefani, Lee Yun Kyung, and Maynard Craig L.. 2018. “Intestinal Microbiota and the Immune System in Metabolic Diseases.” Journal of Microbiology 56: 154–162. 10.1007/s12275-018-7548-y [DOI] [PubMed] [Google Scholar]
- 10. Xue, Kaiyang , Li Jiawei, and Huang Ruijie. 2024. “The Immunoregulatory Role of Gut Microbiota in the Incidence, Progression, and Therapy of Breast Cancer.” Frontiers in Cellular and Infection Microbiology 14: 1411249. 10.3389/fcimb.2024.1411249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Enaud, Raphaël , Prevel Renaud, Ciarlo Eleonora, Beaufils Fabien, Wieërs Gregoire, Guery Benoit, and Delhaes Laurence. 2020. “The Gut‐Lung Axis in Health and Respiratory Diseases: A Place for Inter‐Organ and Inter‐Kingdom Crosstalks.” Frontiers in Cellular and Infection Microbiology 10: 9. 10.3389/fcimb.2020.00009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marsland, Benjamin J. , Trompette Aurélien, and Gollwitzer Eva S.. 2015. “The Gut‐Lung Axis in Respiratory Disease.” Annals of the American Thoracic Society 12(Suppl 2): S150–S156. 10.1513/AnnalsATS.201503-133AW [DOI] [PubMed] [Google Scholar]
- 13. Harris, Nicola L. , and Marsland Benjamin J.. 2025. “The Gut‐Lung Axis: Protozoa Join the Party.” Cell 188: 275–277. 10.1016/j.cell.2024.12.027 [DOI] [PubMed] [Google Scholar]
- 14. Park, Jin‐Sun , Lee Eun‐Jung, Lee Jae‐Chul, Kim Won‐Ki, and Kim Hee‐Sun. 2007. “Anti‐Inflammatory Effects of Short Chain Fatty Acids in IFN‐γ‐Stimulated RAW 264.7 Murine Macrophage Cells: Involvement of NF‐κB and ERK Signaling Pathways.” International Immunopharmacology 7: 70–77. 10.1016/j.intimp.2006.08.015 [DOI] [PubMed] [Google Scholar]
- 15. Chen, Lingming , Zhang Guoliang, Li Guobao, Wang Wei, Ge Zhenhuang, Yang Yi, He Xing, et al. 2022. “ Ifnar Gene Variants Influence Gut Microbial Production of Palmitoleic Acid and Host Immune Responses to Tuberculosis.” Nature Metabolism 4: 359–373. 10.1038/s42255-022-00547-3 [DOI] [PubMed] [Google Scholar]
- 16. Yunusbaeva, Milyausha , Borodina Liliya, Terentyeva Darya, Bogdanova Anna, Zakirova Aigul, Bulatov Shamil, Altinbaev Radick, Bilalov Fanil, and Yunusbayev Bayazit. 2024. “Excess Fermentation and Lactic Acidosis as Detrimental Functions of the Gut Microbes in Treatment‐Naive TB Patients.” Frontiers in Cellular and Infection Microbiology 14: 1331521. 10.3389/fcimb.2024.1331521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Enjeti, Aditya , Sathkumara Harindra Darshana, and Kupz Andreas. 2023. “Impact of the Gut‐Lung Axis on Tuberculosis Susceptibility and Progression.” Frontiers in Microbiology 14: 1209932. 10.3389/fmicb.2023.1209932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cao, Demin , Liu Weihua, Lyu Na, Li Boxing, Song Weibo, Yang Yanxiao, Zhu Jianliang, Zhang Zhiguo, and Zhu Baoli. 2021. “Gut Mycobiota Dysbiosis in Pulmonary Tuberculosis Patients Undergoing Anti‐Tuberculosis Treatment.” Microbiology Spectrum 9: e0061521. 10.1128/spectrum.00615-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang, Shiang‐Fen , Yang Ying‐Ying, Chou Kun‐Ta, Fung Chang‐Phone, Wang Fu‐Der, and Su Wei‐Juin. 2019. “Systemic Proinflammation After Mycobacterium Tuberculosis Infection Was Correlated to the Gut Microbiome in HIV‐Uninfected Humans.” European Journal of Clinical Investigation 49: e13068. 10.1111/eci.13068 [DOI] [PubMed] [Google Scholar]
- 20. Zhuo, Qiqi , Zhang Xianyi, Zhang Kehong, Chen Chan, Huang Zhen, and Xu Yuzhong. 2023. “The Gut and Lung Microbiota in Pulmonary Tuberculosis: Susceptibility, Function, and New Insights Into Treatment.” Expert Review of Anti‐infective Therapy 21: 1355–1364. 10.1080/14787210.2023.2283036 [DOI] [PubMed] [Google Scholar]
- 21. Namasivayam, Sivaranjani , Kauffman Keith D., McCulloch John A., Yuan Wuxing, Thovarai Vishal, Mittereder Lara R., Trinchieri Giorgio, Barber Daniel L., and Sher Alan. 2019. “Correlation Between Disease Severity and the Intestinal Microbiome in Mycobacterium Tuberculosis‐Infected Rhesus Macaques.” mBio 10: e01018–e01019. 10.1128/mBio.01018-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Naidoo, Charissa C. , Nyawo Georgina R., Wu Benjamin G., Walzl Gerhard, Warren Robin M., Segal Leopoldo N., and Theron Grant. 2019. “The Microbiome and Tuberculosis: State of the Art, Potential Applications, and Defining the Clinical Research Agenda.” The Lancet Respiratory Medicine 7: 892–906. 10.1016/S2213-2600(18)30501-0 [DOI] [PubMed] [Google Scholar]
- 23. Yuan, Zongxiang , Kang Yiwen, Mo Chuye, Huang Shihui, Qin Fang, Zhang Junhan, Wang Fengyi, et al. 2024. “Causal Relationship Between Gut Microbiota and Tuberculosis: A Bidirectional Two‐Sample Mendelian Randomization Analysis.” Respiratory Research 25: 16. 10.1186/s12931-023-02652-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Han, MeiQing , Wang Xia, Zhang JiaMin, Su Lin, Ishaq Hafiz Muhammad, Li Duan, Cui JunWei, Zhao HuaJie, and Yang Fan. 2024. “Gut Bacterial and Fungal Dysbiosis in Tuberculosis Patients.” BMC Microbiology 24: 141. 10.1186/s12866-024-03275-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang, Shuting , Yang Liya, Hu Haiyang, Lv Longxian, Ji Zhongkang, Zhao Yanming, Zhang Hua, et al. 2022. “Characteristic Gut Microbiota and Metabolic Changes in Patients With Pulmonary Tuberculosis.” Microbial Biotechnology 15: 262–275. 10.1111/1751-7915.13761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang, Yue , Deng Yali, Liu Nianqiang, Chen Yanggui, Jiang Yuandong, Teng Zihao, Ma Zhi, Chang Yuxue, and Xiang Yang. 2022. “Alterations in the Gut Microbiome of Individuals With Tuberculosis of Different Disease States.” Frontiers in Cellular and Infection Microbiology 12: 836987. 10.3389/fcimb.2022.836987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hu, Yongfei , Feng Yuqing, Wu Jiannan, Liu Fei, Zhang Zhiguo, Hao Yanan, Liang Shihao, et al. 2019. “The Gut Microbiome Signatures Discriminate Healthy From Pulmonary Tuberculosis Patients.” Frontiers in Cellular and Infection Microbiology 9: 90. 10.3389/fcimb.2019.00090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maji, Abhijit , Misra Richa, Dhakan Darshan B., Gupta Vipin, Mahato Nitish K., Saxena Rituja, Mittal Parul, et al. 2018. “Gut Microbiome Contributes to Impairment of Immunity in Pulmonary Tuberculosis Patients by Alteration of Butyrate and Propionate Producers.” Environmental Microbiology 20: 402–419. 10.1111/1462-2920.14015 [DOI] [PubMed] [Google Scholar]
- 29. Namasivayam, Sivaranjani , Maiga Mamoudou, Yuan Wuxing, Thovarai Vishal, Costa Diego L., Mittereder Lara R., Wipperman Matthew F., et al. 2017. “Longitudinal Profiling Reveals a Persistent Intestinal Dysbiosis Triggered by Conventional Anti‐Tuberculosis Therapy.” Microbiome 5: 71. 10.1186/s40168-017-0286-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Luo, Mei , Liu Yong, Wu Pengfei, Luo Dong‐Xia, Sun Qun, Zheng Han, Hu Richard, et al. 2017. “Alternation of Gut Microbiota in Patients With Pulmonary Tuberculosis.” Frontiers in Physiology 8: 822. 10.3389/fphys.2017.00822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Winglee, Kathryn , Eloe‐Fadrosh Emiley, Gupta Shashank, Guo Haidan, Fraser Claire, and Bishai William. 2014. “Aerosol Mycobacterium Tuberculosis Infection Causes Rapid Loss of Diversity in Gut Microbiota.” PLOS One 9: e97048. 10.1371/journal.pone.0097048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alvarado‐Peña, Néstor , Galeana‐Cadena David, Gómez‐García Itzel Alejandra, Mainero Xavier Soberón, and Silva‐Herzog Eugenia. 2023. “The Microbiome and the Gut‐Lung Axis in Tuberculosis: Interplay in the Course of Disease and Treatment.” Frontiers in Microbiology 14: 1237998. 10.3389/fmicb.2023.1237998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Diallo, Dramane , Sun Shan, Somboro Anou Moise, Baya Bocar, Kone Amadou, Diarra Bassirou, Nantoume Mohamed, et al. 2023. “Metabolic and Immune Consequences of Antibiotic Related Microbiome Alterations During First‐Line Tuberculosis Treatment in Bamako, Mali.” Research square Preprint: 3232670. 10.21203/rs.3.rs-3232670/v1 [DOI] [PMC free article] [PubMed]
- 34. Namasivayam, Sivaranjani , Diarra Bassirou, Diabate Seydou, Sarro Yeya dit Sadio, Kone Amadou, Kone Bourahima, Tolofoudie Mohamed, et al. 2020. “Patients Infected With Mycobacterium Africanum Versus Mycobacterium Tuberculosis Possess Distinct Intestinal Microbiota.” PLoS Neglected Tropical Diseases 14: e0008230. 10.1371/journal.pntd.0008230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Majlessi, L. , Sayes F., Bureau J. F., Pawlik A., Michel V., Jouvion G., Huerre M., et al. 2017. “Colonization With Helicobacter Is Concomitant With Modified Gut Microbiota and Drastic Failure of the Immune Control of Mycobacterium Tuberculosis .” Mucosal Immunology 10: 1178–1189. 10.1038/mi.2016.140 [DOI] [PubMed] [Google Scholar]
- 36. Arias, Lilibeth , Goig Galo Adrián, Cardona Paula, Torres‐Puente Manuela, Díaz Jorge, Rosales Yaiza, Garcia Eric, et al. 2019. “Influence of Gut Microbiota on Progression to Tuberculosis Generated by High Fat Diet‐Induced Obesity in C3HeB/FeJ Mice.” Frontiers in Immunology 10: 2464. 10.3389/fimmu.2019.02464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pott, Johanna , and Hornef Mathias. 2012. “Innate Immune Signalling at the Intestinal Epithelium in Homeostasis and Disease.” EMBO Reports 13: 684–698. 10.1038/embor.2012.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rivière, Audrey , Selak Marija, Lantin David, Leroy Frédéric, and De Vuyst Luc. 2016. “Bifidobacteria and Butyrate‐Producing Colon Bacteria: Importance and Strategies for Their Stimulation in the Human Gut.” Frontiers in Microbiology 7: 979. 10.3389/fmicb.2016.00979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mori, Giorgia , Morrison Mark, and Blumenthal Antje. 2021. “Microbiome‐Immune Interactions in Tuberculosis.” PLOS Pathogens 17: e1009377. 10.1371/journal.ppat.1009377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Comberiati, Pasquale , Di Cicco Maria, Paravati Francesco, Pelosi Umberto, Di Gangi Alessandro, Arasi Stefania, Barni Simona, et al. 2021. “The Role of Gut and Lung Microbiota in Susceptibility to Tuberculosis.” International Journal of Environmental Research and Public Health 18: 12220. 10.3390/ijerph182212220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Usuda, Haruki , Okamoto Takayuki, and Wada Koichiro. 2021. “Leaky Gut: Effect of Dietary Fiber and Fats on Microbiome and Intestinal Barrier.” International Journal of Molecular Sciences 22: 7613. 10.3390/ijms22147613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wood, Madeleine R. , Yu Elaine A., and Mehta Saurabh. 2017. “The Human Microbiome in the Fight Against Tuberculosis.” American Journal of Tropical Medicine and Hygiene 96: 1274–1284. 10.4269/ajtmh.16-0581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Osei Sekyere, John , Maningi Nontuthuko E., and Fourie Petrus B.. 2020. “ Mycobacterium Tuberculosis, Antimicrobials, Immunity, and Lung‐Gut Microbiota Crosstalk: Current Updates and Emerging Advances.” Annals of the New York Academy of Sciences 1467: 21–47. 10.1111/nyas.14300 [DOI] [PubMed] [Google Scholar]
- 44. Duarte, S. M. B. , Stefano J. T., Miele L., Ponziani F. R., Souza‐Basqueira M., Okada Lsrr, de Barros Costa F. G., et al. 2018. “Gut Microbiome Composition in Lean Patients With Nash Is Associated With Liver Damage Independent of Caloric Intake: A Prospective Pilot Study.” Nutrition, Metabolism & Cardiovascular Diseases 28: 369–384. 10.1016/j.numecd.2017.10.014 [DOI] [PubMed] [Google Scholar]
- 45. Li, Yue , Zhao Liangjie, Hou Meiling, Gao Tianlin, Sun Jin, Luo Hao, Wang Fengdan, et al. 2022. “ Lactobacillus Casei Improve Anti‐Tuberculosis Drugs‐Induced Intestinal Adverse Reactions in Rat by Modulating Gut Microbiota and Short‐Chain Fatty Acids.” Nutrients 14: 1668. 10.3390/nu14081668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Segal, Leopoldo N. , Clemente Jose C., Li Yonghua, Ruan Chunhai, Cao Jane, Danckers Mauricio, Morris Alison, et al. 2017. “Anaerobic Bacterial Fermentation Products Increase Tuberculosis Risk in Antiretroviral‐Drug‐Treated HIV Patients.” Cell Host & Microbe 21: 530–537.e534. 10.1016/j.chom.2017.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lachmandas, Ekta , van den Heuvel Corina N. A. M., Damen Michelle S. M. A., Cleophas Maartje C. P., Netea Mihai G., and van Crevel Reinout. 2016. “Diabetes Mellitus and Increased Tuberculosis Susceptibility: The Role of Short‐Chain Fatty Acids.” Journal of Diabetes Research 2016: 6014631. 10.1155/2016/6014631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yu, Ziqi , Shen Xiang, Wang Aiyao, Hu Chong, and Chen Jianyong. 2023. “The Gut Microbiome: A Line of Defense Against Tuberculosis Development.” Frontiers in Cellular and Infection Microbiology 13: 1149679. 10.3389/fcimb.2023.1149679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Khan, Nargis , Mendonca Laura, Dhariwal Achal, Fontes Ghislaine, Menzies Dick, Xia Jianguo, Divangahi Maziar, and King Irah L.. 2019. “Intestinal Dysbiosis Compromises Alveolar Macrophage Immunity to Mycobacterium Tuberculosis .” Mucosal Immunology 12: 772–783. 10.1038/s41385-019-0147-3 [DOI] [PubMed] [Google Scholar]
- 50. Mayer‐Barber, Katrin D. , and Barber Daniel L.. 2015. “Innate and Adaptive Cellular Immune Responses to Mycobacterium Tuberculosis Infection.” Cold Spring Harbor Perspectives in Medicine 5(12): a018424. 10.1101/cshperspect.a018424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang, Fang , Yang Yi, Chen Lingming, Zhang Zhiyi, Liu Linna, Zhang Chunmin, Mai Qiongdan, et al. 2022. “The Gut Microbiota Mediates Protective Immunity Against Tuberculosis via Modulation of lncRNA.” Gut Microbes 14: 2029997. 10.1080/19490976.2022.2029997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Roy, Supriya , and Dhaneshwar Suneela. 2023. “Role of Prebiotics, Probiotics, and Synbiotics in Management of Inflammatory Bowel Disease: Current Perspectives.” World Journal of Gastroenterology 29: 2078–2100. 10.3748/wjg.v29.i14.2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Negi, Shikha , Pahari Susanta, Bashir Hilal, and Agrewala Javed N.. 2019. “Gut Microbiota Regulates Mincle Mediated Activation of Lung Dendritic Cells to Protect Against Mycobacterium Tuberculosis .” Frontiers in Immunology 10: 1142. 10.3389/fimmu.2019.01142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu, Yue , Wang Jiaqi, and Wu Changxin. 2021. “Microbiota and Tuberculosis: A Potential Role of Probiotics, and Postbiotics.” Frontiers in Nutrition 8: 626254. 10.3389/fnut.2021.626254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Azad, Md. Abul Kalam , Sarker Manobendro, Li Tiejun, and Yin Jie. 2018. “Probiotic Species in the Modulation of Gut Microbiota: An Overview.” BioMed Research International 2018: 9478630. 10.1155/2018/9478630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Huey, Samantha L. , Yu Elaine A., Finkelstein Julia L., Glesby Marshall J., Bonam Wesley, Russell David G., and Mehta Saurabh. 2021. “Nutrition, Inflammation, and the Gut Microbiota Among Outpatients With Active Tuberculosis Disease in India.” American Journal of Tropical Medicine and Hygiene 105: 1645–1656. 10.4269/ajtmh.21-0310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cheung, Man Kit , Lam Wai Yip, Fung Wendy Yin Wan, Law Patrick Tik Wan, Au Chun Hang, Nong Wenyan, Kam Kai Man, Kwan Hoi Shan, and Tsui Stephen Kwok Wing. 2013. “Sputum Microbiota in Tuberculosis as Revealed by 16S rRNA Pyrosequencing.” PLoS One 8: e54574. 10.1371/journal.pone.0054574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Caradonna, L. , Amati L., Magrone T., Pellegrino N. M., Jirillo E., Caccavo D., Caradonna L., et al. 2000. “Invited Review: Enteric Bacteria, Lipopolysaccharides and Related Cytokines in Inflammatory Bowel Disease: Biological and Clinical Significance.” Journal of Endotoxin Research 6: 205–214. 10.1177/09680519000060030101. https://www.ncbi.nlm.nih.gov/pubmed/11052175 [DOI] [PubMed] [Google Scholar]
- 59. Kamdar, Karishma , Khakpour Samira, Chen Jingyu, Leone Vanessa, Brulc Jennifer, Mangatu Thomas, Antonopoulos Dionysios A., et al. 2016. “Genetic and Metabolic Signals during Acute Enteric Bacterial Infection Alter the Microbiota and Drive Progression to Chronic Inflammatory Disease.” Cell Host & Microbe 19: 21–31. 10.1016/j.chom.2015.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dumas, Alexia , Corral Dan, Colom André, Levillain Florence, Peixoto Antonio, Hudrisier Denis, Poquet Yannick, and Neyrolles Olivier. 2018. “The Host Microbiota Contributes to Early Protection Against Lung Colonization by Mycobacterium Tuberculosis .” Frontiers in Immunology 9: 2656. 10.3389/fimmu.2018.02656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hauptmann, M. , Kalsdorf B., Akoh‐Arrey J. E., Lange C., and Schaible U. E.. 2024. “Microbiota Alterations in Patients Treated for Susceptible or Drug‐Resistant TB.” International Journal of Tuberculosis and Lung Disease Open 1: 355–361. 10.5588/ijtldopen.24.0325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu, Na , Liu Jinfeng, Zheng Binjie, Zeng Xiangchang, Ye Zixin, Huang Xinyi, Liu Wenhui, et al. 2023. “Gut Microbiota Affects Sensitivity to Immune‐Mediated Isoniazid‐Induced Liver Injury.” Biomedicine & Pharmacotherapy 160: 114400. 10.1016/j.biopha.2023.114400 [DOI] [PubMed] [Google Scholar]
- 63. Namasivayam, Sivaranjani , Zimmerman Matthew, Oland Sandra, Wang Han, Mittereder Lara R., Dartois Véronique, and Sher Alan. 2023. “The Dysbiosis Triggered by First‐Line Tuberculosis Antibiotics Fails to Reduce Their Bioavailability.” mBio 14: e0035323. 10.1128/mbio.00353-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Negi, Shikha , Pahari Susanta, Bashir Hilal, and Agrewala Javed N.. 2020. “Intestinal Microbiota Disruption Limits the Isoniazid Mediated Clearance of Mycobacterium Tuberculosis in Mice.” European Journal of Immunology 50: 1976–1987. 10.1002/eji.202048556 [DOI] [PubMed] [Google Scholar]
- 65. Hu, Yongfeng , Yang Qianting, Liu Bo, Dong Jie, Sun Lilian, Zhu Yafang, Su Haoxiang, et al. 2019. “Gut Microbiota Associated With Pulmonary Tuberculosis and Dysbiosis Caused by Anti‐Tuberculosis Drugs.” Journal of Infection 78: 317–322. 10.1016/j.jinf.2018.08.006 [DOI] [PubMed] [Google Scholar]
- 66. Wipperman, Matthew F. , Fitzgerald Daniel W., Juste Marc Antoine Jean, Taur Ying, Namasivayam Sivaranjani, Sher Alan, Bean James M., Bucci Vanni, and Glickman Michael S.. 2017. “Antibiotic Treatment for Tuberculosis Induces a Profound Dysbiosis of the Microbiome That Persists Long After Therapy Is Completed.” Scientific Reports 7: 10767. 10.1038/s41598-017-10346-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Khan, Nargis , Vidyarthi Aurobind, Nadeem Sajid, Negi Shikha, Nair Girish, and Agrewala Javed N.. 2016. “Alteration in the Gut Microbiota Provokes Susceptibility to Tuberculosis.” Frontiers in Immunology 7: 529. 10.3389/fimmu.2016.00529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang, Chi , Ouyang Qi, Zhou Xianyuan, Huang Yingfeng, Zeng Yu, Deng Li, Lin Dachuan, and Zheng Weidong. 2023. “In Vitro Activity of Tetracycline Analogs Against Multidrug‐Resistant and Extensive Drug Resistance Clinical Isolates of Mycobacterium Tuberculosis .” Tuberculosis 140: 102336. 10.1016/j.tube.2023.102336 [DOI] [PubMed] [Google Scholar]
- 69. Rothstein, David M . 2016. “Rifamycins, Alone and in Combination.” Cold Spring Harbor Perspectives in Medicine 6: a027011. 10.1101/cshperspect.a027011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wipperman, Matthew F. , Bhattarai Shakti K., Vorkas Charles Kyriakos, Maringati Venkata Suhas, Taur Ying, Mathurin Laurent, McAulay Katherine, et al. 2021. “Gastrointestinal Microbiota Composition Predicts Peripheral Inflammatory State During Treatment of Human Tuberculosis.” Nature Communications 12: 1141. 10.1038/s41467-021-21475-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Weersma, Rinse K. , Zhernakova Alexandra, and Fu Jingyuan. 2020. “Interaction Between Drugs and the Gut Microbiome.” Gut 69: 1510–1519. 10.1136/gutjnl-2019-320204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hill, D. A. , Hoffmann C., Abt M. C., Du Y., Kobuley D., Kirn T. J., Bushman F. D., and Artis D.. 2010. “Metagenomic Analyses Reveal Antibiotic‐Induced Temporal and Spatial Changes in Intestinal Microbiota With Associated Alterations in Immune Cell Homeostasis.” Mucosal Immunology 3: 148–158. 10.1038/mi.2009.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Keeney, Kristie M. , Yurist‐Doutsch Sophie, Arrieta Marie‐Claire, and Finlay B. Brett. 2014. “Effects of Antibiotics on Human Microbiota and Subsequent Disease.” Annual Review of Microbiology 68: 217–235. 10.1146/annurev-micro-091313-103456 [DOI] [PubMed] [Google Scholar]
- 74. Ramappa, Vidyasagar , and Aithal Guruprasad P.. 2013. “Hepatotoxicity Related to Anti‐Tuberculosis Drugs: Mechanisms and Management.” Journal of Clinical and Experimental Hepatology 3: 37–49. 10.1016/j.jceh.2012.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jia, Baolei , Han Xiao, Kim Kyung Hyun, and Jeon Che Ok. 2022. “Discovery and Mining of Enzymes From the Human Gut Microbiome.” Trends in Biotechnology 40: 240–254. 10.1016/j.tibtech.2021.06.008 [DOI] [PubMed] [Google Scholar]
- 76. Doestzada, Marwah , Vila Arnau Vich, Zhernakova Alexandra, Koonen Debby P. Y., Weersma Rinse K., Touw Daan J., Kuipers Folkert, Wijmenga Cisca, and Fu Jingyuan. 2018. “Pharmacomicrobiomics: A Novel Route Towards Personalized Medicine?” Protein & Cell 9: 432–445. 10.1007/s13238-018-0547-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wu, Chunli , Yi Hengzhong, Hu Yanmei, Luo Danlin, Tang Zhigang, Wen Xinmin, Zhang Yong, et al. 2023. “Effects of Second‐Line Anti‐Tuberculosis Drugs on the Intestinal Microbiota of Patients With Rifampicin‐Resistant Tuberculosis.” Frontiers in Cellular and Infection Microbiology 13: 1127916. 10.3389/fcimb.2023.1127916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pei, Shengfei , Yang Li, Gao Huixia, Liu Yuzhen, Lu Jianhua, Dai Er Hei, Meng Chunyan, Feng Fumin, and Wang Yuling. 2025. “The Association Between the Gut Microbiome and Antituberculosis Drug‐Induced Liver Injury.” Frontiers in Pharmacology 16: 1512815. 10.3389/fphar.2025.1512815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gong, Jin‐Yu , Ren Huan, Chen Hui‐Qing, Xing Kai, Xiao Chen‐Lin, and Luo Jian‐Quan. 2022. “Magnesium Isoglycyrrhizinate Attenuates Anti‐Tuberculosis Drug‐Induced Liver Injury by Enhancing Intestinal Barrier Function and Inhibiting the LPS/TLRs/NF‐κB Signaling Pathway in Mice.” Pharmaceuticals 15: 1130. 10.3390/ph15091130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhuang, Li , Yang Ling, Li Linsheng, Ye Zhaoyang, and Gong Wenping. 2024. “ Mycobacterium Tuberculosis: Immune Response, Biomarkers, and Therapeutic Intervention.” MedComm 5(5): e419. 10.1002/mco2.419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ye, Zhaoyang , Li Linsheng, Yang Ling, Zhuang Li, Aspatwar Ashok, Wang Liang, and Gong Wenping. 2024. “Impact of Diabetes Mellitus on Tuberculosis Prevention, Diagnosis, and Treatment From an Immunologic Perspective.” Exploration 4: 20230138. 10.1002/EXP.20230138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wang, Wei , Li Jingxin, Liang Yuejin, and Gong Wenping. 2024. “Editorial: Immunological Aspects of Emerging and Re‐Emerging Zoonoses.” Frontiers in Immunology 15: 1392382. 10.3389/fimmu.2024.1392382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zhuang, Li , Ye Zhaoyang, Li Linsheng, Yang Ling, and Gong Wenping. 2023. “Next‐Generation TB Vaccines: Progress, Challenges, and Prospects.” Vaccines 11: 1304. 10.3390/vaccines11081304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Frati, Franco , Salvatori Cristina, Incorvaia Cristoforo, Bellucci Alessandro, Di Cara Giuseppe, Marcucci Francesco, and Esposito Susanna. 2018. “The Role of the Microbiome in Asthma: The Gut‐Lung Axis.” International Journal of Molecular Sciences 20: 123. 10.3390/ijms20010123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Thibeault, Charlotte , Suttorp Norbert, and Opitz Bastian. 2021. “The Microbiota in Pneumonia: From Protection to Predisposition.” Science Translational Medicine 13: eaba0501. 10.1126/scitranslmed.aba0501 [DOI] [PubMed] [Google Scholar]
- 86. Silva, Fabiola , Enaud Raphaël, Creissen Elizabeth, Henao‐Tamayo Marcela, Delhaes Laurence, and Izzo Angelo. 2022. “Mouse Subcutaneous BCG Vaccination and Mycobacterium Tuberculosis Infection Alter the Lung and Gut Microbiota.” Microbiology Spectrum 10: e0169321. 10.1128/spectrum.01693-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Chu, Hiutung , and Mazmanian Sarkis K.. 2013. “Innate Immune Recognition of the Microbiota Promotes Host‐Microbial Symbiosis.” Nature Immunology 14: 668–675. 10.1038/ni.2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Rakoff‐Nahoum, Seth , Paglino Justin, Eslami‐Varzaneh Fatima, Edberg Stephen, and Medzhitov Ruslan. 2004. “Recognition of Commensal Microflora by Toll‐Like Receptors Is Required for Intestinal Homeostasis.” Cell 118: 229–241. 10.1016/j.cell.2004.07.002 [DOI] [PubMed] [Google Scholar]
- 89. Danne, Camille , Ryzhakov Grigory, Martínez‐López Maria, Ilott Nicholas Edward, Franchini Fanny, Cuskin Fiona, Lowe Elisabeth C., et al. 2017. “A Large Polysaccharide Produced by Helicobacter Hepaticus Induces an Anti‐Inflammatory Gene Signature in Macrophages.” Cell Host & Microbe 22: 733–745.e735. 10.1016/j.chom.2017.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wolf, Andrea J. , and Underhill David M.. 2018. “Peptidoglycan Recognition by the Innate Immune System.” Nature Reviews Immunology 18: 243–254. 10.1038/nri.2017.136 [DOI] [PubMed] [Google Scholar]
- 91. Brown, Gordon D . 2006. “Dectin‐1: A Signalling Non‐TLR Pattern‐Recognition Receptor.” Nature Reviews Immunology 6: 33–43. 10.1038/nri1745 [DOI] [PubMed] [Google Scholar]
- 92. Föh, Bandik , Buhre Jana Sophia, Lunding Hanna B., Moreno‐Fernandez Maria E., König Peter, Sina Christian, Divanovic Senad, and Ehlers Marc. 2022. “Microbial Metabolite Butyrate Promotes Induction of IL‐10+IgM+ Plasma Cells.” PLoS One 17: e0266071. 10.1371/journal.pone.0266071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Peng, Yi‐Lei , Wang Si‐Han, Zhang Yu‐Long, Chen Man‐Yun, He Kang, Li Qing, Huang Wei‐Hua, and Zhang Wei. 2024. “Effects of Bile Acids on the Growth, Composition and Metabolism of Gut Bacteria.” npj Biofilms Microbiomes 10: 112. 10.1038/s41522-024-00566-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Takiishi, Tatiana , Fenero Camila Ideli Morales, and Câmara Niels Olsen Saraiva. 2017. “Intestinal Barrier and Gut Microbiota: Shaping Our Immune Responses Throughout Life.” Tissue Barriers 5: e1373208. 10.1080/21688370.2017.1373208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Jakobsson, Hedvig E. , Rodríguez‐Piñeiro Ana M., Schütte André, Ermund Anna, Boysen Preben, Bemark Mats, Sommer Felix, et al. 2015. “The Composition of the Gut Microbiota Shapes the Colon Mucus Barrier.” EMBO Reports 16: 164–177. 10.15252/embr.201439263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Quigley, Eamonn M. M . 2013. “Gut Bacteria in Health and Disease.” Gastroenterology & Hepatology 9: 560–569. https://www.ncbi.nlm.nih.gov/pubmed/24729765 [PMC free article] [PubMed] [Google Scholar]
- 97. Lin, Jie , Chen Dongli, Yan Yongen, Pi Jiang, Xu Junfa, Chen Lingming, and Zheng Biying. 2024. “Gut Microbiota: A Crucial Player in the Combat Against Tuberculosis.” Frontiers in Immunology 15: 1442095. 10.3389/fimmu.2024.1442095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Cho, Ilseung , and Blaser Martin J.. 2012. “The Human Microbiome: At the Interface of Health and Disease.” Nature Reviews Genetics 13: 260–270. 10.1038/nrg3182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zheng, Danping , Liwinski Timur, and Elinav Eran. 2020. “Interaction Between Microbiota and Immunity in Health and Disease.” Cell Research 30: 492–506. 10.1038/s41422-020-0332-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Atarashi, Koji , Tanoue Takeshi, Shima Tatsuichiro, Imaoka Akemi, Kuwahara Tomomi, Momose Yoshika, Cheng Genhong, et al. 2011. “Induction of Colonic Regulatory T Cells by Indigenous Clostridium Species.” Science 331: 337–341. 10.1126/science.1198469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Fedele, Giorgio , Stefanelli Paola, Spensieri Fabiana, Fazio Cecilia, Mastrantonio Paola, and Ausiello Clara M.. 2005. “Bordetella Pertussis‐Infected Human Monocyte‐Derived Dendritic Cells Undergo Maturation and Induce Th1 Polarization and Interleukin‐23 Expression.” Infection and Immunity 73: 1590–1597. 10.1128/IAI.73.3.1590-1597.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Atarashi, Koji , Tanoue Takeshi, Ando Minoru, Kamada Nobuhiko, Nagano Yuji, Narushima Seiko, Suda Wataru, et al. 2015. “Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells.” Cell 163: 367–380. 10.1016/j.cell.2015.08.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Loftfield, Erikka , Falk Roni T., Sampson Joshua N., Huang Wen‐Yi, Hullings Autumn, Murphy Gwen, Weinstein Stephanie J., et al. 2022. “Prospective Associations of Circulating Bile Acids and Short‐Chain Fatty Acids With Incident Colorectal Cancer.” JNCI Cancer Spectrum 6: pkac027. 10.1093/jncics/pkac027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Bachem, Annabell , Makhlouf Christina, Binger Katrina J., de Souza David P., Tull Deidra, Hochheiser Katharina, Whitney Paul G., et al. 2019. “Microbiota‐Derived Short‐Chain Fatty Acids Promote the Memory Potential of Antigen‐Activated CD8(+) T Cells.” Immunity 51: 285–297.e285. 10.1016/j.immuni.2019.06.002 [DOI] [PubMed] [Google Scholar]
- 105. Schulthess, Julie , Pandey Sumeet, Capitani Melania, Rue‐Albrecht Kevin C., Arnold Isabelle, Franchini Fanny, Chomka Agnieszka, et al. 2019. “The Short Chain Fatty Acid Butyrate Imprints an Antimicrobial Program in Macrophages.” Immunity 50: 432–445.e437. 10.1016/j.immuni.2018.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Vinolo, Marco A. R. , Rodrigues Hosana G., Nachbar Renato T., and Curi Rui. 2011. “Regulation of Inflammation by Short Chain Fatty Acids.” Nutrients 3: 858–876. 10.3390/nu3100858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Arpaia, Nicholas , Campbell Clarissa, Fan Xiying, Dikiy Stanislav, van der Veeken Joris, deRoos Paul, Liu Hui, et al. 2013. “Metabolites Produced by Commensal Bacteria Promote Peripheral Regulatory T‐Cell Generation.” Nature 504: 451–455. 10.1038/nature12726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Furusawa, Yukihiro , Obata Yuuki, Fukuda Shinji, Endo Takaho A., Nakato Gaku, Takahashi Daisuke, Nakanishi Yumiko, et al. 2013. “Commensal Microbe‐Derived Butyrate Induces the Differentiation of Colonic Regulatory T Cells.” Nature 504: 446–450. 10.1038/nature12721 [DOI] [PubMed] [Google Scholar]
- 109. Smith, Patrick M. , Howitt Michael R., Panikov Nicolai, Michaud Monia, Gallini Carey Ann, Bohlooly‐Y Mohammad, Glickman Jonathan N., and Garrett Wendy S.. 2013. “The Microbial Metabolites, Short‐Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis.” Science 341: 569–573. 10.1126/science.1241165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Peterson, Daniel A. , McNulty Nathan P., Guruge Janaki L., and Gordon Jeffrey I.. 2007. “IgA Response to Symbiotic Bacteria as a Mediator of Gut Homeostasis.” Cell Host & Microbe 2: 328–339. 10.1016/j.chom.2007.09.013 [DOI] [PubMed] [Google Scholar]
- 111. Mazmanian, Sarkis K. , Liu Cui Hua, Tzianabos Arthur O., and Kasper Dennis L.. 2005. “An Immunomodulatory Molecule of Symbiotic Bacteria Directs Maturation of the Host Immune System.” Cell 122: 107–118. 10.1016/j.cell.2005.05.007 [DOI] [PubMed] [Google Scholar]
- 112. Wang, Yulian , Huang Lisi, Huang Tian, Geng Suxia, Chen Xiaomei, Huang Xin, Lai Peilong, Du Xin, and Weng Jianyu. 2022. “The Gut Bacteria Dysbiosis Contributes to Chronic Graft‐Versus‐Host Disease Associated With a Treg/Th1 Ratio Imbalance.” Frontiers in Microbiology 13: 813576. 10.3389/fmicb.2022.813576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Yang, Yicheng , Zhang Hanwen, Wang Yaoyao, Xu Jing, Shu Songren, Wang Peizhi, Ding Shusi, et al. 2024. “Promising Dawn in the Management of Pulmonary Hypertension: The Mystery Veil of Gut Microbiota.” iMeta 3: e159. 10.1002/imt2.159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ashique, Sumel , De Rubis Gabriele, Sirohi Ekta, Mishra Neeraj, Rihan Mohd, Garg Ashish, Reyes Ruby‐Jean, et al. 2022. “Short Chain Fatty Acids: Fundamental Mediators of the Gut‐Lung Axis and Their Involvement in Pulmonary Diseases.” Chemico‐Biological Interactions 368: 110231. 10.1016/j.cbi.2022.110231 [DOI] [PubMed] [Google Scholar]
- 115. Vorkas, Charles Kyriakos , Wipperman Matthew F., Li Kelin, Bean James, Bhattarai Shakti K., Adamow Matthew, Wong Phillip, et al. 2018. “Mucosal‐Associated Invariant and γδ T Cell Subsets Respond to Initial Mycobacterium Tuberculosis Infection.” JCI Insight 3: e121899. 10.1172/jci.insight.121899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Constantinides, Michael G . 2018. “Interactions Between the Microbiota and Innate and Innate‐Like Lymphocytes.” Journal of Leukocyte Biology 103: 409–419. 10.1002/JLB.3RI0917-378R [DOI] [PubMed] [Google Scholar]
- 117. Lee, Naeun , and Kim Wan‐Uk. 2017. “Microbiota in T‐Cell Homeostasis and Inflammatory Diseases.” Experimental & Molecular Medicine 49: e340. 10.1038/emm.2017.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Cadena, Anthony M. , Ma Yixuan, Ding Tao, Bryant MacKenzie, Maiello Pauline, Geber Adam, Lin Philana Ling, Flynn JoAnne L., and Ghedin Elodie. 2018. “Profiling the Airway in the Macaque Model of Tuberculosis Reveals Variable Microbial Dysbiosis and Alteration of Community Structure.” Microbiome 6: 180. 10.1186/s40168-018-0560-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Han, MeiQing , Wang Xia, Su Lin, Pan Shiqi, Liu Ningning, Li Duan, Liu Liang, et al. 2024. “Intestinal Microbiome Dysbiosis Increases Mycobacteria Pulmonary Colonization in Mice by Regulating the Nos2‐Associated Pathways.” eLife 13: RP99282. 10.7554/eLife.99282.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Yinghong, Lu , Wenpei Shi, Yi Hu, Fan Xia, Zhu Ning, Meiying Wu, Cheng Chen, Hu Yue O., and Xu Biao O.. 2021. “A Comparative Study on the Difference of Gut Microbiota and Its Biomarkers Between Patients With Pulmonary Tuberculosis and Healthy Controls.” Chinese Journal of Tuberculosis and Respiratory Diseases 44: 939–946. 10.3760/cma.j.cn112147-20210315-00170 [DOI] [PubMed] [Google Scholar]
- 121. Luo, Dan , Yang Bo‐Yi, Qin Kai, Shi Chong‐Yu, Wei Nian‐Sa, Li Hai, Qin Yi‐Xiang, et al. 2023. “Untargeted Metabolomics of Feces Reveals Diagnostic and Prognostic Biomarkers for Active Tuberculosis and Latent Tuberculosis Infection: Potential Application for Precise and Non‐Invasive Identification.” Infection and Drug Resistance 16: 6121–6138. 10.2147/IDR.S422363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Sahu, Sukanya , Kaushik Sandeep R., Chaudhary Shweta, Mahapatra Amit kumar, Kappa Rukuwe, Kapfo Wetesho, Saha Sourav, et al. 2023. “Gut Microbiota Dysbiosis Observed in Tuberculosis Patients Resolves Partially With Anti‐Tuberculosis Therapy.” medRxiv Preprint: 23291387. 10.1101/2023.06.14.23291387 [DOI]
- 123. Luies, Laneke , van Reenen Mari, Ronacher Katharina, Walzl Gerhard, and Loots Du Toit. 2017. “Predicting Tuberculosis Treatment Outcome Using Metabolomics.” Biomarkers in Medicine 11: 1057–1067. 10.2217/bmm-2017-0133 [DOI] [PubMed] [Google Scholar]
- 124. Nielsen, Stine S. F. , Vibholm Line K., Monrad Ida, Olesen Rikke, Frattari Giacomo S., Pahus Marie H., Højen Jesper F., et al. 2021. “SARS‐CoV‐2 Elicits Robust Adaptive Immune Responses Regardless of Disease Severity.” eBioMedicine 68: 103410. 10.1016/j.ebiom.2021.103410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Zhou, Zheng , Sun Bao, Yu Dongsheng, and Zhu Chunsheng. 2022. “Gut Microbiota: An Important Player in Type 2 Diabetes Mellitus.” Frontiers in Cellular and Infection Microbiology 12: 834485. 10.3389/fcimb.2022.834485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Zhang, Ting , Cheng Jin‐ke, and Hu Yao‐min. 2022. “Gut Microbiota as a Promising Therapeutic Target for Age‐Related Sarcopenia.” Ageing Research Reviews 81: 101739. 10.1016/j.arr.2022.101739 [DOI] [PubMed] [Google Scholar]
- 127. Diallo, Dramane , Somboro Anou M., Diabate Seydou, Baya Bacar, Kone Amadou, Sarro Yeya S., Kone Bourahima, et al. 2021. “Antituberculosis Therapy and Gut Microbiota: Review of Potential Host Microbiota Directed‐Therapies.” Frontiers in Cellular and Infection Microbiology 11: 673100. 10.3389/fcimb.2021.673100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Gavrilova, N. N. , Ratnikova I. A., Sadanov A. K., Bayakisheva K., Tourlibaeva Z. J., and Belikova O. A.. 2014. “Application of Probiotics in Complex Treatment of Tuberculosis.” International Journal of Engineering Research and Applications 4: 13–18. https://www.ijera.com/papers/Vol4_issue11/Part%20-%204/B0411041318.pdf [Google Scholar]
- 129. Trivedi, Riddhi , and Barve Kalyani. 2020. “Gut Microbiome a Promising Target for Management of Respiratory Diseases.” Biochemical Journal 477: 2679–2696. 10.1042/BCJ20200426 [DOI] [PubMed] [Google Scholar]
- 130. Eribo, Osagie A. , du Plessis Nelita, Ozturk Mumin, Guler Reto, Walzl Gerhard, and Chegou Novel N.. 2020. “The Gut Microbiome in Tuberculosis Susceptibility and Treatment Response: Guilty or Not Guilty?” Cellular and Molecular Life Sciences 77: 1497–1509. 10.1007/s00018-019-03370-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Li, Yue , Zhao Liangjie, Sun Changyu, Yang Jingyi, Zhang Xinyue, Dou Sheng, Hua Qinglian, Ma Aiguo, and Cai Jing. 2023. “Regulation of Gut Microflora by Lactobacillus Casei Zhang Attenuates Liver Injury in Mice Caused by Anti‐Tuberculosis Drugs.” International Journal of Molecular Sciences 24: 9444. 10.3390/ijms24119444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Ahmad Khan, Faiz , Gelmanova Irina Y., Franke Molly F., Atwood Sidney, Zemlyanaya Nataliya A., Unakova Irina A., Andreev Yevgeniy G., et al. 2016. “Aggressive Regimens Reduce Risk of Recurrence After Successful Treatment of MDR‐TB.” Clinical Infectious Diseases 63: 214–220. 10.1093/cid/ciw276 [DOI] [PubMed] [Google Scholar]
- 133. Motta, Ilaria , Boeree Martin, Chesov Dumitru, Dheda Keertan, Gunar Günther, Jr. , Horsburgh Charles Robert, Kherabi Yousra, et al. 2024. “Recent Advances in the Treatment of Tuberculosis.” Clinical Microbiology and Infection 30: 1107–1114. 10.1016/j.cmi.2023.07.013 [DOI] [PubMed] [Google Scholar]
- 134. Martineau, Adrian R. , Jolliffe David A., Hooper Richard L., Greenberg Lauren, Aloia John F., Bergman Peter, Dubnov‐Raz Gal, et al. 2017. “Vitamin D Supplementation to Prevent Acute Respiratory Tract Infections: Systematic Review and Meta‐Analysis of Individual Participant Data.” BMJ 356: i6583. 10.1136/bmj.i6583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Satam, Heena , Joshi Kandarp, Mangrolia Upasana, Waghoo Sanober, Zaidi Gulnaz, Rawool Shravani, Thakare Ritesh P., et al. 2023. “Next‐Generation Sequencing Technology: Current Trends and Advancements.” Biology 12: 997. 10.3390/biology12070997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Kinde, Isaac , Wu Jian, Papadopoulos Nick, Kinzler Kenneth W., and Vogelstein Bert. 2011. “Detection and Quantification of Rare Mutations With Massively Parallel Sequencing.” Proceedings of the National Academy of Sciences 108: 9530–9535. 10.1073/pnas.1105422108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Margulies, Marcel , Egholm Michael, Altman William E., Attiya Said, Bader Joel S., Bemben Lisa A., Berka Jan, et al. 2005. “Genome Sequencing in Microfabricated High‐Density Picolitre Reactors.” Nature 437: 376–380. 10.1038/nature03959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Frey, Kenneth G. , and Bishop‐Lilly Kimberly A.. 2015. “Next‐Generation Sequencing for Pathogen Detection and Identification.” Methods in Microbiology 42: 525–554. 10.1016/bs.mim.2015.06.004 [DOI] [Google Scholar]
- 139. Eisenstein, Michael . 2023. “Illumina Faces Short‐Read Rivals.” Nature Biotechnology 41: 3–5. 10.1038/s41587-022-01632-4 [DOI] [PubMed] [Google Scholar]
- 140. Szoboszlay, Márton , Schramm Laetitia, Pinzauti David, Scerri Jeanesse, Sandionigi Anna, and Biazzo Manuele. 2023. “Nanopore Is Preferable Over Illumina for 16S Amplicon Sequencing of the Gut Microbiota When Species‐Level Taxonomic Classification, Accurate Estimation of Richness, or Focus on Rare Taxa Is Required.” Microorganisms 11: 804. 10.3390/microorganisms11030804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Jeon, Min‐Seung , Jeong Da Min, Doh Huijeong, Kang Hyun Ah, Jung Hyungtaek, and Eyun Seong‐il. 2023. “A Practical Comparison of the Next‐Generation Sequencing Platform and Assemblers Using Yeast Genome.” Life Science Alliance 6: e202201744. 10.26508/lsa.202201744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Carbo, Ellen C. , Mourik Kees, Boers Stefan A., Munnink Bas Oude, Nieuwenhuijse David, Jonges Marcel, Welkers Matthijs R. A., et al. 2023. “A Comparison of Five Illumina, Ion Torrent, and Nanopore Sequencing Technology‐Based Approaches for Whole Genome Sequencing of SARS‐CoV‐2.” European Journal of Clinical Microbiology & Infectious Diseases 42: 701–713. 10.1007/s10096-023-04590-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Ormandjieva, A. , and Ivanova M.. 2023. “Evaluation of Gendx Protocol for HLA NGS Genotyping Using the Ion Torrent Sequencing Platform.” Acta Medica Bulgarica 50: 11–17. 10.2478/amb-2023-0024 [DOI] [Google Scholar]
- 144. Sharma, Anupma , Luo Guobin, Li Na, Pea Giorgio, and Hyland Fiona. 2024. “Abstract 331: Ion TorrentTm NGS Sequencing of the TERT Promoter Hotspots With Ion AmpliSeqTM HD Technology.” Cancer Research 84: 331. 10.1158/1538-7445.Am2024-331 [DOI] [Google Scholar]
- 145. Ling, Xiaoting , Wang Chenghan, Li Linlin, Pan Liqiu, Huang Chaoyu, Zhang Caixia, Huang Yunhua, et al. 2023. “Third‐Generation Sequencing for Genetic Disease.” Clinica Chimica Acta 551: 117624. 10.1016/j.cca.2023.117624 [DOI] [PubMed] [Google Scholar]
- 146. Ermini, Luca , and Driguez Patrick. 2024. “The Application of Long‐Read Sequencing to Cancer.” Cancers 16: 1275. 10.3390/cancers16071275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Wang, Yusha , Jia Ruikai, Ye Hua, and Ma Xiaoshu. 2024. “A New Method and Application of Pacbio Sequencing for Low Copy and Difficulty Preparation Plasmids.” Journal of Bioinformatics and Systems Biology 07: 129–138. 10.26502/jbsb.5107085 [DOI] [Google Scholar]
- 148. Howorka, Stefan , Cheley Stephen, and Bayley Hagan. 2001. “Sequence‐Specific Detection of Individual DNA Strands Using Engineered Nanopores.” Nature Biotechnology 19: 636–639. 10.1038/90236 [DOI] [PubMed] [Google Scholar]
- 149. Wei, Jiangtao , Hong Hao, Wang Xing, Lei Xin, Ye Minjie, and Liu Zewen. 2024. “Nanopore‐Based Sensors for DNA Sequencing: A Review.” Nanoscale 16: 18732–18766. 10.1039/d4nr01325e [DOI] [PubMed] [Google Scholar]
- 150. Pei, Yang , Tanguy Melanie, Giess Adam, Dixit Abhijit, Wilson Louise C., Gibbons Richard J., Twigg Stephen R. F., Elgar Greg, and Wilkie Andrew O. M.. 2024. “A Comparison of Structural Variant Calling From Short‐Read and Nanopore‐Based Whole‐Genome Sequencing Using Optical Genome Mapping as a Benchmark.” Genes 15: 925. 10.3390/genes15070925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Ross, Michael G. , Russ Carsten, Costello Maura, Hollinger Andrew, Lennon Niall J., Hegarty Ryan, Nusbaum Chad, and Jaffe David B.. 2013. “Characterizing and Measuring Bias in Sequence Data.” Genome Biology 14: R51. 10.1186/gb-2013-14-5-r51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Kumawat, Rameshwar L. , Jena Milan Kumar, Mittal Sneha, and Pathak Biswarup. 2024. “Advancement of Next‐Generation DNA Sequencing through Ionic Blockade and Transverse Tunneling Current Methods.” Small 20: e2401112. 10.1002/smll.202401112 [DOI] [PubMed] [Google Scholar]
- 153. Dahui, Qin . 2019. “Next‐Generation Sequencing and Its Clinical Application.” Cancer Biology & Medicine 16: 4–10. 10.20892/j.issn.2095-3941.2018.0055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Suwinski, Pawel , Ong ChuangKee, Ling Maurice H. T., Poh Yang Ming, Khan Asif M., and Ong Hui San. 2019. “Advancing Personalized Medicine Through the Application of Whole Exome Sequencing and Big Data Analytics.” Frontiers in Genetics 10: 49. 10.3389/fgene.2019.00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Peymani, Fatemeh , Farzeen Aiman, and Prokisch Holger. 2022. “RNA Sequencing Role and Application in Clinical Diagnostic.” Pediatric Investigation 6: 29–35. 10.1002/ped4.12314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Zhang, Hong , He Lin, and Cai Lei. 2018. “Transcriptome Sequencing: RNA‐Seq.” Methods in Molecular Biology 1754: 15–27. 10.1007/978-1-4939-7717-8_2 [DOI] [PubMed] [Google Scholar]
- 157. Meskini, Maryam , Zamani Mohammad Saber, Amanzadeh Amir, Bouzari Saeid, Karimipoor Morteza, Fuso Andrea, Fateh Abolfazl, and Siadat Seyed Davar. 2024. “Epigenetic Modulation of Cytokine Expression in Mycobacterium Tuberculosis‐Infected Monocyte Derived‐Dendritic Cells: Implications for Tuberculosis Diagnosis.” Cytokine 181: 156693. 10.1016/j.cyto.2024.156693 [DOI] [PubMed] [Google Scholar]
- 158. Barros‐Silva, Daniela , Marques C. Joana, Henrique Rui, and Jerónimo Carmen. 2018. “Profiling DNA Methylation Based on Next‐Generation Sequencing Approaches: New Insights and Clinical Applications.” Genes 9: 429. 10.3390/genes9090429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Tiwari, Neha , Bansal Megha, and Sharma Jai Gopal. 2021. “Metagenomics: A Powerful Lens Viewing the Microbial World.” In Wastewater Treatment Reactors, 309–339. Amsterdam: Elsevier. 10.1016/B978-0-12-823991-9.00015-0 [DOI] [Google Scholar]
- 160. Chiu, Charles Y. , and Miller Steven A.. 2019. “Clinical Metagenomics.” Nature Reviews Genetics 20: 341–355. 10.1038/s41576-019-0113-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Hussen, Bashdar Mahmud , Abdullah Sara Tharwat, Salihi Abbas, Sabir Dana Khdr, Sidiq Karzan R., Rasul Mohammed Fatih, Hidayat Hazha Jamal, et al. 2022. “The Emerging Roles of NGS in Clinical Oncology and Personalized Medicine.” Pathology ‐ Research and Practice 230: 153760. 10.1016/j.prp.2022.153760 [DOI] [PubMed] [Google Scholar]
- 162. Qahwaji, Rowaid , Ashankyty Ibraheem, Sannan Naif S., Hazzazi Mohannad S., Basabrain Ammar A., and Mobashir Mohammad. 2024. “Pharmacogenomics: A Genetic Approach to Drug Development and Therapy.” Pharmaceuticals 17: 940. 10.3390/ph17070940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Browne, Hilary P. , Forster Samuel C., Anonye Blessing O., Kumar Nitin, Neville B. Anne, Stares Mark D., Goulding David, and Lawley Trevor D.. 2016. “Culturing of ‘Unculturable’ Human Microbiota Reveals Novel Taxa and Extensive Sporulation.” Nature 533: 543–546. 10.1038/nature17645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Dubey, Anamika , Malla Muneer Ahmad, and Kumar Ashwani. 2022. “Role of Next‐Generation Sequencing (NGS) in Understanding the Microbial Diversity.” In Molecular Genetics and Genomics Tools in Biodiversity Conservation, 307–328. Singapore: Springer. 10.1007/978-981-16-6005-4_16 [DOI] [Google Scholar]
- 165. Kumar, Kishore R. , Cowley Mark J., and Davis Ryan L.. 2024. “Next‐Generation Sequencing and Emerging Technologies.” Seminars in Thrombosis and Hemostasis 50: 1026–1038. 10.1055/s-0044-1786397 [DOI] [PubMed] [Google Scholar]
- 166. Johnson, Jethro S. , Spakowicz Daniel J., Hong Bo‐Young, Petersen Lauren M., Demkowicz Patrick, Chen Lei, Leopold Shana R., et al. 2019. “Evaluation of 16S rRNA Gene Sequencing for Species and Strain‐Level Microbiome Analysis.” Nature Communications 10: 5029. 10.1038/s41467-019-13036-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Hong, Bo‐Young , Maulén Nancy Paula, Adami Alexander J., Granados Hector, Balcells María Elvira, and Cervantes Jorge. 2016. “Microbiome Changes During Tuberculosis and Antituberculous Therapy.” Clinical Microbiology Reviews 29: 915–926. 10.1128/CMR.00096-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Mann, Elizabeth R. , Lam Ying Ka, and Uhlig Holm H.. 2024. “Short‐Chain Fatty Acids: Linking Diet, the Microbiome and Immunity.” Nature Reviews Immunology 24: 577–595. 10.1038/s41577-024-01014-8 [DOI] [PubMed] [Google Scholar]
- 169. Yang, Wenjing , and Cong Yingzi. 2021. “Gut Microbiota‐Derived Metabolites in the Regulation of Host Immune Responses and Immune‐Related Inflammatory Diseases.” Cellular & Molecular Immunology 18: 866–877. 10.1038/s41423-021-00661-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Truong, Duy Tin , Franzosa Eric A., Tickle Timothy L., Scholz Matthias, Weingart George, Pasolli Edoardo, Tett Adrian, Huttenhower Curtis, and Segata Nicola. 2015. “MetaPhlAn2 for Enhanced Metagenomic Taxonomic Profiling.” Nature Methods 12: 902–903. 10.1038/nmeth.3589 [DOI] [PubMed] [Google Scholar]
- 171. Luo, Ying , Xue Ying, Liu Wei, Song Huijuan, Huang Yi, Tang Guoxing, Wang Feng, et al. 2022. “Development of Diagnostic Algorithm Using Machine Learning for Distinguishing Between Active Tuberculosis and Latent Tuberculosis Infection.” BMC Infectious Diseases 22: 965. 10.1186/s12879-022-07954-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Ozdemir, Tanel , Fedorec Alex J. H., Danino Tal, and Barnes Chris P.. 2018. “Synthetic Biology and Engineered Live Biotherapeutics: Toward Increasing System Complexity.” Cell Systems 7: 5–16. 10.1016/j.cels.2018.06.008 [DOI] [PubMed] [Google Scholar]
- 173. Borody, Thomas J. , Paramsothy Sudarshan, and Agrawal Gaurav. 2013. “Fecal Microbiota Transplantation: Indications, Methods, Evidence, and Future Directions.” Current Gastroenterology Reports 15: 337. 10.1007/s11894-013-0337-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Nyirenda, John L. Z. , Bockey Annabelle, Wagner Dirk, and Lange Berit. 2023. “Effect of Tuberculosis (TB) and Diabetes Mellitus (DM) Integrated Healthcare on Bidirectional Screening and Treatment Outcomes Among TB Patients and People Living With DM in Developing Countries: A Systematic Review.” Pathogens and Global Health 117: 36–51. 10.1080/20477724.2022.2046967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Wu, Hao , Forslund Sofia, Wang Zeneng, and Zhao Guoping. 2025. “Human Gut Microbiome Researches Over the Last Decade: Current Challenges and Future Directions.” Phenomics 5: 1–7. 10.1007/s43657-023-00131-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Cameron, Ellen S. , Schmidt Philip J., Tremblay Benjamin J.‐M., Emelko Monica B., and Müller Kirsten M.. 2021. “Enhancing Diversity Analysis by Repeatedly Rarefying Next Generation Sequencing Data Describing Microbial Communities.” Scientific Reports 11: 22302. 10.1038/s41598-021-01636-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Mousavi‐Sagharchi, Seyyed Mohammad Amin , Afrazeh Elina, Seyyedian‐Nikjeh Seyyedeh Fatemeh, Meskini Maryam, Doroud Delaram, and Siadat Seyed Davar. 2024. “New Insight in Molecular Detection of Mycobacterium Tuberculosis .” AMB Express 14: 74. 10.1186/s13568-024-01730-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Meskini, Maryam , Madadi Nahid, Ahmadi Kamal, Vaziri Farzam, Fateh Abolfazl, and Siadat Seyed Davar. 2023. “Tuberculosis Prevention, Diagnosis, and Treatment Financial Profile During 2006‐–2021: PART A.” Cost Effectiveness and Resource Allocation 21: 68. 10.1186/s12962-023-00479-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Pavelescu, Luciana Alexandra , Profir Monica, Enache Robert Mihai, Roşu Oana Alexandra, Creţoiu Sanda Maria, and Gaspar Bogdan Severus. 2024. “A Proteogenomic Approach to Unveiling the Complex Biology of the Microbiome.” International Journal of Molecular Sciences 25: 10467. 10.3390/ijms251910467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Pan, Sheng , and Chen Ru. 2020. “Metaproteomic Analysis of Human Gut Microbiome in Digestive and Metabolic Diseases.” Advances in Clinical Chemistry 97: 1–12. 10.1016/bs.acc.2019.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Saraswat, Istuti , and Goel Anjana. 2025. “Therapeutic Modulation of the Microbiome in Oncology: Current Trends and Future Directions.” Current Pharmaceutical Biotechnology 26: 680–699. 10.2174/0113892010353600241109132441 [DOI] [PubMed] [Google Scholar]
- 182. Gilbert, Jack A. , Blaser Martin J., Caporaso J Gregory, Jansson Janet K., Lynch Susan V., and Knight Rob. 2018. “Current Understanding of the Human Microbiome.” Nature Medicine 24: 392–400. 10.1038/nm.4517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Lai, Lisa A. , Tong Zachary, Chen Ru, and Pan Sheng. 2019. “Metaproteomics Study of the Gut Microbiome.” Functional Proteomics 1871: 123–132. 10.1007/978-1-4939-8814-3_8 [DOI] [PubMed] [Google Scholar]
- 184. Allué‐Guardia, Anna , García Juan I., and Torrelles Jordi B.. 2021. “Evolution of Drug‐Resistant Mycobacterium Tuberculosis Strains and Their Adaptation to the Human Lung Environment.” Frontiers in Microbiology 12: 612675. 10.3389/fmicb.2021.612675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Ullah, Nadeem , Hao Ling, Banga Ndzouboukou Jo‐Lewis, Chen Shiyun, Wu Yaqi, Li Longmeng, Borham Mohamed Eman, Hu Yangbo, and Fan Xionglin. 2021. “Label‐Free Comparative Proteomics of Differentially Expressed Mycobacterium Tuberculosis Protein in Rifampicin‐Related Drug‐Resistant Strains.” Pathogens 10: 607. 10.3390/pathogens10050607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Wu, Zhuhua , Wei Wenjing, Zhou Ying, Guo Huixin, Zhao Jiao, Liao Qinghua, Chen Liang, Zhang Xiaoli, and Zhou Lin. 2020. “Integrated Quantitative Proteomics and Metabolome Profiling Reveal Msmeg_6171 Overexpression Perturbing Lipid Metabolism of Mycobacterium Smegmatis Leading to Increased Vancomycin Resistance.” Frontiers in Microbiology 11: 1572. 10.3389/fmicb.2020.01572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Bendre, Ameya D. , Peters Peter J., and Kumar Janesh. 2021. “Recent Insights Into the Structure and Function of Mycobacterial Membrane Proteins Facilitated by Cryo‐EM.” The Journal of Membrane Biology 254: 321–341. 10.1007/s00232-021-00179-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. An, Yajing , Ni Ruizi, Zhuang Li, Yang Ling, Ye Zhaoyang, Li Linsheng, Parkkila Seppo, Aspatwar Ashok, and Gong Wenping. 2025. “Tuberculosis Vaccines and Therapeutic Drug: Challenges and Future Directions.” Molecular Biomedicine 6: 4. 10.1186/s43556-024-00243-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Arora, Gunjan , Bothra Ankur, Prosser Gareth, Arora Kriti, and Sajid Andaleeb. 2021. “Role of Post‐Translational Modifications in the Acquisition of Drug Resistance in Mycobacterium Tuberculosis .” The FEBS Journal 288: 3375–3393. 10.1111/febs.15582 [DOI] [PubMed] [Google Scholar]
- 190. Kontsevaya, Irina , Lange Christoph, Comella‐Del‐Barrio Patricia, Coarfa Cristian, DiNardo Andrew R., Gillespie Stephen H., Hauptmann Matthias, et al. 2021. “Perspectives for Systems Biology in the Management of Tuberculosis.” European Respiratory Review 30: 200377. 10.1183/16000617.0377-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Pan, Xian , Jiang Shan, Zhang Xinzhuang, Wang Zhenzhong, Wang Xin, Cao Liang, and Xiao Wei. 2024. “Recent Strategies in Target Identification of Natural Products: Exploring Applications in Chronic Inflammation and Beyond.” British Journal of Pharmacology. In press. 10.1111/bph.17356 [DOI] [PubMed] [Google Scholar]
- 192. Ehrt, Sabine , Schnappinger Dirk, and Rhee Kyu Y.. 2018. “Metabolic Principles of Persistence and Pathogenicity in Mycobacterium Tuberculosis .” Nature Reviews Microbiology 16: 496–507. 10.1038/s41579-018-0013-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Saiboonjan, Bhanubong , Roytrakul Sittiruk, Sangka Arunnee, Lulitanond Viraphong, Faksri Kiatichai, and Namwat Wises. 2021. “Proteomic Analysis of Drug‐Susceptible and Multidrug‐Resistant Nonreplicating Beijing Strains of Mycobacterium tuberculosis Cultured In Vitro.” Biochemistry and Biophysics Reports 26: 100960. 10.1016/j.bbrep.2021.100960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Whiston, Emily , and Taylor John W.. 2016. “Comparative Phylogenomics of Pathogenic and Nonpathogenic Species.” G3‐genes Genomes Genetics 6: 235–244. 10.1534/g3.115.022806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Melnik, Andre , Cappelletti Valentina, Vaggi Federico, Piazza Ilaria, Tognetti Marco, Schwarz Carmen, Cereghetti Gea, et al. 2020. “Comparative Analysis of the Intracellular Responses to Disease‐Related Aggregation‐Prone Proteins.” Journal of Proteomics 225: 103862. 10.1016/j.jprot.2020.103862 [DOI] [PubMed] [Google Scholar]
- 196. Sharma, Smriti , Bhat Rahul, Singh Rohit, Sharma Sumit, Wazir Priya, Singh Parvinder Pal, Vishwakarma Ram A., and Khan Inshad Ali. 2020. “High‐Throughput Screening of Compounds Library to Identify Novel Inhibitors Against Latent Mycobacterium tuberculosis Using Streptomycin‐Dependent Mycobacterium Tuberculosis 18b Strain as a Model.” Tuberculosis 124: 101958. 10.1016/j.tube.2020.101958 [DOI] [PubMed] [Google Scholar]
- 197. Robison, Heather M. , Chapman Cole A., Zhou Haowen, Erskine Courtney L., Theel Elitza, Peikert Tobias, Lindestam Arlehamn Cecilia S., et al. 2021. “Risk Assessment of Latent Tuberculosis Infection through a Multiplexed Cytokine Biosensor Assay and Machine Learning Feature Selection.” Scientific Reports 11: 20544. 10.1038/s41598-021-99754-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Jayaraman, Manikandan , Gosu Vijayakumar, Kumar Rajalakshmi, and Jeyaraman Jeyakanthan. 2024. “Computational Insights Into Potential Marine Natural Products as Selective Inhibitors of Mycobacterium Tuberculosis InhA: A Structure‐Based Virtual Screening Study.” Computational Biology and Chemistry 108: 107991. 10.1016/j.compbiolchem.2023.107991 [DOI] [PubMed] [Google Scholar]
- 199. Khan, Muhammad Fayaz , Ali Amjad, Rehman Hafiz Muzzammel, Noor Khan Sadiq, Hammad Hafiz Muhammad, Waseem Maaz, Wu Yurong, Clark Taane G., and Jabbar Abdul. 2024. “Exploring Optimal Drug Targets through Subtractive Proteomics Analysis and Pangenomic Insights for Tailored Drug Design in Tuberculosis.” Scientific Reports 14: 10904. 10.1038/s41598-024-61752-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Wang, Menglong , Pan Wei, Xu Yao, Zhang Jishou, Wan Jun, and Jiang Hong. 2022. “Microglia‐Mediated Neuroinflammation: A Potential Target for the Treatment of Cardiovascular Diseases.” Journal of Inflammation Research 15: 3083–3094. 10.2147/JIR.S350109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Nikitushkin, Vadim , Shleeva Margarita, Loginov Dmitry, Filip Dyčka F., Sterba Jan, and Kaprelyants Arseny. 2022. “Shotgun Proteomic Profiling of Dormant, ‘Non‐Culturable’ Mycobacterium Tuberculosis .” PLoS One 17: e0269847. 10.1371/journal.pone.0269847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Tucci, Paula , Portela Madelón, Chetto Carlos Rivas, González‐Sapienza Gualberto, and Marín Mónica. 2020. “Integrative Proteomic and Glycoproteomic Profiling of Mycobacterium Tuberculosis Culture Filtrate.” PLoS One 15: e0221837. 10.1371/journal.pone.0221837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Mateos, Jesús , Estévez Olivia, González‐Fernández África, Anibarro Luis, Pallarés Ángeles, Reljic Rajko, Gallardo José M, Medina Isabel, and Carrera Mónica. 2019. “High‐Resolution Quantitative Proteomics Applied to the Study of the Specific Protein Signature in the Sputum and Saliva of Active Tuberculosis Patients and Their Infected and Uninfected Contacts.” Journal of Proteomics 195: 41–52. 10.1016/j.jprot.2019.01.010 [DOI] [PubMed] [Google Scholar]
- 204. Villela, Anne Drumond , Eichler Paula, Pinto Antonio Frederico Michel, Rodrigues‐Junior Valnês, Yates John R. III, Bizarro Cristiano Valim, Basso Luiz Augusto, and Santos Diógenes Santiago. 2015. “Gene Replacement and Quantitative Mass Spectrometry Approaches Validate Guanosine Monophosphate Synthetase as Essential for Mycobacterium Tuberculosis Growth.” Biochemistry and Biophysics Reports 4: 277–282. 10.1016/j.bbrep.2015.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Abdullaeva, Yulduzkhon , Ambika Manirajan Binoy, Honermeier Bernd, Schnell Sylvia, and Cardinale Massimiliano. 2021. “Domestication Affects the Composition, Diversity, and Co‐Occurrence of the Cereal Seed Microbiota.” Journal of Advanced Research 31: 75–86. 10.1016/j.jare.2020.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Li, Zheng , Ma Jianqing, Li Xingye, Chan Matthew T. V., Wu William K. K., Wu Zhanyong, and Shen Jianxiong. 2019. “Aberrantly Expressed Long Non‐Coding RNAs in Air Pollution‐Induced Congenital Defects.” Journal of Cellular and Molecular Medicine 23: 7717–7725. 10.1111/jcmm.14645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Mogasale, Vittal , Mogasale Vijayalaxmi V., and Hsiao Amber. 2020. “Economic Burden of Cholera in Asia.” Vaccine 38(Suppl 1): A160–A166. 10.1016/j.vaccine.2019.09.099 [DOI] [PubMed] [Google Scholar]
- 208. Gardner, Alycia , de Mingo Pulido Álvaro, and Ruffell Brian. 2020. “Dendritic Cells and Their Role in Immunotherapy.” Frontiers in Immunology 11: 924. 10.3389/fimmu.2020.00924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Chopra, Hitesh , Mohanta Yugal Kishore, Rauta Pradipta Ranjan, Ahmed Ramzan, Mahanta Saurov, Mishra Piyush Kumar, Panda Paramjot, et al. 2023. “An Insight Into Advances in Developing Nanotechnology Based Therapeutics, Drug Delivery, Diagnostics and Vaccines: Multidimensional Applications in Tuberculosis Disease Management.” Pharmaceuticals 16: 581. 10.3390/ph16040581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Yao, Fusheng , Zhang Ruiqi, Lin Qiao, Xu Hui, Li Wei, Ou Min, Huang Yiting, et al. 2024. “Plasma Immune Profiling Combined With Machine Learning Contributes to Diagnosis and Prognosis of Active Pulmonary Tuberculosis.” Emerging Microbes & Infections 13: 2370399. 10.1080/22221751.2024.2370399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Robak, Aleksandra , Kistowski Michał, Wojtas Grzegorz, Perzanowska Anna, Targowski Tomasz, Michalak Agata, Krasowski Grzegorz, Dadlez Michał, and Domański Dominik. 2022. “Diagnosing Pleural Effusions Using Mass Spectrometry‐Based Multiplexed Targeted Proteomics Quantitating Mid‐ to High‐Abundance Markers of Cancer, Infection/Inflammation and Tuberculosis.” Scientific Reports 12: 3054. 10.1038/s41598-022-06924-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Chen, Jing , Han Yu‐Shuai, Yi Wen‐Jing, Huang Huai, Li Zhi‐Bin, Shi Li‐Ying, Wei Li‐Liang, et al. 2020. “Serum sCD14, PGLYRP2 and FGA as Potential Biomarkers for Multidrug‐Resistant Tuberculosis Based on Data‐Independent Acquisition and Targeted Proteomics.” Journal of Cellular and Molecular Medicine 24: 12537–12549. 10.1111/jcmm.15796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Fulcher, James M. , Markillie Lye Meng, Mitchell Hugh D., Williams Sarah M., Engbrecht Kristin M., Degnan David J., Bramer Lisa M., et al. 2024. “Parallel Measurement of Transcriptomes and Proteomes From Same Single Cells Using Nanodroplet Splitting.” Nature Communications 15: 10614. 10.1038/s41467-024-54099-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Ahmad, Rushdy , and Budnik Bogdan. 2023. “A Review of the Current State of Single‐Cell Proteomics and Future Perspective.” Analytical and Bioanalytical Chemistry 415: 6889–6899. 10.1007/s00216-023-04759-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Albrethsen, Jakob , Agner Jeppe, Piersma Sander R., Højrup Peter, Pham Thang V., Weldingh Karin, Jimenez Connie R., Andersen Peter, and Rosenkrands Ida. 2013. “Proteomic Profiling of Mycobacterium Tuberculosis Identifies Nutrient‐Starvation‐Responsive Toxin‐Antitoxin Systems.” Molecular & Cellular Proteomics 12: 1180–1191. 10.1074/mcp.M112.018846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Hua, Yongfeng , Cheng Min, Liu Bo, Dong Jie, Sun Lilian, Yang Jian, Yang Fan, Chen Xinchun, and Jin Qi. 2020. “Metagenomic Analysis of the Lung Microbiome in Pulmonary Tuberculosis−A Pilot Study.” Emerging Microbes & Infections 9: 1444–1452. 10.1080/22221751.2020.1783188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Kruh‐Garcia, Nicole A. , Wolfe Lisa M., Chaisson Lelia H., Worodria William O., Nahid Payam, Schorey Jeff S., Davis J. Lucian, and Dobos Karen M.. 2014. “Detection of Mycobacterium tuberculosis Peptides in the Exosomes of Patients With Active and Latent M. Tuberculosis Infection Using MRM‐MS.” PLoS One 9: e103811. 10.1371/journal.pone.0103811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Saleh, Sara , Staes An, Deborggraeve Stijn, and Gevaert Kris. 2019. “Targeted Proteomics for Studying Pathogenic Bacteria.” Proteomics 19: e1800435. 10.1002/pmic.201800435 [DOI] [PubMed] [Google Scholar]
- 219. Zheng, Weihao , Borja Michael, Dorman Leah C., Liu Jonathan, Zhou Andy, Seng Amanda, Arjyal Ritwicq, et al. 2025. “Single‐Cell Analysis Reveals Mycobacterium Tuberculosis Esx‐1‐Mediated Accumulation of Permissive Macrophages in Infected Mouse Lungs.” Science Advances 11: eadq8158. 10.1126/sciadv.adq8158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Geraldes, Inês , Fernandes Mónica, Fraga Alexandra G., and Osório Nuno S.. 2022. “The Impact of Single‐Cell Genomics on the Field of Mycobacterial Infection.” Frontiers in Microbiology 13: 989464. 10.3389/fmicb.2022.989464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Bespyatykh, Ju. A. , Shitikov E. A., and Ilina E. N.. 2017. “Proteomics for the Investigation of Mycobacteria.” Acta Naturae 9: 15–25. 10.32607/20758251-2017-9-1-15-25. https://www.ncbi.nlm.nih.gov/pubmed/28461970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Chung, Eun Seon , Kar Prathitha, Kamkaew Maliwan, Amir Ariel, and Aldridge Bree B.. 2024. “Single‐Cell Imaging of the Mycobacterium Tuberculosis Cell Cycle Reveals Linear and Heterogenous Growth.” Nature Microbiology 9: 3332–3344. 10.1038/s41564-024-01846-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Kaur, Simran , Angrish Nupur, Vasudevan Madavan, and Khare Garima. 2024. “Global Proteomics Reveals Pathways of Mesenchymal Stem Cells Altered by Mycobacterium Tuberculosis .” Scientific Reports 14: 30677. 10.1038/s41598-024-75722-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Devasundaram, Santhi , Gopalan Akilandeswari, Das Sulochana D., and Raja Alamelu. 2016. “Proteomics Analysis of Three Different Strains of Mycobacterium Tuberculosis Under In Vitro Hypoxia and Evaluation of Hypoxia Associated Antigen's Specific Memory T Cells in Healthy Household Contacts.” Frontiers in Microbiology 7: 1275. 10.3389/fmicb.2016.01275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Forrellad, Marina A. , Klepp Laura I, Gioffré Andrea, Sabio y García Julia, Morbidoni Hector R., Santangelo María de la Paz, Cataldi Angel A., and Bigi Fabiana. 2013. “Virulence Factors of the Mycobacterium Tuberculosis Complex.” Virulence 4: 3–66. 10.4161/viru.22329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Goldberg, Michael F. , Saini Neeraj K., and Porcelli Steven A.. 2014. “Evasion of Innate and Adaptive Immunity by Mycobacterium Tuberculosis .” Microbiology Spectrum 2: 1–24. 10.1128/microbiolspec.MGM2-0005-2013 [DOI] [PubMed] [Google Scholar]
- 227. Soto‐Heredero, Gonzalo , Gómez de las Heras Manuel M., Gabandé‐Rodríguez Enrique, Oller Jorge, and Mittelbrunn María. 2020. “Glycolysis−A Key Player in the Inflammatory Response.” The FEBS Journal 287: 3350–3369. 10.1111/febs.15327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Dar, Hamza Arshad , Zaheer Tahreem, Ullah Nimat, Bakhtiar Syeda Marriam, Zhang Tianyu, Yasir Muhammad, Azhar Esam I., and Ali Amjad. 2020. “Pangenome Analysis of Mycobacterium Tuberculosis Reveals Core‐Drug Targets and Screening of Promising Lead Compounds for Drug Discovery.” Antibiotics 9: 819. 10.3390/antibiotics9110819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Bagcchi, Sanjeet . 2023. “WHO's Global Tuberculosis Report 2022.” The Lancet Microbe 4: e20. 10.1016/S2666-5247(22)00359-7 [DOI] [PubMed] [Google Scholar]
- 230. Curreli, Sabrina , Benedetti Francesca, Yuan Weirong, Munawwar Arshi, Cocchi Fiorenza, Gallo Robert C., Sherman Nicholas E., and Zella Davide. 2022. “Characterization of the Interactome Profiling of Mycoplasma Fermentans DnaK in Cancer Cells Reveals Interference With Key Cellular Pathways.” Frontiers in Microbiology 13: 1022704. 10.3389/fmicb.2022.1022704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. McKinney, John D. , zu Bentrup Kerstin Höner, Muñoz‐Elías Ernesto J., Miczak Andras, Chen Bing, Chan Wai‐Tsing, Swenson Dana, et al. 2000. “Persistence of Mycobacterium Tuberculosis in Macrophages and Mice Requires the Glyoxylate Shunt Enzyme Isocitrate Lyase.” Nature 406: 735–738. 10.1038/35021074 [DOI] [PubMed] [Google Scholar]
- 232. Ma, Y. W. , Lu Y. H., Yi J. B., Feng Y. P., Herng T. S., Liu X., Gao D. Q., et al. 2012. “Room Temperature Ferromagnetism in Teflon Due to Carbon Dangling Bonds.” Nature Communications 3: 727. 10.1038/ncomms1689 [DOI] [PubMed] [Google Scholar]
- 233. Miyoshi‐Akiyama, Tohru , Zhao Jizi, Kato Hidehito, Kikuchi Ken, Totsuka Kyoichi, Kataoka Yasushi, Katsumi Masanori, and Uchiyama Takehiko. 2003. “Streptococcus Dysgalactiae‐Derived Mitogen (SDM), a Novel Bacterial Superantigen: Characterization of Its Biological Activity and Predicted Tertiary Structure.” Molecular Microbiology 47: 1589–1599. 10.1046/j.1365-2958.2003.03411.x [DOI] [PubMed] [Google Scholar]
- 234. Paroha, Ruchi , Wang Jia, and Lee Sunhee. 2024. “PDCD4 as a Marker of mTOR Pathway Activation and Therapeutic Target in Mycobacterial Infections.” Microbiology Spectrum 12: e0006224. 10.1128/spectrum.00062-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Cole, S. T. , Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., et al. 1998. “Deciphering the Biology of Mycobacterium Tuberculosis From the Complete Genome Sequence.” Nature 393: 537–544. 10.1038/31159 [DOI] [PubMed] [Google Scholar]
- 236. Lu, Qian , Zhang Wei, Fang Jun, Zheng Jianjian, Dong Chunsheng, and Xiong Sidong. 2020. “Mycobacterium Tuberculosis Rv1096, Facilitates Mycobacterial Survival by Modulating the NF‐κB/MAPK Pathway as Peptidoglycan N‐Deacetylase.” Molecular Immunology 127: 47–55. 10.1016/j.molimm.2020.08.005 [DOI] [PubMed] [Google Scholar]
- 237. Gehring, Adam J. , Dobos Karen M., Belisle John T., Harding Clifford V., and Boom W Henry. 2004. “Mycobacterium tuberculosis LprG (Rv1411c): A Novel TLR‐2 Ligand That Inhibits Human Macrophage Class II MHC Antigen Processing.” Journal of Immunology 173: 2660–2668. 10.4049/jimmunol.173.4.2660. [DOI] [PubMed] [Google Scholar]
- 238. Walburger, Anne , Koul Anil, Ferrari Giorgio, Nguyen Liem, Prescianotto‐Baschong Cristina, Huygen Kris, Klebl Bert, et al. 2004. “Protein Kinase G From Pathogenic Mycobacteria Promotes Survival Within Macrophages.” Science 304: 1800–1804. 10.1126/science.1099384 [DOI] [PubMed] [Google Scholar]
- 239. Voskuil, Martin I. , Schnappinger Dirk, Visconti Kevin C., Harrell Maria I., Dolganov Gregory M., Sherman David R., and Schoolnik Gary K.. 2003. “Inhibition of Respiration by Nitric Oxide Induces a Mycobacterium Tuberculosis Dormancy Program.” Journal of Experimental Medicine 198: 705–713. 10.1084/jem.20030205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Zahrt, Thomas C. , and Deretic Vojo. 2001. “ Mycobacterium Tuberculosis Signal Transduction System Required for Persistent Infections.” Proceedings of the National Academy of Sciences 98: 12706–12711. 10.1073/pnas.221272198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Akhter, Yusuf , Ehebauer Matthias T., Mukhopadhyay Sangita, and Hasnain Seyed E.. 2012. “The PE/PPE Multigene Family Codes for Virulence Factors and Is a Possible Source of Mycobacterial Antigenic Variation: Perhaps More?” Biochimie 94: 110–116. 10.1016/j.biochi.2011.09.026 [DOI] [PubMed] [Google Scholar]
- 242. Campbell, Elizabeth A. , Korzheva Nataliya, Mustaev Arkady, Murakami Katsuhiko, Nair Satish, Goldfarb Alex, and Darst Seth A.. 2001. “Structural Mechanism for Rifampicin Inhibition of Bacterial RNA Polymerase.” Cell 104: 901–912. 10.1016/s0092-8674(01)00286-0 [DOI] [PubMed] [Google Scholar]
- 243. Domenech, Pilar , Reed Michael B., and Clifton E. Barry, III . 2005. “Contribution of the Mycobacterium Tuberculosis MmpL Protein Family to Virulence and Drug Resistance.” Infection and Immunity 73: 3492–3501. 10.1128/IAI.73.6.3492-3501.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Banerjee, Asesh , Dubnau Eugenie, Quemard Annaik, Balasubramanian V., Um Kyung Sun, Wilson Theresa, Collins Des, De Lisle Geoffrey, and Jacobs William R.. 1994. “ inhA, a Gene Encoding a Target for Isoniazid and Ethionamide in Mycobacterium Tuberculosis .” Science 263: 227–230. 10.1126/science.8284673 [DOI] [PubMed] [Google Scholar]
- 245. Takayama, Kuni , Wang Cindy, and Besra Gurdyal S.. 2005. “Pathway to Synthesis and Processing of Mycolic Acids in Mycobacterium Tuberculosis .” Clinical Microbiology Reviews 18: 81–101. 10.1128/CMR.18.1.81-101.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Beier, Sara , and Bertilsson Stefan. 2013. “Bacterial Chitin Degradation‐Mechanisms and Ecophysiological Strategies.” Frontiers in Microbiology 4: 149. 10.3389/fmicb.2013.00149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Sachdeva, Preeti , Misra Richa, Tyagi Anil K., and Singh Yogendra. 2010. “The Sigma Factors of Mycobacterium Tuberculosis: Regulation of the Regulators.” The FEBS Journal 277: 605–626. 10.1111/j.1742-4658.2009.07479.x [DOI] [PubMed] [Google Scholar]
- 248. Rodrigue, Sébastien , Provvedi Roberta, Jacques Pierre‐Étienne, Gaudreau Luc, and Manganelli Riccardo. 2006. “The σ Factors Ofmycobacterium Tuberculosis.” FEMS Microbiology Reviews 30: 926–941. 10.1111/j.1574-6976.2006.00040.x [DOI] [PubMed] [Google Scholar]
- 249. Sala, Claudia , Odermatt Nina T., Soler‐Arnedo Paloma, Gülen Muhammet F., von Schultz Sofia, Benjak Andrej, and Cole Stewart T.. 2018. “EspL Is Essential for Virulence and Stabilizes EspE, EspF and EspH Levels in Mycobacterium Tuberculosis .” PLOS Pathogens 14: e1007491. 10.1371/journal.ppat.1007491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Su, Haibo , Zhu Shenglin, Zhu Lin, Huang Wei, Wang Honghai, Zhang Zhi, and Xu Ying. 2016. “Recombinant Lipoprotein Rv1016c Derived From Mycobacterium tuberculosis Is a TLR‐2 Ligand That Induces Macrophages Apoptosis and Inhibits MHC II Antigen Processing.” Frontiers in cellular and infection microbiology 6: 147. 10.3389/fcimb.2016.00147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Aagaard, Claus , Hoang Truc, Dietrich Jes, Cardona Pere‐Joan, Izzo Angelo, Dolganov Gregory, Schoolnik Gary K., et al. 2011. “A Multistage Tuberculosis Vaccine That Confers Efficient Protection Before and After Exposure.” Nature Medicine 17: 189–194. 10.1038/nm.2285 [DOI] [PubMed] [Google Scholar]
- 252. Champion, Matthew M. , Williams Emily A., Pinapati Richard S., and Champion Patricia A. DiGiuseppe. 2014. “Correlation of Phenotypic Profiles Using Targeted Proteomics Identifies Mycobacterial Esx‐1 Substrates.” Journal of Proteome Research 13: 5151–5164. 10.1021/pr500484w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Mukamolova, Galina V. , Turapov Obolbek A., Young Danielle I., Kaprelyants Arseny S., Kell Douglas B., and Young Michael. 2002. “A Family of Autocrine Growth Factors in Mycobacterium Tuberculosis .” Molecular Microbiology 46: 623–635. 10.1046/j.1365-2958.2002.03184.x [DOI] [PubMed] [Google Scholar]
- 254. Rivas‐Santiago, Bruno , Rivas Santiago Cesar E., Castañeda‐Delgado Julio E., León–Contreras Juan C., Hancock Robert E. W., and Hernandez‐Pando Rogelio. 2013. “Activity of LL‐37, CRAMP and Antimicrobial Peptide‐Derived Compounds E2, E6 and CP26 Against Mycobacterium Tuberculosis .” International Journal of Antimicrobial Agents 41: 143–148. 10.1016/j.ijantimicag.2012.09.015 [DOI] [PubMed] [Google Scholar]
- 255. Troy, Amber , Esparza‐Gonzalez Sandra C., Bartek Alicia, Creissen Elizabeth, Izzo Linda, and Izzo Angelo A.. 2020. “Pulmonary Mucosal Immunity Mediated through CpG Provides Adequate Protection Against Pulmonary Mycobacterium Tuberculosis Infection in the Mouse Model. A Role for Type I Interferon.” Tuberculosis 123: 101949. 10.1016/j.tube.2020.101949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Matsumoto, Takehisa , Hashimoto Masayuki, Teng Ching‐Hao, Hsu Po‐Chuen, Ota Yusuke, Takamizawa Masaru, Kato Ryosuke, and Negishi Tatsuya. 2020. “Molecular Characterization of a Carbon Dioxide‐Dependent Escherichia Coli Small‐Colony Variant Isolated From Blood Cultures.” International Journal of Medical Microbiology 310: 151431. 10.1016/j.ijmm.2020.151431 [DOI] [PubMed] [Google Scholar]
- 257. Herrera Ramírez, J. C. , la Mora A Ch De, De la Mora Valle A., Lopez‐Valencia G., Hurtado R. M. B., Rentería Evangelista T. B., Rodríguez Castillo J. L., et al. 2017. “Immunopathological Evaluation of Recombinant Mycobacterial Antigen Hsp65 Expressed in Lactococcus Lactis as a Novel Vaccine Candidate.” Iranian Journal of Veterinary Research 18: 197–202. https://pubmed.ncbi.nlm.nih.gov/29163649/ [PMC free article] [PubMed] [Google Scholar]
- 258. Kubinak, Jason L. , and Round June L.. 2016. “Do Antibodies Select a Healthy Microbiota?” Nature Reviews Immunology 16: 767–774. 10.1038/nri.2016.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Ratledge, Colin , and Dover Lynn G.. 2000. “Iron Metabolism in Pathogenic Bacteria.” Annual Review of Microbiology 54: 881–941. 10.1146/annurev.micro.54.1.881 [DOI] [PubMed] [Google Scholar]
- 260. Hoang, Truc , Aagaard Claus, Dietrich Jes, Cassidy Joseph P., Dolganov Gregory, Schoolnik Gary K., Lundberg Carina Vingsbo, Agger Else Marie, and Andersen Peter. 2013. “ESAT‐6 (EsxA) and TB10.4 (EsxH) Based Vaccines for Pre‐ and Post‐Exposure Tuberculosis Vaccination.” PLOS One 8: e80579. 10.1371/journal.pone.0080579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Fraga‐Corral, M. , Carpena M., Garcia‐Oliveira P., Pereira A. G., Prieto M. A., and Simal‐Gandara J.. 2022. “Analytical Metabolomics and Applications in Health, Environmental and Food Science.” Critical Reviews in Analytical Chemistry 52: 712–734. 10.1080/10408347.2020.1823811 [DOI] [PubMed] [Google Scholar]
- 262. Krautkramer, Kimberly A. , Fan Jing, and Bäckhed Fredrik. 2021. “Gut Microbial Metabolites as Multi‐Kingdom Intermediates.” Nature Reviews Microbiology 19: 77–94. 10.1038/s41579-020-0438-4 [DOI] [PubMed] [Google Scholar]
- 263. Folz, Jacob , Culver Rebecca Neal, Morales Juan Montes, Grembi Jessica, Triadafilopoulos George, Relman David A., Huang Kerwyn Casey, Shalon Dari, and Fiehn Oliver. 2023. “Human Metabolome Variation Along the Upper Intestinal Tract.” Nature Metabolism 5: 777–788. 10.1038/s42255-023-00777-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Daliri, Eric Banan‐Mwine , Wei Shuai, Oh Deog H., and Lee Byong H.. 2017. “The Human Microbiome and Metabolomics: Current Concepts and Applications.” Critical Reviews in Food Science and Nutrition 57: 3565–3576. 10.1080/10408398.2016.1220913 [DOI] [PubMed] [Google Scholar]
- 265. Hu, Yangyang , Jiang Guangyu, Wen Yalun, Shao Yuchen, Yang Ge, and Qu Feng. 2025. “Selection of Aptamers Targeting Small Molecules by Capillary Electrophoresis: Advances, Challenges, and Prospects.” Biotechnology Advances 78: 108491. 10.1016/j.biotechadv.2024.108491 [DOI] [PubMed] [Google Scholar]
- 266. Fernández‐García, Miguel , Rey‐Stolle Fernanda, Boccard Julien, Reddy Vineel P., García Antonia, Cumming Bridgette M., Steyn Adrie J. C., Rudaz Serge, and Barbas Coral. 2020. “Comprehensive Examination of the Mouse Lung Metabolome Following Mycobacterium Tuberculosis Infection Using a Multiplatform Mass Spectrometry Approach.” Journal of Proteome Research 19: 2053–2070. 10.1021/acs.jproteome.9b00868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Bartels, Benjamin , and Svatoš Aleš. 2015. “Spatially Resolved In Vivo Plant Metabolomics by Laser Ablation‐Based Mass Spectrometry Imaging (MSI) Techniques: LDI‐MSI and LAESI.” Frontiers in Plant Science 6: 471. 10.3389/fpls.2015.00471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Takáts, Zoltán , Wiseman Justin M., Gologan Bogdan, and Cooks R Graham. 2004. “Mass Spectrometry Sampling Under Ambient Conditions With Desorption Electrospray Ionization.” Science 306: 471–473. 10.1126/science.1104404 [DOI] [PubMed] [Google Scholar]
- 269. Prideaux, Brendan , Via Laura E., Zimmerman Matthew D., Eum Seokyong, Sarathy Jansy, O'Brien Paul, Chen Chao, et al. 2015. “The Association Between Sterilizing Activity and Drug Distribution Into Tuberculosis Lesions.” Nature Medicine 21: 1223–1227. 10.1038/nm.3937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Blanc, Landry , Lenaerts Anne, Dartois Véronique, and Prideaux Brendan. 2018. “Visualization of Mycobacterial Biomarkers and Tuberculosis Drugs in Infected Tissue by MALDI‐MS Imaging.” Analytical Chemistry 90: 6275–6282. 10.1021/acs.analchem.8b00985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Andreas, Nicholas J. , Basu Roy Robindra, Gomez‐Romero Maria, Horneffer‐van der Sluis Verena, Lewis Matthew R., Camuzeaux Stephane S. M., Jiménez Beatriz, et al. 2020. “Performance of Metabonomic Serum Analysis for Diagnostics in Paediatric Tuberculosis.” Scientific Reports 10: 7302. 10.1038/s41598-020-64413-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Olivier, Cara , and Luies Laneke. 2024. “Metabolic Insights Into HIV/TB Co‐Infection: An Untargeted Urinary Metabolomics Approach.” Metabolomics 20: 78. 10.1007/s11306-024-02148-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Mason, Shayne , Van Furth A. Marceline Tutu, Solomons Regan, Wevers Ron A., Van Reenen Mari, and Reinecke Carolus J.. 2016. “A Putative Urinary Biosignature for Diagnosis and Follow‐Up of Tuberculous Meningitis in Children: Outcome of a Metabolomics Study Disclosing Host‐Pathogen Responses.” Metabolomics 12: 110. 10.1007/s11306-016-1053-2 [DOI] [Google Scholar]
- 274. Isaiah, Simon , Loots Du Toit, van Furth A. Marceline Tutu, Davoren Elmarie, van Elsland Sabine, Solomons Regan, van der Kuip Martijn, and Mason Shayne. 2024. “Urinary Markers of Mycobacterium Tuberculosis and Dysbiosis in Paediatric Tuberculous Meningitis Cases Undergoing Treatment.” Gut Pathogens 16: 14. 10.1186/s13099-024-00609-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Ren, Sheng , Hinzman Anna A., Kang Emily L., Szczesniak Rhonda D., and Lu Long Jason. 2015. “Computational and Statistical Analysis of Metabolomics Data.” Metabolomics 11: 1492–1513. 10.1007/s11306-015-0823-6 [DOI] [Google Scholar]
- 276. Anwardeen, Najeha R. , Diboun Ilhame, Mokrab Younes, Althani Asma A., and Elrayess Mohamed A.. 2023. “Statistical Methods and Resources for Biomarker Discovery Using Metabolomics.” BMC Bioinformatics 24: 250. 10.1186/s12859-023-05383-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Anh, Nguyen Ky , Yen Nguyen Thi Hai, Tien Nguyen Tran Nam, Phat Nguyen Ky, Park Young Jin, Kim Ho‐Sook, Vu Dinh Hoa, et al. 2024. “Metabolic Phenotyping and Global Functional Analysis Facilitate Metabolic Signature Discovery for Tuberculosis Treatment Monitoring.” Biochimica et Biophysica Acta (BBA) ‐ Molecular Basis of Disease 1870: 167064. 10.1016/j.bbadis.2024.167064 [DOI] [PubMed] [Google Scholar]
- 278. Yi, Wen‐Jing , Han Yu‐Shuai, Wei Li‐Liang, Shi Li‐Ying, Huang Huai, Jiang Ting‐Ting, Li Zhi‐Bin, et al. 2019. “L‐Histidine, Arachidonic Acid, Biliverdin, and L‐Cysteine‐Glutathione Disulfide as Potential Biomarkers for Cured Pulmonary Tuberculosis.” Biomedicine & Pharmacotherapy 116: 108980. 10.1016/j.biopha.2019.108980 [DOI] [PubMed] [Google Scholar]
- 279. Ding, Yi , Raterink Robert‐Jan, Marín‐Juez Rubén, Veneman Wouter J., Egbers Koen, van den Eeden Susan, Haks Mariëlle C., et al. 2020. “Tuberculosis Causes Highly Conserved Metabolic Changes in Human Patients, Mycobacteria‐Infected Mice and Zebrafish Larvae.” Scientific Reports 10: 11635. 10.1038/s41598-020-68443-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Albors‐Vaquer, A. , Rizvi A., Matzapetakis M., Lamosa P., Coelho A. V., Patel A. B., Mande S. C., et al. 2020. “Active and Prospective Latent Tuberculosis Are Associated With Different Metabolomic Profiles: Clinical Potential for the Identification of Rapid and Non‐Invasive Biomarkers.” Emerging Microbes & Infections 9: 1131–1139. 10.1080/22221751.2020.1760734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Yang, Boyi , Guo Xiaojing, Shi Chongyu, Liu Gang, Qin Xiaoling, Chen Shiyi, Gan Li, et al. 2024. “Alterations in Purine and Pyrimidine Metabolism Associated With Latent Tuberculosis Infection: Insights From Gut Microbiome and Metabolomics Analyses.” mSystems 9: e0081224. 10.1128/msystems.00812-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Chen, Siyu , Li Chunyan, Qin Zhonghua, Song Lili, Zhang Shiyuan, Sun Chongxiang, Zhuang Pengwei, et al. 2023. “Serum Metabolomic Profiles for Distinguishing Lung Cancer From Pulmonary Tuberculosis: Identification of Rapid and Noninvasive Biomarker.” Journal of Infectious Diseases 228: 1154–1165. 10.1093/infdis/jiad175 [DOI] [PubMed] [Google Scholar]
- 283. Anh, Nguyen Ky , Phat Nguyen Ky, Thu Nguyen Quang, Tien Nguyen Tran Nam, Eunsu Cho, Kim Ho‐Sook, Nguyen Duc Ninh, et al. 2024. “Discovery of Urinary Biosignatures for Tuberculosis and Nontuberculous Mycobacteria Classification Using Metabolomics and Machine Learning.” Scientific Reports 14: 15312. 10.1038/s41598-024-66113-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Weiner January 3rd, Jeroen, Maertzdorf , Sutherland Jayne S., Duffy Fergal J., Thompson Ethan, Suliman Sara, McEwen Gayle, Thiel Bonnie, et al. 2018. “Metabolite Changes in Blood Predict the Onset of Tuberculosis.” Nature Communications 9: 5208. 10.1038/s41467-018-07635-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Jeyanathan, Mangalakumari , Vaseghi‐Shanjani Maryam, Afkhami Sam, Grondin Jensine A., Kang Alisha, D'Agostino Michael R., Yao Yushi, et al. 2022. “Parenteral BCG Vaccine Induces Lung‐Resident Memory Macrophages and Trained Immunity via the Gut‐Lung Axis.” Nature Immunology 23: 1687–1702. 10.1038/s41590-022-01354-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Anh, Nguyen Ky , Phat Nguyen Ky, Yen Nguyen Thi Hai, Jayanti Rannissa Puspita, Anh Thu Vo Thuy, Park Young Jin, and Cho Yong‐Soon, et al. 2023. “Comprehensive Lipid Profiles Investigation Reveals Host Metabolic and Immune Alterations During Anti‐Tuberculosis Treatment: Implications for Therapeutic Monitoring.” Biomedicine & Pharmacotherapy 158: 114187. 10.1016/j.biopha.2022.114187 [DOI] [PubMed] [Google Scholar]
- 287. Opperman, Monique , Loots Du Toit, van Reenen Mari, Ronacher Katharina, Walzl Gerhard, and du Preez Ilse. 2021. “Chronological Metabolic Response to Intensive Phase TB Therapy in Patients With Cured and Failed Treatment Outcomes.” ACS Infectious Diseases 7: 1859–1869. 10.1021/acsinfecdis.1c00162 [DOI] [PubMed] [Google Scholar]
- 288. Suster, Carl J. E. , Pham David, Kok Jen, and Sintchenko Vitali. 2024. “Emerging Applications of Artificial Intelligence in Pathogen Genomics.” Frontiers in Bacteriology 3: 1326958. 10.3389/fbrio.2024.1326958 [DOI] [Google Scholar]
- 289. D'Urso, Fabiana , and Broccolo Francesco. 2024. “Applications of Artificial Intelligence in Microbiome Analysis and Probiotic Interventions—An Overview and Perspective Based on the Current State of the Art.” Applied Sciences 14: 8627. 10.3390/app14198627 [DOI] [Google Scholar]
- 290. Bai, Defeng , Chen Tong, Xun Jiani, Ma Chuang, Luo Hao, Yang Haifei, Cao Chen, et al. 2025. “EasyMetagenome: A User‐Friendly and Flexible Pipeline for Shotgun Metagenomic Analysis in Microbiome Research.” iMeta 4: e70001. 10.1002/imt2.70001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Turnbaugh, Peter J. , Ley Ruth E., Hamady Micah, Fraser‐Liggett Claire M., Knight Rob, and Gordon Jeffrey I.. 2007. “The Human Microbiome Project.” Nature 449: 804–810. 10.1038/nature06244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. King, Charles H. , Desai Hiral, Sylvetsky Allison C., LoTempio Jonathan, Ayanyan Shant, Carrie Jill, Crandall Keith A., et al. 2019. “Baseline Human Gut Microbiota Profile in Healthy People and Standard Reporting Template.” PLOS One 14: e0206484. 10.1371/journal.pone.0206484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Markowitz, V. M. , Chen I.‐M. A., Palaniappan K., Chu K., Szeto E., Grechkin Y., Ratner A., et al. 2012. “IMG: The Integrated Microbial Genomes Database and Comparative Analysis System.” Nucleic Acids Research 40: D115–D122. 10.1093/nar/gkr1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Wishart, David S. , Guo AnChi, Oler Eponine, Wang Fei, Anjum Afia, Peters Harrison, Dizon Raynard, et al. 2022. “HMDB 5.0: The Human Metabolome Database for 2022.” Nucleic Acids Research 50: D622–D631. 10.1093/nar/gkab1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Guijas, Carlos , Montenegro‐Burke J Rafael, Domingo‐Almenara Xavier, Palermo Amelia, Warth Benedikt, Hermann Gerrit, Koellensperger Gunda, et al. 2018. “METLIN: A Technology Platform for Identifying Knowns and Unknowns.” Analytical Chemistry 90: 3156–3164. 10.1021/acs.analchem.7b04424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Kanehisa, M . 2000. “KEGG: Kyoto Encyclopedia of Genes and Genomes.” Nucleic Acids Research 28: 27–30. 10.1093/nar/28.1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Karp, Peter D. , Billington Richard, Caspi Ron, Fulcher Carol A., Latendresse Mario, Kothari Anamika, Keseler Ingrid M., et al. 2019. “The BioCyc Collection of Microbial Genomes and Metabolic Pathways.” Briefings in Bioinformatics 20: 1085–1093. 10.1093/bib/bbx085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Szklarczyk, Damian , Kirsch Rebecca, Koutrouli Mikaela, Nastou Katerina, Mehryary Farrokh, Hachilif Radja, Gable Annika L., et al. 2023. “The STRING Database in 2023: Protein‐Protein Association Networks and Functional Enrichment Analyses for Any Sequenced Genome of Interest.” Nucleic Acids Research 51: D638–D646. 10.1093/nar/gkac1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Hunter, Sarah , Corbett Matthew, Denise Hubert, Fraser Matthew, Gonzalez‐Beltran Alejandra, Hunter Christopher, Jones Philip, et al. 2014. “EBI Metagenomics—A New Resource for the Analysis and Archiving of Metagenomic Data.” Nucleic Acids Research 42: D600–D606. 10.1093/nar/gkt961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Dai, Die , Zhu Jiaying, Sun Chuqing, Li Min, Liu Jinxin, Wu Sicheng, Ning Kang, et al. 2022. “GMrepo V2: A Curated Human Gut Microbiome Database With Special Focus on Disease Markers and Cross‐Dataset Comparison.” Nucleic Acids Research 50: D777–D784. 10.1093/nar/gkab1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. DeSantis, T. Z. , Hugenholtz P., Larsen N., Rojas M., Brodie E. L., Keller K., Huber T., et al. 2006. “Greengenes, a Chimera‐Checked 16S rRNA Gene Database and Workbench Compatible With ARB.” Applied and Environmental Microbiology 72: 5069–5072. 10.1128/AEM.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Quast, Christian , Pruesse Elmar, Yilmaz Pelin, Gerken Jan, Schweer Timmy, Yarza Pablo, Peplies Jörg, and Glöckner Frank Oliver. 2012. “The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web‐Based Tools.” Nucleic Acids Research 41: D590–D596. 10.1093/nar/gks1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Cole, James R. , Wang Qiong, Fish Jordan A., Chai Benli, McGarrell Donna M., Sun Yanni, Brown C. Titus, et al. 2014. “Ribosomal Database Project: Data and Tools for High Throughput rRNA Analysis.” Nucleic Acids Research 42: D633–D642. 10.1093/nar/gkt1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Qi, Changlu , Cai Yiting, Qian Kai, Li Xuefeng, Ren Jialiang, Wang Ping, Fu Tongze, et al. 2023. “gutMDisorder V2.0: A Comprehensive Database for Dysbiosis of Gut Microbiota in Phenotypes and Interventions.” Nucleic Acids Research 51: D717–D722. 10.1093/nar/gkac871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Koren, Omry , Knights Dan, Gonzalez Antonio, Waldron Levi, Segata Nicola, Knight Rob, Huttenhower Curtis, and Ley Ruth E.. 2013. “A Guide to Enterotypes Across the Human Body: Meta‐Analysis of Microbial Community Structures in Human Microbiome Datasets.” PLOS Computational Biology 9: e1002863. 10.1371/journal.pcbi.1002863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Novielli, Pierfrancesco , Romano Donato, Magarelli Michele, Bitonto Pierpaolo Di, Diacono Domenico, Chiatante Annalisa, Lopalco Giuseppe, et al. 2024. “Explainable Artificial Intelligence for Microbiome Data Analysis in Colorectal Cancer Biomarker Identification.” Frontiers in Microbiology 15: 1348974. 10.3389/fmicb.2024.1348974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Wang, Ming‐Gui , Wu Shou‐Quan, Zhang Meng‐Meng, and He Jian‐Qing. 2022. “Urine Metabolomics and Microbiome Analyses Reveal the Mechanism of Anti‐Tuberculosis Drug‐Induced Liver Injury, as Assessed for Causality Using the Updated RUCAM: A Prospective Study.” Frontiers in Immunology 13: 1002126. 10.3389/fimmu.2022.1002126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Lv, Xinna , Li Ye, Cai Botao, He Wei, Wang Ren, Chen Minghui, Pan Junhua, and Hou Dailun. 2023. “Utility of Machine Learning and Radiomics Based on Cavity for Predicting the Therapeutic Response of MDR‐TB.” Infection and Drug Resistance 16: 6893–6904. 10.2147/IDR.S435984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Chen, Jingfang , Jiang Youli, Li Zhihuan, Zhang Mingshu, Liu Linlin, Li Ao, and Lu Hongzhou. 2024. “Predictive Machine Learning Models for Anticipating Loss to Follow‐Up in Tuberculosis Patients Throughout Anti‐TB Treatment Journey.” Scientific Reports 14: 24685. 10.1038/s41598-024-74942-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Peetluk, Lauren S. , Rebeiro Peter F., Ridolfi Felipe M., Andrade Bruno B., Cordeiro‐Santos Marcelo, Kritski Afranio, Durovni Betina, et al. 2022. “A Clinical Prediction Model for Unsuccessful Pulmonary Tuberculosis Treatment Outcomes.” Clinical Infectious Diseases 74: 973–982. 10.1093/cid/ciab598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Sambarey, Awanti , Smith Kirk, Chung Carolina, Arora Harkirat Singh, Yang Zhenhua, Agarwal Prachi P., and Chandrasekaran Sriram. 2024. “Integrative Analysis of Multimodal Patient Data Identifies Personalized Predictors of Tuberculosis Treatment Prognosis.” iScience 27: 109025. 10.1016/j.isci.2024.109025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Liao, Kuang‐Ming , Liu Chung‐Feng, Chen Chia‐Jung, Feng Jia‐Yih, Shu Chin‐Chung, and Ma Yu‐Shan. 2023. “Using an Artificial Intelligence Approach to Predict the Adverse Effects and Prognosis of Tuberculosis.” Diagnostics 13: 1075. 10.3390/diagnostics13061075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Gupta, Ankit , Dhakan Darshan B., Maji Abhijit, Saxena Rituja, PK Vishnu Prasoodanan, Mahajan Shruti, Mahajan Shruti, Pulikkan Joby, et al. 2019. “Association of Flavonifractor Plautii, a Flavonoid‐Degrading Bacterium, With the Gut Microbiome of Colorectal Cancer Patients in India.” mSystems 4: e00438–19. 10.1128/mSystems.00438-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Shi, Yushu , Zhang Liangliang, Peterson Christine B., Do Kim‐Anh, and Jenq Robert R.. 2022. “Performance Determinants of Unsupervised Clustering Methods for Microbiome Data.” Microbiome 10: 25. 10.1186/s40168-021-01199-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Lipton, Zachary C . 2018. “The Mythos of Model Interpretability: In Machine Learning, the Concept of Interpretability Is Both Important and Slippery.” Queue 16: 31–57. 10.1145/3236386.3241340 [DOI] [Google Scholar]
- 316. Lee, Sang‐Mok , Park Hong‐Tae, Park Seojoung, Lee Jun Ho, Kim Danil, Yoo Han Sang, and Kim Donghyuk. 2023. “A Machine Learning Approach Reveals a Microbiota Signature for Infection With Mycobacterium Avium Subsp. Paratuberculosis in Cattle.” Microbiology Spectrum 11: e0313422. 10.1128/spectrum.03134-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Hosu, Mojisola Clara , Faye Lindiwe Modest, and Apalata Teke. 2024. “Predicting Treatment Outcomes in Patients With Drug‐Resistant Tuberculosis and Human Immunodeficiency Virus Coinfection, Using Supervised Machine Learning Algorithm.” Pathogens 13: 923. 10.3390/pathogens13110923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Ahamed Fayaz, Shaik , Babu Lakshmanan, Paridayal Loganathan, Vasantha Mahalingam, Paramasivam Palaniyandi, Sundarakumar Karuppasamy, and Ponnuraja Chinnaiyan. 2024. “Machine Learning Algorithms to Predict Treatment Success for Patients With Pulmonary Tuberculosis.” PLoS One 19: e0309151. 10.1371/journal.pone.0309151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Larkins‐Ford, Jonah , Degefu Yonatan N., Van Nhi, Sokolov Artem, and Aldridge Bree B.. 2022. “Design Principles to Assemble Drug Combinations for Effective Tuberculosis Therapy Using Interpretable Pairwise Drug Response Measurements.” Cell Reports Medicine 3: 100737. 10.1016/j.xcrm.2022.100737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Xiong, Yan , Ba Xiaojun, Hou Ao, Zhang Kaiwen, Chen Longsen, and Li Ting. 2018. “Automatic Detection of Mycobacterium Tuberculosis Using Artificial Intelligence.” Journal of Thoracic Disease 10: 1936–1940. 10.21037/jtd.2018.01.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Sharma, Anshu , Sharma Anurag, Malhotra Rahul, Singh Parulpreet, Chakrabortty Ripon K., Mahajan Shubham, and Pandit Amit Kant. 2021. “An Accurate Artificial Intelligence System for the Detection of Pulmonary and Extra Pulmonary Tuberculosis.” Tuberculosis 131: 102143. 10.1016/j.tube.2021.102143 [DOI] [PubMed] [Google Scholar]
- 322. Baur, Sebastien , Nabulsi Zaid, Weng Wei‐Hung, Garrison Jake, Blankemeier Louis, Fishman Sam, Chen Christina, et al. 2024. “HeAR‐‐Health Acoustic Representations.” arXiv Preprint: 02522. 10.48550/arXiv.2403.02522 [DOI]
- 323. Callahan, Benjamin J. , McMurdie Paul J., Rosen Michael J., Han Andrew W., Johnson Amy Jo A., and Holmes Susan P.. 2016. “DADA2: High‐Resolution Sample Inference From Illumina Amplicon Data.” Nature Methods 13: 581–583. 10.1038/nmeth.3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Yellapu, Gayatri Devi , Rudraraju Gowrisree, Sripada Narayana Rao, Mamidgi Baswaraj, Jalukuru Charan, Firmal Priyanka, Yechuri Venkat, et al. 2023. “Development and Clinical Validation of Swaasa AI Platform for Screening and Prioritization of Pulmonary TB.” Scientific Reports 13: 4740. 10.1038/s41598-023-31772-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Gholami, Amir , Kim Sehoon, Dong Zhen, Yao Zhewei, Mahoney Michael W., and Keutzer Kurt. 2022. “A Survey of Quantization Methods for Efficient Neural Network Inference.” arXiv Preprint 13630. [preprint] 10.48550/arXiv.2103.13630 [DOI] [Google Scholar]
- 326. Liang, Shufan , Ma Jiechao, Wang Gang, Shao Jun, Li Jingwei, Deng Hui, Wang Chengdi, and Li Weimin. 2022. “The Application of Artificial Intelligence in the Diagnosis and Drug Resistance Prediction of Pulmonary Tuberculosis.” Frontiers in Medicine 9: 935080. 10.3389/fmed.2022.935080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Du, Jingli , Su Yue, Qiao Juan, Gao Shang, Dong Enjun, Wang Ruilan, Nie Yanhui, et al. 2024. “Application of Artificial Intelligence in Diagnosis of Pulmonary Tuberculosis.” Chinese Medical Journal 137: 559–561. 10.1097/CM9.0000000000003018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Jamal, Salma , Khubaib Mohd., Gangwar Rishabh, Grover Sonam, Grover Abhinav, and Hasnain Seyed E.. 2020. “Artificial Intelligence and Machine Learning Based Prediction of Resistant and Susceptible Mutations in Mycobacterium Tuberculosis .” Scientific Reports 10: 5487. 10.1038/s41598-020-62368-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Shevtsova, Daria , Ahmed Anam, Boot Iris W. A., Sanges Carmen, Hudecek Michael, Jacobs John J. L., Hort Simon, and Vrijhoef Hubertus J. M.. 2024. “Trust in and Acceptance of Artificial Intelligence Applications in Medicine: Mixed Methods Study.” JMIR Human Factors 11: e47031. 10.2196/47031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Leikas, Jaana , Johri Aditya, Latvanen Marko, Wessberg Nina, and Hahto Antti. 2022. “Governing Ethical AI Transformation: A Case Study of AuroraAI.” Frontiers in Artificial Intelligence 5: 836557. 10.3389/frai.2022.836557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Kuziemsky, Craig E. , Chrimes Dillon, Minshall Simon, Mannerow Michael, and Lau Francis. 2024. “AI Quality Standards in Health Care: Rapid Umbrella Review.” Journal of Medical Internet Research 26: e54705. 10.2196/54705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Mardis, Elaine R . 2008. “Next‐Generation DNA Sequencing Methods.” Annual Review of Genomics and Human Genetics 9: 387–402. 10.1146/annurev.genom.9.081307.164359 [DOI] [PubMed] [Google Scholar]
- 333. Ansorge, Wilhelm J . 2009. “Next‐Generation DNA Sequencing Techniques.” New Biotechnology 25: 195–203. 10.1016/j.nbt.2008.12.009 [DOI] [PubMed] [Google Scholar]
- 334. Jovel, Juan , Patterson Jordan, Wang Weiwei, Hotte Naomi, O'Keefe Sandra, Mitchel Troy, Perry Troy, et al. 2016. “Characterization of the Gut Microbiome Using 16S or Shotgun Metagenomics.” Frontiers in Microbiology 7: 459. 10.3389/fmicb.2016.00459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. Namasivayam, Sivaranjani , Sher Alan, Glickman Michael S., and Wipperman Matthew F.. 2018. “The Microbiome and Tuberculosis: Early Evidence for Cross Talk.” mBio 9: e01420‐18. 10.1128/mBio.01420-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Wu, Xuangao , Zhang Ting, Zhang TianShun, and Park Sunmin. 2024. “The Impact of Gut Microbiome Enterotypes on Ulcerative Colitis: Identifying Key Bacterial Species and Revealing Species Co‐Occurrence Networks Using Machine Learning.” Gut Microbes 16: 2292254. 10.1080/19490976.2023.2292254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337. He, Ruiqiao , Li Pan, Wang Jinfeng, Cui Bota, Zhang Faming, and Zhao Fangqing. 2022. “The Interplay of Gut Microbiota Between Donors and Recipients Determines the Efficacy of Fecal Microbiota Transplantation.” Gut Microbes 14: 2100197. 10.1080/19490976.2022.2100197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Huang, Zhiqiang , Liu Kun, Ma Wenwen, Li Dezhi, Mo Tianlu, and Liu Qing. 2022. “The Gut Microbiome in Human Health and Disease−Where Are We and Where Are We Going? A Bibliometric Analysis.” Frontiers in Microbiology 13: 1018594. 10.3389/fmicb.2022.1018594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Janda, J. Michael , and Abbott Sharon L.. 2007. “16S rRNA Gene Sequencing for Bacterial Identification in the Diagnostic Laboratory: Pluses, Perils, and Pitfalls.” Journal of Clinical Microbiology 45: 2761–2764. 10.1128/JCM.01228-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340. Leek, Jeffrey T. , Scharpf Robert B., Bravo Héctor Corrada, Simcha David, Langmead Benjamin, Johnson W. Evan, Geman Donald, Baggerly Keith, and Irizarry Rafael A.. 2010. “Tackling the Widespread and Critical Impact of Batch Effects in High‐Throughput Data.” Nature Reviews Genetics 11: 733–739. 10.1038/nrg2825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Johnson, W. Evan , Li Cheng, and Rabinovic Ariel. 2007. “Adjusting Batch Effects in Microarray Expression Data Using Empirical Bayes Methods.” Biostatistics 8: 118–127. 10.1093/biostatistics/kxj037 [DOI] [PubMed] [Google Scholar]
- 342. Smyth, G. K. 2005. “Limma: Linear Models for Microarray Data.” Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Springer New York, 397–420. 10.1007/0-387-29362-0_23 [DOI]
- 343. Valdes, Camilo , Stebliankin Vitalii, and Narasimhan Giri. 2019. “Large Scale Microbiome Profiling in the Cloud.” Bioinformatics 35: i13–i22. 10.1093/bioinformatics/btz356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Manconia, Andrea , Gnocchia Matteo, Milanesia Luciano, Marullob Osvaldo, and Armano Giuliano. 2023. “Framing Apache Spark in Life Sciences.” Heliyon 9: e13368. 10.1016/j.heliyon.2023.e13368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Brown, Joseph , Pirrung Meg, and McCue Lee Ann. 2017. “FQC Dashboard: Integrates FastQC Results Into a Web‐Based, Interactive, and Extensible FASTQ Quality Control Tool.” Bioinformatics 33: 3137–3139. 10.1093/bioinformatics/btx373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346. Sewe, Steven O. , Silva Gonçalo, Sicat Paulo, Seal Susan E., and Visendi Paul. 2022. “Trimming and Validation of Illumina Short Reads Using Trimmomatic, Trinity Assembly, and Assessment of RNA‐Seq Data.” Methods in Molecular Biology 2443: 211–232. 10.1007/978-1-0716-2067-0_11 [DOI] [PubMed] [Google Scholar]
- 347. Liu, Yong‐Xin , Chen Lei, Ma Tengfei, Li Xiaofang, Zheng Maosheng, Zhou Xin, Chen Liang, et al. 2023. “EasyAmplicon: An Easy‐to‐Use, Open‐Source, Reproducible, and Community‐Based Pipeline for Amplicon Data Analysis in Microbiome Research.” iMeta 2: e83. 10.1002/imt2.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Hall, Michael , and Beiko Robert G.. 2018. “16S rRNA Gene Analysis With QIIME2.” Methods in Molecular Biology 1849: 113–129. 10.1007/978-1-4939-8728-3_8 [DOI] [PubMed] [Google Scholar]
- 349. Blanco‐Míguez, Aitor , Beghini Francesco, Cumbo Fabio, McIver Lauren J., Thompson Kelsey N., Zolfo Moreno, Manghi Paolo, et al. 2023. “Extending and Improving Metagenomic Taxonomic Profiling With Uncharacterized Species Using MetaPhlAn 4.” Nature Biotechnology 41: 1633–1644. 10.1038/s41587-023-01688-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Lu, Jennifer , Rincon Natalia, Wood Derrick E., Breitwieser Florian P., Pockrandt Christopher, Langmead Ben, Salzberg Steven L., and Steinegger Martin. 2022. “Metagenome Analysis Using the Kraken Software Suite.” Nature Protocols 17: 2815–2839. 10.1038/s41596-022-00738-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Hua, Xinwei , McGoldrick Jessica, Nakrour Nour, Staller Kyle, Chung Daniel Chulyong, Xavier Ramnik Joseph, and Khalili Hamed. 2024. “Gut Microbiome Structure and Function in Asymptomatic Diverticulosis.” Genome Medicine 16: 105. 10.1186/s13073-024-01374-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Douglas, Gavin M. , Maffei Vincent J., Zaneveld Jesse R., Yurgel Svetlana N., Brown James R., Taylor Christopher M., Huttenhower Curtis, and Langille Morgan G. I.. 2020. “PICRUSt2 for Prediction of Metagenome Functions.” Nature Biotechnology 38: 685–688. 10.1038/s41587-020-0548-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. McMurdie, Paul J. , and Holmes Susan. 2013. “Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data.” PLoS One 8: e61217. 10.1371/journal.pone.0061217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Zemke, Anna C. , Hilliam Yasmin, Stapleton Amanda L., Kimple Adam J., Goralski Jennifer L., Shaffer Amber D., Pilewski Joseph M., et al. 2024. “Elexacaftor‐Tezacaftor‐Ivacaftor Decreases Pseudomonas Abundance in the Sinonasal Microbiome in Cystic Fibrosis.” International Forum of Allergy & Rhinology 14: 928–938. 10.1002/alr.23288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Dhariwal, Achal , Chong Jasmine, Habib Salam, King Irah L., Agellon Luis B., and Xia Jianguo. 2017. “MicrobiomeAnalyst: A Web‐Based Tool for Comprehensive Statistical, Visual and Meta‐Analysis of Microbiome Data.” Nucleic Acids Research 45: W180–W188. 10.1093/nar/gkx295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. Uritskiy, Gherman V. , DiRuggiero Jocelyne, and Taylor James. 2018. “MetaWRAP−A Flexible Pipeline for Genome‐Resolved Metagenomic Data Analysis.” Microbiome 6: 158. 10.1186/s40168-018-0541-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357. Edgar, Robert C . 2013. “UPARSE: Highly Accurate OTU Sequences From Microbial Amplicon Reads.” Nature Methods 10: 996–998. 10.1038/nmeth.2604 [DOI] [PubMed] [Google Scholar]
- 358. Sanschagrin, Sylvie , and Yergeau Etienne. 2014. “Next‐Generation Sequencing of 16S Ribosomal RNA Gene Amplicons.” Journal of Visualized Experiments 29: 51709. 10.3791/51709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. Jing, Gongchao , Sun Zheng, Wang Honglei, Gong Yanhai, Huang Shi, Ning Kang, Xu Jian, and Su Xiaoquan. 2017. “Parallel‐META 3: Comprehensive Taxonomical and Functional Analysis Platform for Efficient Comparison of Microbial Communities.” Scientific Reports 7: 40371. 10.1038/srep40371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Kohl, Thomas Andreas , Utpatel Christian, Schleusener Viola, De Filippo Maria Rosaria, Beckert Patrick, Cirillo Daniela Maria, and Niemann Stefan. 2018. “MTBseq: A Comprehensive Pipeline for Whole Genome Sequence Analysis of Mycobacterium Tuberculosis Complex Isolates.” PeerJ 6: e5895. 10.7717/peerj.5895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361. Rognes, Torbjørn , Flouri Tomáš, Nichols Ben, Quince Christopher, and Mahé Frédéric. 2016. “VSEARCH: A Versatile Open Source Tool for Metagenomics.” PeerJ 4: e2584. 10.7717/peerj.2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362. Zhou, Yuanping , Liu Yong‐Xin, and Li Xuemeng. 2024. “USEARCH 12: Open‐Source Software for Sequencing Analysis in Bioinformatics and Microbiome.” iMeta 3: e236. 10.1002/imt2.236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. Zhou, Ruwen , Ng Siu Kin, Sung Joseph Jao Yiu, Goh Wilson Wen Bin, and Wong Sunny Hei. 2023. “Data Pre‐Processing for Analyzing Microbiome Data−A Mini Review.” Computational and Structural Biotechnology Journal 21: 4804–4815. 10.1016/j.csbj.2023.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364. Bolger, Anthony M. , Lohse Marc, and Usadel Bjoern. 2014. “Trimmomatic: A Flexible Trimmer for Illumina Sequence Data.” Bioinformatics 30: 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Bolyen, Evan , Rideout Jai Ram, Dillon Matthew R., Bokulich Nicholas A., Abnet Christian C., Al‐Ghalith Gabriel A., Alexander Harriet, et al. 2019. “Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2.” Nature Biotechnology 37: 852–857. 10.1038/s41587-019-0209-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Wood, Derrick E. , Lu Jennifer, Langmead Ben. 2019. “Improved Metagenomic Analysis With Kraken 2.” Genome Biology 20: 257. 10.1186/s13059-019-1891-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367. Flandrois, Jean‐Pierre , Lina Gérard, and Dumitrescu Oana. 2014. “MUBII‐TB‐DB: A Database of Mutations Associated With Antibiotic Resistance in Mycobacterium Tuberculosis .” BMC Bioinformatics 15: 107. 10.1186/1471-2105-15-107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368. Beghini, Francesco , McIver Lauren J., Blanco‐Míguez Aitor, Dubois Leonard, Asnicar Francesco, Maharjan Sagun, Mailyan Ana, et al. 2021. “Integrating Taxonomic, Functional, and Strain‐Level Profiling of Diverse Microbial Communities With bioBakery 3.” eLife 10: e65088. 10.7554/eLife.65088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369. Langille, Morgan G. I. , Zaneveld Jesse, Caporaso J Gregory, McDonald Daniel, Knights Dan, Reyes Joshua A., Clemente Jose C., et al. 2013. “Predictive Functional Profiling of Microbial Communities Using 16S rRNA Marker Gene Sequences.” Nature Biotechnology 31: 814–821. 10.1038/nbt.2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Lu, Yao , Zhou Guangyan, Ewald Jessica, Pang Zhiqiang, Shiri Tanisha, and Xia Jianguo. 2023. “MicrobiomeAnalyst 2.0: Comprehensive Statistical, Functional and Integrative Analysis of Microbiome Data.” Nucleic Acids Research 51: W310–W318. 10.1093/nar/gkad407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Ewels, Philip , Magnusson Måns, Lundin Sverker, and Käller Max. 2016. “MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report.” Bioinformatics 32: 3047–3048. 10.1093/bioinformatics/btw354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372. Chong, Jasmine , Wishart David S., and Xia Jianguo. 2019. “Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis.” Current Protocols 68: e86. 10.1002/cpbi.86 [DOI] [PubMed] [Google Scholar]
- 373. Tian, Suyan , and Wang Chi. 2021. “An Ensemble of the Icluster Method to Analyze Longitudinal lncRNA Expression Data for Psoriasis Patients.” Human Genomics 15: 23. 10.1186/s40246-021-00323-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Rohart, Florian , Gautier Benoît, Singh Amrit, and Lê Cao Kim‐Anh. 2017. “Mixomics: An R Package for ‘Omics Feature Selection and Multiple Data Integration.” PLoS Computational Biology 13: e1005752. 10.1371/journal.pcbi.1005752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375. Argelaguet, Ricard , Velten Britta, Arnol Damien, Dietrich Sascha, Zenz Thorsten, Marioni John C., Buettner Florian, Huber Wolfgang, and Stegle Oliver. 2018. “Multi‐Omics Factor Analysis‐A Framework for Unsupervised Integration of Multi‐Omics Data Sets.” Molecular Systems Biology 14: e8124. 10.15252/msb.20178124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376. Zhou, Guangyan , Pang Zhiqiang, Lu Yao, Ewald Jessica, and Xia Jianguo. 2022. “OmicsNet 2.0: A Web‐Based Platform for Multi‐Omics Integration and Network Visual Analytics.” Nucleic Acids Research 50: W527–W533. 10.1093/nar/gkac376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377. Shannon, Paul , Markiel Andrew, Ozier Owen, Baliga Nitin S., Wang Jonathan T., Ramage Daniel, Amin Nada, Schwikowski Benno, and Ideker Trey. 2003. “Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks.” Genome Research 13: 2498–2504. 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378. Abueg, Linelle Ann L. , Afgan Enis, Allart Olivier, Awan Ahmed H., Bacon Wendi A., Baker Dannon, Bassetti Madeline, et al. 2024. “The Galaxy Platform for Accessible, Reproducible, and Collaborative Data Analyses: 2024 Update.” Nucleic Acids Research 52: W83–W94. 10.1093/nar/gkae410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Gonzalez, Antonio , Navas‐Molina Jose A., Kosciolek Tomasz, McDonald Daniel, Vázquez‐Baeza Yoshiki, Ackermann Gail, DeReus Jeff, et al. 2018. “Qiita: Rapid, Web‐Enabled Microbiome Meta‐Analysis.” Nature Methods 15: 796–798. 10.1038/s41592-018-0141-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Wilke, Andreas , Bischof Jared, Gerlach Wolfgang, Glass Elizabeth, Harrison Travis, Keegan Kevin P., Paczian Tobias, et al. 2016. “The MG‐RAST Metagenomics Database and Portal in 2015.” Nucleic Acids Research 44: D590–D594. 10.1093/nar/gkv1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381. Manghi, Paolo , Blanco‐Míguez Aitor, Manara Serena, NabiNejad Amir, Cumbo Fabio, Beghini Francesco, Armanini Federica, et al. 2023. “MetaPhlAn 4 Profiling of Unknown Species‐Level Genome Bins Improves the Characterization of Diet‐Associated Microbiome Changes in Mice.” Cell Reports 42: 112464. 10.1016/j.celrep.2023.112464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382. Xiao, Liwen , Zhang Fengyi, and Zhao Fangqing. 2022. “Large‐Scale Microbiome Data Integration Enables Robust Biomarker Identification.” Nature Computational Science 2: 307–316. 10.1038/s43588-022-00247-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383. Langfelder, Peter , and Horvath Steve. 2008. “WGCNA: An R Package for Weighted Correlation Network Analysis.” BMC Bioinformatics 9: 559. 10.1186/1471-2105-9-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384. Saenz, Carmen , Nigro Eleonora, Gunalan Vithiagaran, and Arumugam Manimozhiyan. 2022. “MIntO: A Modular and Scalable Pipeline for Microbiome Metagenomic and Metatranscriptomic Data Integration.” Frontiers in Bioinformatics 2: 846922. 10.3389/fbinf.2022.846922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385. Kuleshov, Maxim V. , Jones Matthew R., Rouillard Andrew D., Fernandez Nicolas F., Duan Qiaonan, Wang Zichen, Koplev Simon, et al. 2016. “Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update.” Nucleic Acids Research 44: W90–W97. 10.1093/nar/gkw377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386. Tuncbag, Nurcan , Gosline Sara J. C., Kedaigle Amanda, Soltis Anthony R., Gitter Anthony, and Fraenkel Ernest. 2016. “Network‐Based Interpretation of Diverse High‐Throughput Datasets Through the Omics Integrator Software Package.” PLoS Computational Biology 12: e1004879. 10.1371/journal.pcbi.1004879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Baysoy, Alev , Bai Zhiliang, Satija Rahul, and Fan Rong. 2023. “The Technological Landscape and Applications of Single‐Cell Multi‐Omics.” Nature Reviews Molecular Cell Biology 24: 695–713. 10.1038/s41580-023-00615-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388. Bock, Christoph , Farlik Matthias, and Sheffield Nathan C.. 2016. “Multi‐Omics of Single Cells: Strategies and Applications.” Trends in Biotechnology 34: 605–608. 10.1016/j.tibtech.2016.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389. Stuart, Tim , Butler Andrew, Hoffman Paul, Hafemeister Christoph, Papalexi Efthymia, Mauck William M., Hao Yuhan, et al. 2019. “Comprehensive Integration of Single‐Cell Data.” Cell 177: 1888–1902.e1821. 10.1016/j.cell.2019.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390. Greenacre, Michael , Groenen Patrick J. F., Hastie Trevor, D'Enza Alfonso Iodice, Markos Angelos, and Tuzhilina Elena. 2022. “Principal Component Analysis.” Nature Reviews Methods Primers 2: 100. 10.1038/s43586-022-00184-w [DOI] [Google Scholar]
- 391. Cieslak, Matthew C. , Castelfranco Ann M., Roncalli Vittoria, Lenz Petra H., and Hartline Daniel K.. 2020. “t‐Distributed Stochastic Neighbor Embedding (t‐SNE): A Tool for Eco‐Physiological Transcriptomic Analysis.” Marine Genomics 51: 100723. 10.1016/j.margen.2019.100723 [DOI] [PubMed] [Google Scholar]
- 392. Xu, Yunpei , Li Hong‐Dong, Lin Cui‐Xiang, Zheng Ruiqing, Li Yaohang, Xu Jinhui, and Wang Jianxin. 2023. “CellBRF: A Feature Selection Method for Single‐Cell Clustering Using Cell Balance and Random Forest.” Bioinformatics 39: i368–i376. 10.1093/bioinformatics/btad216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393. Missarova, Alsu , Jain Jaison, Butler Andrew, Ghazanfar Shila, Stuart Tim, Brusko Maigan, Wasserfall Clive, et al. 2021. “geneBasis: An Iterative Approach for Unsupervised Selection of Targeted Gene Panels From scRNA‐Seq.” Genome Biology 22: 333. 10.1186/s13059-021-02548-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394. Senghor, Bruno , Sokhna Cheikh, Ruimy Raymond, and Lagier Jean‐Christophe. 2018. “Gut Microbiota Diversity According to Dietary Habits and Geographical Provenance.” Human Microbiome Journal 7–8: 1–9. 10.1016/j.humic.2018.01.001 [DOI] [Google Scholar]
- 395. Pant, Archana , Das Bhabatosh, and Arimbasseri Gopalakrishnan Aneeshkumar. 2023. “Host Microbiome in Tuberculosis: Disease, Treatment, and Immunity Perspectives.” Frontiers in Microbiology 14: 1236348. 10.3389/fmicb.2023.1236348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396. Arya, Rakesh , Shakya Hemlata, Chaurasia Reetika, Kumar Surendra, Vinetz Joseph M., and Kim Jong Joo. 2024. “Computational Reassessment of RNA‐Seq Data Reveals Key Genes in Active Tuberculosis.” PLoS One 19: e0305582. 10.1371/journal.pone.0305582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397. Verma, Anjali , Bhagchandani Tannu, Rai Ankita, Nikita I., Sardarni Urvinder Kaur, Bhavesh Neel Sarovar, Gulati Sameer, Malik Rupali, and Tandon Ravi. 2024. “Short‐Chain Fatty Acid (SCFA) as a Connecting Link Between Microbiota and Gut‐Lung Axis−A Potential Therapeutic Intervention to Improve Lung Health.” ACS Omega 9: 14648–14671. 10.1021/acsomega.3c05846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398. Zheng, Yuanting , Liu Yaqing, Yang Jingcheng, Dong Lianhua, Zhang Rui, Tian Sha, Yu Ying, et al. 2024. “Multi‐Omics Data Integration Using Ratio‐Based Quantitative Profiling With Quartet Reference Materials.” Nature Biotechnology 42: 1133–1149. 10.1038/s41587-023-01934-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399. Guha, Priyanka , Dutta Siddhartha, Murti Krishna, Charan Jay Karan, Pandey Krishna, Ravichandiran V., and Dhingra Sameer. 2024. “The Integration of Omics: A Promising Approach to Personalized Tuberculosis Treatment.” Medicine in Omics 12: 100033. 10.1016/j.meomic.2024.100033 [DOI] [Google Scholar]
- 400. Liu, Pinyi , Wang Yanbing, Yang Ge, Zhang Qihe, Meng Lingbin, Xin Ying, and Jiang Xin. 2021. “The Role of Short‐Chain Fatty Acids in Intestinal Barrier Function, Inflammation, Oxidative Stress, and Colonic Carcinogenesis.” Pharmacological Research 165: 105420. 10.1016/j.phrs.2021.105420 [DOI] [PubMed] [Google Scholar]
- 401. Sun, Mingming , Wu Wei, Chen Liang, Yang Wenjing, Huang Xiangsheng, Ma Caiyun, Chen Feidi, et al. 2018. “Microbiota‐Derived Short‐Chain Fatty Acids Promote Th1 Cell IL‐10 Production to Maintain Intestinal Homeostasis.” Nature Communications 9: 3555. 10.1038/s41467-018-05901-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402. Dikeocha, Ifeoma Julieth , Al‐Kabsi Abdelkodose Mohammed, Chiu Hsien‐Tai, and Alshawsh Mohammed Abdullah. 2022. “ Faecalibacterium Prausnitzii Ameliorates Colorectal Tumorigenesis and Suppresses Proliferation of HCT116 Colorectal Cancer Cells.” Biomedicines 10: 1128. 10.3390/biomedicines10051128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403. Dong, Chen . 2021. “Cytokine Regulation and Function in T Cells.” Annual Review of Immunology 39: 51–76. 10.1146/annurev-immunol-061020-053702 [DOI] [PubMed] [Google Scholar]
- 404. Noecker, Cecilia , Eng Alexander, Srinivasan Sujatha, Theriot Casey M., Young Vincent B., Jansson Janet K., Fredricks David N., and Borenstein Elhanan. 2016. “Metabolic Model‐Based Integration of Microbiome Taxonomic and Metabolomic Profiles Elucidates Mechanistic Links Between Ecological and Metabolic Variation.” mSystems 1: e00013–e00015. 10.1128/mSystems.00013-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405. Singh, Vineet , Lee GyuDae, Son HyunWoo, Koh Hong, Kim Eun Soo, Unno Tatsuya, and Shin Jae‐Ho. 2022. “Butyrate Producers, “The Sentinel of Gut”: Their Intestinal Significance With and Beyond Butyrate, and Prospective Use as Microbial Therapeutics.” Frontiers in Microbiology 13: 1103836. 10.3389/fmicb.2022.1103836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406. Duncan, Sylvia H. , Barcenilla Adela, Stewart Colin S., Pryde Susan E., and Flint Harry J.. 2002. “Acetate Utilization and Butyryl Coenzyme A (CoA):Acetate‐CoA Transferase in Butyrate‐Producing Bacteria From the Human Large Intestine.” Applied and Environmental Microbiology 68: 5186–5190. 10.1128/AEM.68.10.5186-5190.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407. Joshi, Lavanya , Ponnana Meenakshi, Sivangala Ramya, Chelluri Lakshmi Kiran, Nallari Prathiba, Penmetsa Sitaramaraju, Valluri Vijayalakshmi, and Gaddam Sumanlatha. 2015. “Evaluation of TNF‐ɑ, IL‐10 and IL‐6 Cytokine Production and Their Correlation With Genotype Variants Amongst Tuberculosis Patients and Their Household Contacts.” PLOS One 10: e0137727. 10.1371/journal.pone.0137727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408. Subramanian, Indhupriya , Verma Srikant, Kumar Shiva, Jere Abhay, and Anamika Krishanpal. 2020. “Multi‐Omics Data Integration, Interpretation, and Its Application.” Bioinformatics and Biology Insights 14: 1177932219899051. 10.1177/1177932219899051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409. Liu, Xi , Chen Yuanli, Ouyang Hui, Liu Jian, Luo Xiaoqing, Huang Yayi, Chen Yan, et al. 2021. “Tuberculosis Diagnosis by Metagenomic Next‐Generation Sequencing on Bronchoalveolar Lavage Fluid: A Cross‐Sectional Analysis.” International Journal of Infectious Diseases 104: 50–57. 10.1016/j.ijid.2020.12.063 [DOI] [PubMed] [Google Scholar]
- 410. Wu, Jiayu , Wang Kai, Wang Xuemei, Pang Yanli, and Jiang Changtao. 2021. “The Role of the Gut Microbiome and Its Metabolites in Metabolic Diseases.” Protein & Cell 12: 360–373. 10.1007/s13238-020-00814-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411. Ney, Lisa‐Marie , Wipplinger Maximilian, Grossmann Martha, Engert Nicole, Wegner Valentin D., and Mosig Alexander S.. 2023. “Short Chain Fatty Acids: Key Regulators of the Local and Systemic Immune Response in Inflammatory Diseases and Infections.” Open Biology 13: 230014. 10.1098/rsob.230014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412. Creasy, Heather Huot , Felix Victor, Aluvathingal Jain, Crabtree Jonathan, Ifeonu Olukemi, Matsumura James, McCracken Carrie, et al. 2021. “HMPDACC: A Human Microbiome Project Multi‐Omic Data Resource.” Nucleic Acids Research 49: D734–D742. 10.1093/nar/gkaa996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413. Chen, I‐Min A , Chu Ken, Palaniappan Krishna, Pillay Manoj, Ratner Anna, Huang Jinghua, Huntemann Marcel, et al. 2019. “IMG/M V.5.0: An Integrated Data Management and Comparative Analysis System for Microbial Genomes and Microbiomes.” Nucleic Acids Research 47: D666–D677. 10.1093/nar/gky901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414. Galagan, James E. , Sisk Peter, Stolte Christian, Weiner Brian, Koehrsen Michael, Wymore Farrell, Reddy T. B. K., et al. 2010. “TB Database 2010: Overview and Update.” Tuberculosis 90: 225–235. 10.1016/j.tube.2010.03.010 [DOI] [PubMed] [Google Scholar]
- 415. Kapopoulou, Adamandia , Lew Jocelyne M., and Cole Stewart T.. 2011. “The MycoBrowser Portal: A Comprehensive and Manually Annotated Resource for Mycobacterial Genomes.” Tuberculosis 91: 8–13. 10.1016/j.tube.2010.09.006 [DOI] [PubMed] [Google Scholar]
- 416. Perez‐Riverol, Yasset , Zorin Andrey, Dass Gaurhari, Vu Manh‐Tu, Xu Pan, Glont Mihai, Vizcaíno Juan Antonio, et al. 2019. “Quantifying the Impact of Public Omics Data.” Nature Communications 10: 3512. 10.1038/s41467-019-11461-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417. Pang, Zhiqiang , Chong Jasmine, Zhou Guangyan, de Lima Morais David Anderson, Chang Le, Barrette Michel, Gauthier Carol, et al. 2021. “MetaboAnalyst 5.0: Narrowing the Gap Between Raw Spectra and Functional Insights.” Nucleic Acids Research 49: W388–W396. 10.1093/nar/gkab382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418. Overbeek, Ross , Olson Robert, Pusch Gordon D., Olsen Gary J., Davis James J., Disz Terry, Edwards Robert A., et al. 2014. “The SEED and the Rapid Annotation of Microbial Genomes Using Subsystems Technology (RAST).” Nucleic Acids Research 42: D206–D214. 10.1093/nar/gkt1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419. Heusden, Peter , van Ziphozakhe, Mashologu Thoba, Lose Robin, Warren Alan, and Christoffels. 2022. “The COMBAT‐TB Workbench: Making Powerful Mycobacterium Tuberculosis Bioinformatics Accessible.” mSphere 7: e0099121. 10.1128/msphere.00991-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420. Carithers, Latarsha J. , and Moore Helen M.. 2015. “The Genotype‐Tissue Expression (GTEx) Project.” Biopreservation and Biobanking 13: 307–308. 10.1089/bio.2015.29031.hmm [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421. Lamb, Justin . 2007. “The Connectivity Map: A New Tool for Biomedical Research.” Nature Reviews Cancer 7: 54–60. 10.1038/nrc2044 [DOI] [PubMed] [Google Scholar]
- 422. Reddy, T. B. K. , Riley R., Wymore F., Montgomery P., DeCaprio D., Engels R., Gellesch M., et al. 2009. “TB Database: An Integrated Platform for Tuberculosis Research.” Nucleic Acids Research 37: D499–D508. 10.1093/nar/gkn652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423. Metri, Rahul , Hariharaputran Sridhar, Ramakrishnan Gayatri, Anand Praveen, Raghavender Upadhyayula S., Ochoa‐Montaño Bernardo, Higueruelo Alicia P., et al. 2015. “SInCRe‐Structural Interactome Computational Resource for Mycobacterium Tuberculosis .” Database 2015: bav060. 10.1093/database/bav060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424. Perez‐Riverol, Yasset , Bai Mingze, da Veiga Leprevost Felipe, Squizzato Silvano, Park Young Mi, Haug Kenneth, Carroll Adam J., et al. 2017. “Discovering and Linking Public Omics Data Sets Using the Omics Discovery Index.” Nature Biotechnology 35: 406–409. 10.1038/nbt.3790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425. Xia, Jianguo , and Wishart David S.. 2016. “Using MetaboAnalyst 3.0 for Comprehensive Metabolomics Data Analysis.” Current Protocols 55: 14.10.11–14.10.91. 10.1002/cpbi.11 [DOI] [PubMed] [Google Scholar]
- 426. Sequeira, João C. , Pereira Vítor, Alves M. Madalena, Pereira M. Alcina, Rocha Miguel, and Salvador Andreia F.. 2024. “MOSCA 2.0: A Bioinformatics Framework for Metagenomics, Metatranscriptomics and Metaproteomics Data Analysis and Visualization.” Molecular Ecology Resources 24: e13996. 10.1111/1755-0998.13996 [DOI] [PubMed] [Google Scholar]
- 427. Muñoz‐Benavent, Maria , Hartkopf Felix, Van Den Bossche Tim, Piro Vitor C., García‐Ferris Carlos, Latorre Amparo, Renard Bernhard Y., and Muth Thilo. 2020. “gNOMO: A Multi‐Omics Pipeline for Integrated Host and Microbiome Analysis of Non‐Model Organisms.” NAR Genomics and Bioinformatics 2: lqaa058. 10.1093/nargab/lqaa083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428. Davis, James J. , Wattam Alice R., Aziz Ramy K., Brettin Thomas, Butler Ralph, Butler Rory M., and Chlenski Philippe, et al. 2020. “The Patric Bioinformatics Resource Center: Expanding Data and Analysis Capabilities.” Nucleic Acids Research 48: D606–D612. 10.1093/nar/gkz943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429. Sinkov, Viacheslav , Ogarkov Oleg, Mokrousov Igor, Bukin Yuri, Zhdanova Svetlana, and Heysell Scott K.. 2018. “New Epidemic Cluster of Pre‐Extensively Drug Resistant Isolates of Mycobacterium Tuberculosis Ural Family Emerging in Eastern Europe.” BMC Genomics 19: 762. 10.1186/s12864-018-5162-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430. Zoppi, Johanna , Guillaume Jean‐François, Neunlist Michel, and Chaffron Samuel. 2021. “MiBiOmics: An Interactive Web Application for Multi‐Omics Data Exploration and Integration.” BMC Bioinformatics 22: 6. 10.1186/s12859-020-03921-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431. Schuster, Viktoria , Dann Emma, Krogh Anders, and Teichmann Sarah A.. 2024. “MultiDGD: A Versatile Deep Generative Model for Multi‐Omics Data.” Nature Communications 15: 10031. 10.1038/s41467-024-53340-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432. Dhanda, Sandeep Kumar , Vir Pooja, Singla Deepak, Gupta Sudheer, Kumar Shailesh, and Raghava Gajendra P. S.. 2016. “A Web‐Based Platform for Designing Vaccines Against Existing and Emerging Strains of Mycobacterium Tuberculosis .” PLOS One 11: e0153771. 10.1371/journal.pone.0153771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433. Sandgren, Andreas , Strong Michael, Muthukrishnan Preetika, Weiner Brian K., Church George M., and Murray Megan B.. 2009. “Tuberculosis Drug Resistance Mutation Database.” PLOS Medicine 6: e2. 10.1371/journal.pmed.1000002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434. Cansdale, Annabel , and Chong James P. J.. 2024. “MAGqual: A Stand‐Alone Pipeline to Assess the Quality of Metagenome‐Assembled Genomes.” Microbiome 12: 226. 10.1186/s40168-024-01949-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435. Mulder, Nicola , Schwartz Russell, Brazas Michelle D., Brooksbank Cath, Gaeta Bruno, Morgan Sarah L., Pauley Mark A., et al. 2018. “The Development and Application of Bioinformatics Core Competencies to Improve Bioinformatics Training and Education.” PLoS Computational Biology 14: e1005772. 10.1371/journal.pcbi.1005772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436. Attwood, Teresa K. , Blackford Sarah, Brazas Michelle D., Davies Angela, and Schneider Maria Victoria. 2019. “A Global Perspective on Evolving Bioinformatics and Data Science Training Needs.” Briefings in Bioinformatics 20: 398–404. 10.1093/bib/bbx100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437. Searls, David B . 2012. “An Online Bioinformatics Curriculum.” PLoS Computational Biology 8: e1002632. 10.1371/journal.pcbi.1002632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438. Cantelli, Gaia , Cochrane Guy, Brooksbank Cath, McDonagh Ellen, Flicek Paul, McEntyre Johanna, Birney Ewan, and Apweiler Rolf. 2021. “The European Bioinformatics Institute: Empowering Cooperation in Response to a Global Health Crisis.” Nucleic Acids Research 49: D29–D37. 10.1093/nar/gkaa1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439. Stajich, J. E . 2006. “Open Source Tools and Toolkits for Bioinformatics: Significance, and Where Are We?” Briefings in Bioinformatics 7: 287–296. 10.1093/bib/bbl026 [DOI] [PubMed] [Google Scholar]
- 440. Dubilier, Nicole , McFall‐Ngai Margaret, and Zhao Liping. 2015. “Microbiology: Create a Global Microbiome Effort.” Nature 526: 631–634. 10.1038/526631a [DOI] [PubMed] [Google Scholar]
- 441. Li, Jing‐Woei , Schmieder Robert, Ward R. Matthew, Delenick Joann, Olivares Eric C., and Mittelman David. 2012. “SEQanswers: An Open Access Community for Collaboratively Decoding Genomes.” Bioinformatics 28: 1272–1273. 10.1093/bioinformatics/bts128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442. Arkin, Adam P. , Cottingham Robert W., Henry Christopher S., Harris Nomi L., Stevens Rick L., Maslov Sergei, Dehal Paramvir, et al. 2018. “KBase: The United States Department of Energy Systems Biology Knowledgebase.” Nature Biotechnology 36: 566–569. 10.1038/nbt.4163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443. Ye, Shiqing , Wang Liang, Li Shengkai, Ding Qingyong, Wang Yu, Wan Xinxin, Ji Xiaoyun, Lou Yongliang, and Li Xiang. 2022. “The Correlation Between Dysfunctional Intestinal Flora and Pathology Feature of Patients With Pulmonary Tuberculosis.” Frontiers in Cellular and Infection Microbiology 12: 1090889. 10.3389/fcimb.2022.1090889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444. Zhang, Meng , Shen Li, Zhou Xia, and Chen Huidong. 2022. “The Microbiota of Human Lung of Pulmonary Tuberculosis and the Alteration Caused by Anti‐Tuberculosis Drugs.” Current Microbiology 79: 321. 10.1007/s00284-022-03019-9 [DOI] [PubMed] [Google Scholar]
- 445. Fontana, Andrea , Panebianco Concetta, Picchianti‐Diamanti Andrea, Laganà Bruno, Cavalieri Duccio, Potenza Adele, Pracella Riccardo, et al. 2019. “Gut Microbiota Profiles Differ Among Individuals Depending on Their Region of Origin: an Italian Pilot Study.” International Journal of Environmental Research and Public Health 16: 4065. 10.3390/ijerph16214065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446. Lawal, Samuel Adefisoye , Voisin Athalia, Olof Hana, Bording‐Jorgensen Michael, and Armstrong Heather. 2023. “Diversity of the Microbiota Communities Found in the Various Regions of the Intestinal Tract in Healthy Individuals and Inflammatory Bowel Diseases.” Frontiers in Immunology 14: 1242242. 10.3389/fimmu.2023.1242242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447. Moon, Ji‐Hoi , Roh Dae‐Hyun, Kwack Kyu Hwan, and Lee Jae‐Hyung. 2023. “Bacterial Single‐Cell Transcriptomics: Recent Technical Advances and Future Applications in Dentistry.” Japanese Dental Science Review 59: 253–262. 10.1016/j.jdsr.2023.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448. McShane, Helen , and Williams Ann. 2014. “A Review of Preclinical Animal Models Utilised for TB Vaccine Evaluation in the Context of Recent Human Efficacy Data.” Tuberculosis 94: 105–110. 10.1016/j.tube.2013.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449. Ivanov, Ivaylo I. , Tuganbaev Timur, Skelly Ashwin N., and Honda Kenya. 2022. “T Cell Responses to the Microbiota.” Annual Review of Immunology 40: 559–587. 10.1146/annurev-immunol-101320-011829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450. Yu, You , Wang Weihong, and Zhang Faming. 2023. “The Next Generation Fecal Microbiota Transplantation: To Transplant Bacteria or Virome.” Advanced Science 10: e2301097. 10.1002/advs.202301097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451. Monette, Anne , Aguilar‐Mahecha Adriana, Altinmakas Emre, Angelos Mathew G., Assad Nima, Batist Gerald, Bommareddy Praveen K., et al. 2025. “The Society for Immunotherapy of Cancer Perspective on Tissue‐Based Technologies for Immuno‐Oncology Biomarker Discovery and Application.” Clinical Cancer Research 31: 439–456. 10.1158/1078-0432.CCR-24-2469 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data were generated or analyzed in this review. Supplementary materials (graphical abstract, slides, videos, Chinese translated version, and update materials) may be found in the online DOI or iMeta Science http://www.imeta.science/.