

Abstract
The unique three-dimensional structure of both monomeric and oligomeric proteins is encoded in their sequence. The biological functions of proteins are dependent on their tertiary and quaternary structures, and hence it is important to understand the determinants of quaternary association in proteins. Although a large number of investigations have been carried out in this direction, the underlying principles of protein oligomerization are yet to be completely understood. Recently, new insights into this problem have been gained from the analysis of structure graphs of proteins belonging to the legume lectin family. The legume lectins are an interesting family of proteins with very similar tertiary structures but varied quaternary structures. Hence they have become a very good model with which to analyse the role of primary structures in determining the modes of quaternary association. The present review summarizes the results of a legume lectin study as well as those obtained from a similar analysis carried out here on the animal lectins, namely galectins, pentraxins, calnexin, calreticulin and rhesus rotavirus Vp4 sialic-acid-binding domain. The lectin structure graphs have been used to obtain clusters of non-covalently interacting amino acid residues at the intersubunit interfaces. The present study, performed along with traditional sequence alignment methods, has provided the signature sequence motifs for different kinds of quaternary association seen in lectins. Furthermore, the network representation of the lectin oligomers has enabled us to detect the residues which make extensive interactions (‘hubs’) across the oligomeric interfaces that can be targetted for interface-destabilizing mutations. The present review also provides an overview of the methodology involved in representing oligomeric protein structures as connected networks of amino acid residues. Further, it illustrates the potential of such a representation in elucidating the structural determinants of protein–protein association in general and will be of significance to protein chemists and structural biologists.
Keywords: galectin, graph-spectral method, interface amino acid clusters and hubs, legume lectin, oligomeric-protein structure graph, pentraxin
Abbreviations: ConA, concanavalin A; CRP, C-reactive protein; DB58 and and DBL, Dolichos biflorus (horse gram) stem-and-leaf and seed lectins respectively; EcorL, Erythrina corallodendron (coral tree) lectin; GS1 and GS4, Griffonia simplicifolia (griffonia) lectins 1 and 4; PNA, peanut (Arachis hypogaea) agglutinin; SAP, serum amyloid P component
INTRODUCTION
Protein–protein association and oligomerization have been found to be extremely important for the functioning of many proteins found in Nature. Understanding the factors determining the nature and states of oligomerization or quaternary associations in proteins has been the aim of numerous studies. Various investigations have been carried out to dissect the structural features of protein interfaces. For instance, the nature and type of amino acid interactions constituting the protein interfaces [1–5], accessible surface area calculations [3–5], conservation of amino acid residues at interfaces [6,7], the geometry and nature of surface patches in monomers constituting the oligomers [3,4,8], conformational entropies of side chains at protein interfaces [9], docking of one monomer on to the other based on empirical methods [10,11], computational design and prediction of protein–protein interactions [12–14], and many more such investigations [5] have all been carried out in the past to understand and elucidate the principles underlying protein associations. Though many of these investigations have provided insights into the factors responsible for protein association, the understanding of the role of both sequence and structure in protein oligomerization is far from complete. The proteins belonging to the lectin family are an excellent model with which to investigate this problem, since they have very similar tertiary structure characterized by the ‘jelly-roll’ fold, and yet they have very different modes of quaternary associations. They are known to exist as monomers, dimers and higher oligomers where the dimers and the higher oligomers comprise many different types of protein interfaces and topologically different quaternary associations. The present review provides a detailed perspective on the proteins of the lectin family as an example with which to understand the determinants of protein quaternary association.
THE LECTIN FAMILY
Lectins are carbohydrate-binding proteins that have varied applications in the field of biochemical and biomedical research [15,16]. They are found in almost all organisms, ranging from viruses to vertebrates, and have been implicated in a variety of cellular functions such as cell–cell interactions, cell-surface recognition, the innate immune system etc. [15,16]. The lectin superfamily as classified by SCOP (structural classification of proteins [17]) comprises 15 families that include the legume lectins, β-glucanases, endoglucanases, sialidases, galectins, pentraxins and calnexin/calreticulin, among others. All of them belong to the all-β class and have the ConA (concanavalin-A-like) jelly-roll fold that can be seen in Figure 1. The jelly-roll motif consists of three sets of anti-parallel β-sheets, as can also be seen from the legume lectins shown in Figure 1. There is a six-stranded flat ‘back’ sheet, a curved seven-stranded ‘front’ sheet and a short five-member sheet at the ‘top’ of the molecule. The sheets are connected by several loops of various lengths [18–19].
Figure 1. Different types of dimeric interfaces in plant and animal lectins.

The three-dimensional structure of the dimeric interfaces of the legume lectins (II, X4, X3, X1 and X2), galectins (galectin-1, galectin-2 and galectin-7) and pentraxins (SAP and CRP) are shown in cartoon representation with the monomers differentiated using different colours. The jelly-roll fold that characterizes the tertiary structures of these lectin can be clearly seen in the Figure. The monomeric lectins, such as arcelin-5, galectin-3, Charcot–Leyden protein, calnexin and calreticulin, which do have the jelly-roll tertiary structure, are not shown.
The legume lectins are classified under one family since they have high sequence similarities, very similar tertiary structure and biophysical properties. Further, most of these legume lectins exist as oligomers in nature, and all of them require metal ions for their carbohydrate-binding activities [15,16]. The metal-binding and carbohydrate-binding sites have been identified in various legume lectins, and they are found to be structurally overlapping [15,16]. However, these lectins do differ widely in their carbohydrate specificities and in their quaternary associations [18,19]. The other lectins in this superfamily, such as the galectins, pentraxins, calnexin and calreticulin, have very much less sequence similarity with the legume lectins. However, they do share the same jelly-roll tertiary structure and show different quaternary associations. The reasons for the varied quaternary association in lectins were investigated previously using traditional methods. For example, preliminary analysis of the legume lectin oligomers, including the determination of chemical characteristics of the different legume lectin interfaces [20], correlation with the phylogenetic trees [21], identification of conserved residues from multiple sequence alignment [22] or inspection of pairwise interactions at the intersubunit interface(s) [22], have been carried out. Though these analyses were able to elucidate some of the factors behind the lectin quaternary structures, the exact residues, sequence motifs or structural features responsible for the quaternary association were not deduced from such studies, since the overall similarity in both sequence and tertiary structure is very high in these proteins. Hence, one requires newer methods of analysis to understand protein quaternary association and the choice of the method should be such that it takes a global view of the amino acid interactions at the interfaces and not at the pairwise level. A recent study of the amino acid clusters at the protein interfaces of legume lectin structures using a graph-spectral method has aided significantly in obtaining signature sequences required for a specific type of interface in legume lectins [23]. The present review consolidates these features by detailing similar investigations on several other lectins. Furthermore, all these lectins have been investigated from a network perspective, which takes a global view of the oligomeric structures. The characterization of the amino acid clusters and hubs at the protein interfaces, as elucidated by the network representation of the lectin oligomers, is presented here. The results pertaining to the legume lectin family are taken from [23], whereas the analysis of other lectins, as well as the network perspective of lectins, are previously unpublished results presented here for the first time. Since the concept of protein structure network is relatively new, an overview of the network based methodology and its significance in biology with special emphasis to the protein structure networks is discussed below.
THE NETWORK/GRAPH PERSPECTIVE IN BIOLOGY
In the past decade, the network model has been applied in different aspects of biology to obtain useful insights into protein structure, folding, stability, regulation and evolution, and in genome analysis and comparison [24]. First, a network representation requires consideration of the system under study as a set of nodes (points) and edges (links) that constitute the graph representing the system. For example, the complete protein interaction network, or the regulatory network, or the metabolic pathway of an organism, say yeast, can be represented as a network graph. In these networks, each protein, ligand or metabolite can be the nodes in the graph and the interaction they make with the other nodes in the graph or the way they biologically regulate the other nodes in the graph can become the edges in these networks. Such networks have recently been constructed and analysed for yeast, yielding valuable insights into the organization of biological networks in genomes [24,25]. Interestingly, all these biological networks are found to have similar overall graphical properties and hence seem to follow some common rules. They belong to a class of real-world networks termed ‘scale-free networks’, which show a power-law degree distribution of nodes and are characterized by the presence of a small number of highly connected nodes called ‘hubs’, which yield robustness to these networks [24]. Similarly, the protein domain network has also been constructed by connecting the domains based on structural similarity score, which has been analysed from the domain organization and the evolutionary perspective [26]. An interesting concept relevant to the present review is to consider each protein structure as a network of non-covalent interactions between amino acid residues, and the efforts made in this direction are summarized below.
Networks in protein structures
Studies on protein structure networks have been carried out where the protein structure itself is considered as a network of atoms, amino acids or secondary structural elements according to the requirement, and the edges are constructed based on the interaction between these structural elements. A summary of such investigations is given in Table 1. The studies include the use of graph theory in protein structure comparison [27], where the protein structures to be compared are represented as graphs using their secondary structures as nodes and the interactions between them as edges. The graphs thus generated are compared using standard graph theoretical techniques, such as graph isomorphism, to obtain insights into the structural similarities of the proteins involved (Table 1). The graph representation of protein structures have also been used to understand protein structure and folding at different levels [28–33], dynamics [34,35], comparative modelling [36,37], for the identification of structural domains in proteins [38] and in identifying modular clusters in protein interfaces [39] as given in Table 1. In some of these studies, the amino acid residues in the protein structures or their Cα atoms have been considered as the nodes of the graph. In some others, the secondary structure elements or all the atoms in the protein structure are considered as independent nodes. However, edge-forming criteria vary according to the aim of the study and can vary from sequential proximity to spatial interactions between the nodes.
Table 1. Summary of protein structure networks.
| Group and reference(s) | Network | Analysis |
|---|---|---|
| Grindley et al. [27] | Protein structure graphs based on secondary-structural elements | Subgraph isomorphism for structure comparison |
| Dokholyan et al. [33] | Protein structure graphs based on Cα atoms | Network properties and hubs relating to protein folding using graphs of transition states and intermediates |
| Vendruscolo et al. [31,32] | Protein structure graphs based on Cα atoms | Network properties and hubs relating to protein folding using graphs of transition states and intermediates |
| Atilgan et al. [29] | Protein structure graphs based on Cα atoms | Shortest path length and clustering coefficients, network properties |
| Greene and Higman [30] | Protein structure graphs based on atomic details | Network properties, scale-free nature of protein structure graphs |
| Brinda and Vishveshwara (unpublished work) | Residue-based protein structure graphs | Network properties, scale-free nature, role of hubs in protein structures |
| Vishveshwara and co-workers [40–45] | Residue-based protein structure graphs | Graph-spectral parameters for detection of amino acid clusters at protein core, protein/protein and protein/DNA interfaces |
| Sistla et al. [38] | Residue-based protein structure graphs | Identification of structural domains in proteins structures from graph spectra |
| Reichman et al. [39] | Residue-based protein structure graphs | Modular clusters at protein/protein interfaces |
| Przytycka et al. [28] | Protein structure graphs based on secondary-structural elements | Rules of protein folding in β-proteins by basic graph manipulations |
| Bahar et al. [34,35] | Protein structure graphs based on Cα atoms | Graph-spectral parameters for elucidating protein dynamics using the Gaussian network model (GNM) and the anisotropic network model (ANM) |
| Samudrala and Moult [36,37] | Protein graphs based on steriacally allowed conformations of protein side chains (rotamers) | Clique-detecting algorithm used for modelling the side chains in proteins with unknown structures |
Another set of analyses that have aided our understanding of protein structure and stability include the identification and analysis of clusters of amino acid residues in protein structures from a protein structure network perspective, using graph-spectral methods [40] (Table 1). Here, the amino acid residues are the nodes in the graph and the edges are determined on the basis of the strength of the non-covalent interactions between them. The spectral parameters of such a graph give the cluster-forming residues in the protein structure [40,41]. This cluster information can be used to identify active-site clusters, folding clusters or intersubunit interface clusters. The main advantage of this method is that it requires a single numeric computation and also involves the global topology of the protein, because the complete protein structure is represented in the form of a connected network [41]. This kind of graph representation has yielded very useful results regarding protein stability [42], protein–protein interactions [43] and protein–DNA interactions [44]. The interface analysis of a set of functional homodimers using this method gave the amino acid clusters and their cluster centres at the interfaces, which aided the identification of interface ‘hot spots’ and also provided a method to predict the interactive surfaces on monomers [43]. It has also helped in the identification of residues important in the formation of the α–α dimer in RNA polymerase, where a single mutation at the α/α interface predicted using this method, led to the de-activation of the enzyme due to the destabilization of the α–α dimer [45]. A similar graph-spectral analysis of multidomain proteins has yielded an elegant and easy method to identify structural domains in proteins and also the residues forming the interface between the domains [38]. Recently, Reichmann et al. [39] used a residue-based interaction graph of proteins to identify modular clusters at protein/protein interfaces aiding our understanding of the architecture of intersubunit interfaces. Thus the concept of representing protein structures as networks and the identification of amino acid clusters in protein structures have, in many ways, enhanced our understanding of protein structures, folding, stability and interactions and hence has the potential to address various other aspects related to protein structural biology. In the following sections, we present the necessary description of this method and its application to the analysis of various kinds of quaternary associations seen in legume lectins, galectins and pentraxins.
METHODOLOGY
Recently, the concept of using protein structure graphs to determine amino acid clusters at protein interfaces has been applied to a set of legume lectin oligomers and the analysis yielded the sequence motifs characteristic of each type of quaternary association seen in them [23]. We have further extended this study to other proteins exhibiting the legume lectin fold, ranging from plants to vertebrates. These analyses underscore the power of such an algorithm in dissecting protein/protein interfaces across the diverse members of this highly complex family of proteins with strikingly disparate modes of oligomerization. The algorithm consists of representing protein structures as graphs comprising of a set of nodes and edges, where the amino acid residues are nodes and the strength of the non-covalent interactions between them determines the edges as explained below [40]. Such a graph can then be analysed in various ways to obtain information regarding clusters of amino acid residues and highly connected amino acid residues (known as hubs) involved in these protein structure networks as explained below.
Representation of protein graphs
The atomic co-ordinates of the protein structures were obtained from the Protein Data Bank [46]. Each amino acid in the protein structure is represented as a node and the non-covalent interactions between their side-chains are evaluated for edge-formation [40], as follows:
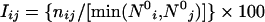 |
(1) |
where nij is the number of distinct side-chain atoms pairs of i and j coming within a distance of 0.45 nm (4.5 Å), evaluated from the crystal structures. N0i and N0j are the normalization values for residue type i and j, which were evaluated previously from a non-redundant set of proteins [40] and are given in Table 2. It can be noted from Table 2 that these normalization values correlate well with the size of the residues.
Table 2. Normalization values of amino acids.
For further details, see the text.
| Amino acid | Normalization value |
|---|---|
| Ala | 55.76 |
| Arg | 93.79 |
| Asn | 73.41 |
| Asp | 75.15 |
| Cys | 54.95 |
| Gln | 78.13 |
| Glu | 78.83 |
| Gly | 47.31 |
| His | 83.74 |
| Ile | 67.95 |
| Leu | 72.25 |
| Lys | 69.61 |
| Met | 69.26 |
| Phe | 93.31 |
| Pro | 51.33 |
| Ser | 61.39 |
| Thr | 63.71 |
| Trp | 106.70 |
| Tyr | 100.72 |
| Val | 62.37 |
A cut-off value of interaction, Imin, is then considered, and any ij pair that has Iij greater than Imin is connected in the graph. For example, when an Imin of 6% is used, all the connected residues in the graph interact with a value more than 6%. Figure 2(a) shows an example of high interaction (8%) between two aromatic residues. The variable parameter in the construction of a protein structure graph is the interaction cut-off, Imin, which can be varied rationally to obtain interesting results, as elucidated below.
Figure 2. Interaction strength and hubs.

(a) Two aromatic residues (shown in ball-and-stick representation) interacting with high value of interaction strength (Iij=8%). The non-covalent atom–atom contacts occuring within a distance of 0.45 nm (4.5 Å) are indicated by thin lines. (b) Hubs: a phenylalanine residue (shown in van der Waals representation) interacting with seven other residues (shown in ball-and-stide representation), thus acting as a hub in the protein structure.
Cluster analysis
The protein structure graph obtained as explained above can be represented in the form of a matrix called the ‘Laplacian matrix’, which has the connectivity information among the residues [40]. This matrix is an N×N matrix, where N is the number of residues in the protein structure. From the matrix we obtain the spectra of the graph comprising eigenvalues and vector components, which provide information regarding the residues forming clusters in the protein structures and those that form the centres of these clusters [40,41]. The clusters can be obtained at different interaction cut-offs (Imin), which would yield clusters comprising residues interacting at various strengths. A higher Imin indicates stronger interaction among the residues forming the cluster, whereas a lower Imin indicates weaker interaction among the same. The typical working cut-off that is used for cluster analysis varies from 4 to 12% [40,41]. The clusters obtained using this clustering method can be further combined with other traditional multiple sequence-alignment methods to obtain information regarding conserved clusters [43]. Although these conserved clusters are a set of residues that are far apart in sequence, they interact in the three-dimensional structure of the protein and are also conserved across different species. The analysis of the conserved clusters at the interfaces of some plant and animal lectin oligomers has been carried out and are discussed in detail below.
The graph-spectral method has the distinct advantage over traditional clustering methods in dealing with weighted graphs, in identifying the cluster centres and in detecting subclusters in a connected graph [41]. Hence this method has been useful in protein structure analysis where clusters of biological significance, such as active-site clusters, hydrophobic-core-forming clusters, clusters imparting thermal stability to proteins, clusters at the protein/protein and protein/DNA interface [40–45] have been identified. This has aided in the understanding of the role of noncovalent interactions in the folding, stability and interaction of protein structures.
Hubs
Hubs are defined as highly connected nodes in a graph. In a protein structure graph, those nodes with more than four connections (links, edges), are termed the hubs. The presence of hubs is a characteristic feature of many real-world networks, where it has been found that there are a limited number of nodes acting as hubs in these networks, which help in reducing the distance between any two nodes in the network and thus form highly connected, compact networks [24]. It has also been found that these hubs provide robustness to the networks, because random attacks on the non-hubs do not affect the network organization or its stability, whereas targeted attacks on the few hubs present can cause severe damage to the network [24]. Applying the same principle to the protein structure networks, one would expect the hubs (residues) in these residue-based protein structure networks to play an important role in stabilizing the folded structure of the protein, and hence a targeted mutation of the hub residue may destabilize the protein structure. An earlier analysis of hubs in protein structures showed that the hubs identified at different Imin values correlated with experimentally available thermodynamic and kinetic parameters (K. V. Brinda and S. Vishveshwara, unpublished work) and also showed a preference for phenylalanine, tyrosine, tryptophan and arginine as hubs at higher Imin values, whereas leucine and isoleucine were preferred at lower Imin values (K. V. Brinda and S. Vishveshwara, unpublished work).
As explained above, the evaluation of interaction between two residues in a protein structure involves the normalization values of both the residue types. However, for the identification of hubs in a protein structure, it would be accurate to use the normalization value of the hub-forming residue alone. Hence, the interaction equation given in eqn (1) reduces to the following for hub identification:
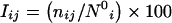 |
(2) |
where, Iij, nij and N0i are the same as in eqn (1), with i being the residue whose hub character is being evaluated. An example of a hub-forming residue in the residue-based protein structure graph is shown in Figure 2(b), where a single phenylalanine residue interacts with many other residues. The concept of hubs is applied to some of the lectin oligomers and is presented in the subsection below entitled ‘Interface hubs’.
Size of the largest cluster
The size of the largest cluster in a network is one of the important parameters that are generally used to understand the nature and properties of the network [24,26]. In the case of the protein structure graphs described above, most of the residues in the protein exist as a part of the largest cluster at lower Imin and the size of the largest cluster decreases with increasing Imin. A characteristic profile of the protein structure graphs is observed when the size of the largest cluster is plotted as a function of Imin. The analysis of a non-redundant set of monomeric proteins showed that a plot of the size of the largest cluster versus Imin exhibits a sigmoidal profile with a transition around Imin=4% in proteins of all sizes and folds, indicating a universal behaviour in protein structures (K. V. Brinda and S. Vishveshwara, unpublished work). This transition in the size of the largest cluster around Imin=4% is found to occur due to the loss of numerous contacts made mainly by the hydrophobic residues such as leucine and isoleucine. In physical terms this change refers to a structural transition that occurs when one large connected cluster (as seen at Imin=0%) splits into smaller distinct clusters (as seen at Imin greater than 4%). The analysis of the size of the largest cluster in the lectin oligomers is also presented in the subsection ‘Size of the largest cluster as a function of interaction cut-off (Imin)’ below.
INSIGHTS INTO LEGUME LECTIN QUATERNARY ASSOCIATION
The legume lectins are known to have varied types of quaternary associations in spite of very similar tertiary structures. What precisely determines their mode of quaternary association remained unresolved until the protein-structure-graph approach was used [23]. The key results of the analysis are presented here.
Classification of legume lectin interfaces and quaternary structures
The legume lectins can be structurally classified into nine types on the basis of their overall quaternary structure, and these nine quaternary structure types consist of seven known dimeric interface types. The different kinds of dimeric interfaces seen in legume lectin oligomers include types II (canonical), X1 [DB58 (Dolichos biflorus stem-and-leaf lectin)-type], X2 (non-canonical interface of ConA), X3 [EcorL (Erythrina corallodendron lectin)-type, handshake], X4 [GS4 (Griffonia simplicifolia lectin 4)-type, back-to-back] and the unusual interfaces of PNA [peanut (Arachis hypogaea) agglutinin] and GS1 (G. simplicifolia lectin 1) (Figure 1). The higher oligomers, mainly tetramers, are generally dimers of dimers and hence have combinations of these dimeric interfaces. The nine different kinds of legume lectin quaternary structures made up of these seven dimeric interface types comprise of canonical (II dimer), EcorL-type (X3 dimer), GS4-type (X4 dimer), DB58-type (X1 dimer), DBL (D. biflorus seed lectin)-type (II+X1 tetramer), ConA-type (II+X2 tetramer), the open quaternary structure of PNA (II+X4+unusual tetramer), GS1-type (X4+unusual tetramer) and arcelin-5-type (monomer) [arcelin-5 is a lectin-like defence protein from Phaseolus vulgaris (French bean)]. As can be seen, some of the interface types are found to be present in the dimeric as well as the tetrameric legume lectins (like II, X1 and X4), whereas some of them are seen exclusively in dimeric legume lectins (like X3) and some are seen exclusively in the tetramers (like X2 and the unusual interfaces of PNA and GS1). The legume lectins with known structures are thus classified into different types of quaternary structures, and the higher oligomers are separated into pairs of dimers so as to classify their interface types.
Consensus signature sequence motifs
The method for obtaining the signature sequences determining each of the legume lectin interface types actually involves a combination of the cluster-detecting algorithm and the traditional sequence-alignment methods. The amino acid clusters in the legume lectin structures are first identified using the graph-spectral method explained above. The interface clusters in these oligomers are then identified by the fact that these clusters comprise residues from both the monomers forming the interface. The interaction cut-off (Imin) used for cluster identification has been optimized for each lectin so as to obtain distinct interface clusters differentiated from the bulk of the protein [23]. These interface cluster-forming residues are then mapped on to the multiple sequence alignments (obtained using ClustalW; [48]) of all those legume lectins that belong to a particular interface type. Those residues that are conserved in the multiple sequence alignments and are also present in the interface clusters of all the legume lectins belonging to a particular interface type constitute the consensus signature sequence for each interface type. The consensus patterns thus obtained for five of the prominent legume lectin interface types, namely the type II, X1, X2, X3 and X4 are given in Figure 3. Further, this algorithm also works to an extent even when there is only one known structure of a particular interface type. In such a case the mapping on to multiple sequence alignments cannot be carried out, since there is only one known example. However, the information regarding the cluster-forming residues at the interface can still be obtained to give some idea regarding the residues required for the interface formation, though it might not be conclusive. This was carried out in the two unusual interfaces of PNA and GS1 where there are only single structures available. The unusual interface of GS1 is characterized by residues W10, T26, G28, Q31, T35, F75, Y226 and L228 and that of PNA is characterized by residues L27, Q33, S28, V160, R221, N31, E72, K74 and G158 (for brevity the one-letter amino acid notation is used). The residues present in the interface clusters of the legume lectin interface types II, X1, X2, X3, X4 and the unusual interfaces of PNA and GS1 are shown in Figure 4. It is clear from the Figure 4 that the different types have different clustering patterns, which further lead to their specific consensus signature motif. The analysis thus provides the signature sequence motifs for each legume lectin interface type, and the higher oligomers with multiple interfaces were found to contain the signatures of all the interface types that they comprise.
Figure 3. Multiple sequence alignments of different legume lectins with galectins, pentraxins, calnexin, calreticulin and Vp4 sialic-acid-binding domain.

Only a few representatives of each type, and the sequence fragments involving the signature motifs, are shown. Types II, X1, X2, X3 and X4 interface types of the legume lectins are shown. The first two sequences in each type belong to the characteristic legume lectins of the particular type. The residues forming the signature sequence motifs of each legume lectin interface type are highlighted in bold and underlined in the first two legume lectin sequences of each interface type. The other lectins included in the alignments are, galectin-1 (1a78), congerin [conger-eel (Conger conger) galectin; 1c1f], galectin-2 (1uld), galectin-3 (1a3k), Charcot–Leyden protein (1lcl), galectin-7 (2gal), human CRP (pentraxin, 1b09), human SAP (pentraxin, 1gyk), calnexin (1jhn), calreticulin (1gv9) and Vp4 sialic-acid-binding domain (1kqr). The residues in these lectins, which are conserved or conservatively mutated at the signature motif positions of legume lectins, are also shown in bold and underlined. The residue numbers are indicated at the end of each line. It can be seen from these alignments that although some residues from the signature motifs are conserved in the galectins, pentraxins, calnexin, calreticulin and Vp4 sialic-acid-binding domain, most of the residues required for any of the legume lectin interfaces are absent, thereby excluding these legume lectin interface types in these lectins.
Figure 4. Interface clusters in the seven types of interfaces in legume lectins.

(a)–(g) give the cartoon representation of the three-dimensional structure of the legume lectins belonging to these seven types of dimeric interfaces. The monomer chains are represented by blue- and red-coloured cartoons respectively. Although the tertiary structures are very similar in all the seven cases, their quaternary associations are different, as seen in the Figure. The interface cluster-forming residues are represented as van der Waal's spheres. Each cluster is coloured differently to differentiate them in the three-dimensional space. (a) Canonical, type II (1fnyAB at 8% cut-off); (b) EcorL-type, X3 (1axy at 5% cut-off); (c) GS4-type, X4 (1gsl at 6% cut-off); (d) DB58-type, X1 (1qnwAC at 4% cut-off); (e) non-canonical interface of ConA-type, X2 (1dglAC at 4% cut-off); (f) unusual interface of PNA (2pelBD at 6% cut-off); (g) unusual interface of GS1 (1hqlAC at 6% cut-off).
Comparison of different legume lectin interfaces based on the signature motifs
The comparison of the consensus patterns of all the interface types showed that they were localized in a few sequence regions, namely the N-terminus and the C-terminus and the region near residues 70–80 and 160–200 (Figure 3). The type II interface and the unusual interfaces of PNA and GS1 are characterized mainly by the N-terminal and the C-terminal regions, whereas X1, X2, X3 and X4 have their consensus patterns in the 70–80 and 160–200 regions. Hence, X1, X2, X3 and X4 mutually exclude each other and therefore cannot co-exist with each other in the higher oligomers. Therefore X1, X2, X3 and X4 can only combine with either type II or the unusual interfaces. Theoretically, any of the X1, X2, X3 and X4 dimeric interfaces can combine with the type II or the unusual interfaces of PNA and GS1 to form tetramers. However, not all these combinations are observed in Nature, and there are specific preferences to the combinations seen as explained above. A look at the multiple sequence alignments indicates that these preferences are mainly due to the absence of the residues required for the other interface types, which leads to the exclusion of the other interface types. For example, in the X4-forming legume lectins GS1 and GS4, the type II residues are found to be absent, and for that reason they cannot form type II interfaces. However, in PNA, the II+X4 combination is seen because the residues required for both II and X4 are present in PNA (which also has an unusual interface and an open quaternary structure). Moreover, GS4 exists as an X4 dimer only, whereas GS1 forms the X4+unusual tetramer. When the sequences of the two were compared, it was found that although six out of the eight residues required for the unusual interface formation of GS1 are present in GS4 also, two of the significant residues are mutated (T26I and F75L), leading to the loss of some important interactions across the interface. Hence the unusual interface type in GS4 could be considerably destabilized, thus explaining why GS4 remains an X4 dimer and does not form a tetramer like GS1.
Arcelin-5 is the only known monomeric legume lectin to be crystallized as a monomer. The sequence of arcelin-5 was checked for the presence of the consensus residues of all the seven interface types. It was found that it has the patterns required for type II as well as X3 interfaces, though most of the X3-forming residues are conservatively mutated. Hence, X3 might be highly destabilized in arcelin-5. Moreover, a close homologue of arcelin-5, namely arcelin-1, is known to exist as a type II dimer. Arcelin-5 is also known to exist as dimer under specific conditions in solution. Since it has both the X3 and type II patterns, both interface types are theoretically feasible, although the type II dimer might be preferred over X3 in solution.
Type II seems to be a more basic and general type of interface in these legume lectins, since it is preferred in many legume lectin dimers and tetramers. Moreover, the type II interfaces in both dimers and tetramers are stronger than the others, since the interface clusters in type II are formed at higher Imin values when compared with the others. Hence, in lectins with multiple oligomerization states, such as DB58, MAL [Maackia amurensis (amur maackia) lectin] and FRIL [tyrosine kinase Flt3 interacting lectin], which are known to exist as dimers in solution and as II+X1/X2 tetramers in crystal structures, the solution dimers are more likely to be of type II rather than X1 or X2, since the type II interface is stronger than the X1/X2 in these cases. Further, the unusual interfaces are generally found to substitute for the type II interfaces and occur when type II is sterically or sequentially disallowed, as in the case of PNA or GS1. Hence the unusual interfaces involve the same sequence regions as type II. Thus the graph-spectra-based interface cluster analysis of legume lectins has answered most of the questions regarding specificities of interfaces in the legume lectin quaternary structures.
Prediction of quaternary association for lectins with unknown structure
The acid test for the signature sequence motifs of each legume lectin interface type, identified using the conserved interface cluster method, was carried out using the legume lectins with unknown structures. The sequences of six such lectins [from Onobrychis viciifolia (sainfoin), Cytisus scoparius (scotch broom), Lotus tetragonolobus (asparagus pea), Vatairea macrocarpa, Bauhinia purpurea (orchid tree) and Cicer arietinum (chickpea)] were aligned with the others with known interface types and were checked for the presence of any of the signature motifs so that their nature of oligomerization can be predicted from the presence or absence of these consensus patterns. This was then compared with the information about their state of oligomerization available from biochemical experiments and those predicted from phylogenetic tree analysis [21]. The results of this analysis predicted that O. viciifolia lectin should form type II dimers, Cy. scoparius, V. macrocarpa and Ci. arietinum lectins should form type II+X1 tetramers and B. purpurea lectin perhaps forms an X4+unusual tetramer. The results for L. tetragonolobus were inconclusive from this analysis. Thus the method could predict the interfaces in five of the six lectins, which also correlated well with the available experimental evidence and results from the phylogenetic tree analysis, proving that this method has a significant predictive value.
Inferences from the legume lectin study
The analysis of the legume lectin quaternary structure using the graph-spectral method has yielded the signature sequences required for each type of quaternary association seen in the legume lectins. It has also aided in characterizing the different interface types and quaternary associations seen in the legume lectins based on the clusters of amino acids seen at these interfaces, in understanding the different factors responsible for the specific oligomerization type of these lectins and in predicting the quaternary association of legume lectins with unknown structures. One of the important points that need to be highlighted is that the application of the clustering algorithm to the oligomeric interfaces of proteins gives a better understanding of the interface nature and composition, as can be seen from these results, because the clusters involve all types of interactions namely, charged, hydrophobic, polar, hydrogen bonds and van der Waal's interactions. There is no discrimination of interactions based on the nature of residues, which helps in obtaining an overall picture of the interactions at the interfaces rather than the pairwise interactions across the interface. Moreover, the algorithm combines the identification of amino acid clusters in protein structures with sequence alignments, thus enabling us to obtain a consensus of both sequentially and structurally conserved clusters of interacting amino acids. Further, the graph-spectral cluster-determining algorithm gives the information regarding the residues which form the centres of clusters, which often coincide with the highly conserved residues in legume lectins. Hence such an analysis has been found to be more successful in handling the complexity of oligomerization in legume lectins and also has the potential to be applied to address various other aspects of protein structure, stability, folding and interactions.
GALECTINS
The representation and analysis of the structure graphs of galectin and pentraxin oligomers are presented here for the first time.
Background
Galectins, though originally discovered in animals, have recently been found in mushrooms as well [49]. They are lectins that bind β-galactose-containing glycoconjugates and do not require bivalent cations to carry out their function, unlike the legume lectins [15,16]. The proposed biological functions of galectins include roles in cell–cell adhesion, cell–matrix adhesion, direct effects on cell growth and viability, potential intracellular functions in regulating metabolism, induction of apoptosis or programmed cell death, and induction of metabolic changes, such as cellular activation and mitosis [15,16]. Their expression in many types of tumour cells has led to the hypothesis that galectins may also be involved in tumorigenesis and metastasis.
Galectins exist as both monomers and dimers. Galectins 1, 2 and 7 are homodimers, whereas the other galectins are monomers. Interestingly, the tertiary structure of galectins is similar to the jelly-roll fold found in many leguminous plant lectins, although they differ considerably in their primary structures. The crystal structures of galectins 1, 2, 3 and 7, congerin and the Charcot–Leyden protein are available and therefore have been considered in this analysis. Galectin-2 is a fungal galectin (Cgl2 from Coprinopsis cinerea [49]). The homodimeric galectin interfaces, namely those of galectins 1, 2 and 7, are topologically different from each other, as can be seen from Figure 1. These are also significantly different from the legume lectin interfaces mentioned above. Congerin forms a galectin-1 type interface and galectin-3 and the Charcot–Leyden protein remain as monomers.
Characterization of galectin interfaces
A careful examination of the sequences of the monomeric and dimeric galectins shows that they do not contain most of the residues present in the signature sequences of the interface types seen in legume lectins, namely II, X1, X2, X3 and X4 (as can be seen from Figure 3). The galectins have their very own signatures that characterize their quaternary association types. The amino acid clusters at the interfaces of the dimeric galectins were identified using the graph-spectral method for those galectins whose structures are available (galectins 1, 2 and 7, shown in Figure 5, and congerin) and were mapped on to the multiple sequence alignment of these galectins obtained along with the monomeric ones (galectin-3 and the Charcot–Leyden protein). These interface cluster-forming residues are highlighted in the multiple sequence alignment shown in Figure 6(a). A comparison of the interface cluster residues characterizing each galectin interface type gave interesting results regarding the factors determining the mode of quaternary association in these lectins. Galectins 1, 2 and 7 have different sequence patterns that determine their oligomerization type, as can be seen from Figure 6(a). The galectin-1 type interface seen in galectin-1 and congerin involves a few residues from the N-terminus and the C-terminus. The T/K+N (Thr/Lys+Asn) pattern observed at the N-terminus in these galectins is partially conserved in galectin-2 and the F+F/L (Phe+Phe/Leu) at the C terminus is partially conserved in the monomers and galectin-7 (Figure 6a). However, none of them have both patterns conserved, which could render them incapable of forming the galectin-1 type interface. The interface clusters in galectin-1 are shown in Figure 5.
Figure 5. Interface clusters in galectins and pentraxins.

The dimeric interfaces of galectins 1, 2 and 7 and pentraxin, along with the amino acid clusters obtained at the interfaces at about Imin=6% are shown. The lectins are represented in cartoon diagram and the interface clusters in van der Waals representation. Each monomer and interface clusters are coloured differently.
Figure 6. Sequence alignments of (a) galectins and (b) pentraxins.

The galectins include galectin-1 (1a78), congerin (1c1f), galectin-2 (1uld), galectin-3 (1a3k), Charcot–Leyden protein (1lcl) and galectin-7 (2gal). The pentraxins include human CRP (1B09), human SAP (1GYK) and a pentraxin from frog (X. laevis) (Swiss-Prot accession code Q07203), which is known to exist as a homodimer. The residues present in the interface clusters of galectins and pentraxins are highlighted and underlined. Galectin-3 (1a3k) and Charcot–Leyden protein are monomers and so have no interface clusters. The structure of the frog pentraxin is not known, and hence we do not have information regarding the interface clusters in this lectin. It is evident from the Figure that galectin-1-like interfaces (including galectin-1 and congerin), galectin-2, galectin-7 and pentraxins all have completely different residues contributing to the interfaces. These can be used for characterizing these interfaces, since they are mutually exclusive.
The galectin-7 interface is characterized by a number of charged-charged interactions among a set of arginine residues (numbers 14, 20, 22, 133) at the N-terminus and C-terminus, along with His108, Glu87, Asp95 and Asp103. Phe135 and Val100 also contribute to the cluster (Figure 5). The arginine residues from one chain are neutralized by the aspartate residues from the other, with histindine and glutamate also adding to the stability. On examining the other galectin sequences for the presence of these residues (Figure 6a), we find that all the others do have some of these residues, but none of them have all of them. In fact, those that have the negatively charged residues conserved in those positions lack the complementary positively charged residues and vice versa. Therefore none of them have the complete complement of the arginines, histidine, glutamate and aspartate residues, thus rendering them incapable of forming the galectin-7 type interface.
The galectin-2 type interface is also characterized by charged-charged interactions across the interface. However, here the positive charge is contributed by Arg99, Arg103 and His96, and the negative charge is contributed by Asp98, Gln101 and Glu20 (Figure 6a). In contrast with galectin-7, the positive charges in the interface of galectin-2 come from the residues-90–100 region of the sequence instead of the N-terminus and C-terminus as in galectin-7. The interesting feature of this interface (and of galectin-7 interface type) is that the positively charged residues and negative charged residues that neutralize them always come from two different monomers, thus making the interface extremely strong and stable. Further, Ser109 and Tyr111 from both of the chains add to the stability of the cluster. An additional interface cluster comprising His141, Pro145 (from same chain) and Leu147 (from other chain) also stabilize the interface. Figure 5 shows the interface clusters in galectin-2. Although some of these residues are conserved in the other galectins, in none of them is the complete pattern conserved (Figure 6a).
Thus the factors determining the type of quaternary association in galectins can be obtained from this approach. Moreover, the signature sequence motifs that characterize each of them have also become apparent from this analysis.
PENTRAXINS
Background
The earliest described pentraxins, CRP (C-reactive protein) and SAP (serum amyloid P component), are cytokine-inducible acute-phase proteins implicated in innate immunity, whose concentrations in the blood increase dramatically upon infection or trauma [15,16]. The pentraxins are a phylogenetically ancient family of oligomeric plasma proteins, all of which bind Ca2+ ions. The binding of Ca2+ is necessary for the expression of ligand-binding activities. These proteins have evolved very little and hence they are highly conserved. They exhibit remarkable conservation of structure and binding specificities. Since the proteins have evolved conservatively and they are present in vertebrates, this suggests an essential function for these proteins in the body. Within vertebrates, there exist two main branches of the pentraxin family, namely the CRP-like proteins and SAP-like proteins. The pentraxins that bind phosphocholine are CRP-like and those that bind carbohydrate moieties are SAP-like pentraxins. All of the pentraxins are oligomers arranged in a discoid-like pentagonal (rarely hexagonal) cyclic symmetry [50], as shown in Figure 7. These proteins were named pentraxins because of their cyclic configuration of five non-covalently bound identical subunits. These proteins consist of a hydrophobic core and have a beta-jelly roll motif as seen in the leguminous-plant lectins.
Figure 7. Discoid arrangement of pentameric pentraxin.

Each monomer is shown in a differently coloured cartoon representation. The interface clusters at each of the dimeric interfaces are shown as van der Waal's spheres.
Characterization of pentraxin interfaces
The pentraxin pentamer consists of five similar dimeric interfaces contributed by five monomers. The dimeric interface of the pentraxin is shown in Figure 1. Each monomer makes two types of interactions with the two other monomers using two different faces coming from different sequential regions. Face 1 of monomer 1 interacts with face 2 of monomer 2 and the face 1 of monomer 2 interacts with face 2 of monomer 3 and so on and so forth till the pentamer is formed (Figure 7). Hence, the dimers themselves are asymmetric, since the interacting monomeric regions at the dimeric interface are different. However, the same pattern is followed subsequently in all five dimeric interfaces, to give rise to a symmetric pentamer. An example each of CRP and SAP, both from humans and whose crystal structures are available, have been considered in this analysis A comparison of the pentraxins with the consensus patterns of all the legume lectin interface types shows that they lack the signature residues required for all these patterns (Figure 3). It is evident from Figure 3 that the pentraxins lack the motifs required for II, X1, X2, X3 or X4 interface types of the legume lectin family. The pentrxins also lack the signature motifs required for all the three galectin-like interfaces (1, 2, and 7; sequence alignments not shown).
The interface clusters obtained in the pentraxins (shown in Figure 5) are as usual mapped on to the sequence alignment of CRP with SAP (Figure 6b), and the consensus pattern of residues required for the oligomerization of the pentraxins are thus obtained (highlighted in Figure 6b). The pentraxin dimeric interface signature is contributed by residues Glu108 (Glu106 in SAP), Val117 (Val115 in SAP) and Arg118 (Lys116 in SAP) from one monomer and Tyr40 (Tyr40 in SAP), Glu42 (Asp42 in SAP), Pro93 (Pro91 in SAP), Asp155 (Glu153 in SAP) and Trp205 (Trp203 in SAP) from the other monomer. One of the CRPs from African clawed frog (Xenopus laevis) is known to exist as a homodimer, but its structure is yet unknown (SwissProt accession number Q07203). This sequence was examined for the presence of the residues contributing to the pentraxin interface obtained from the human CRP and SAP analysis (also shown in Figure 6b). It can be seen that this sequence has only two of the three residues required for one of the monomeric faces of the dimer and two out of the five residues required for the other monomeric face of the dimer. Hence, this pentraxin interface could be considerably destabilized, owing to the mutations of some critical residues leading to its dimeric nature rather than the pentameric form normally seen in the other pentraxins. Thus the study of pentraxins shows that they have a characteristic signature sequence motif different from that of the legume lectins or galectins, which determines their unique pentameric nature of association.
Calnexin/calreticulin/Vp4 sialic-acid-binding domain
Calnexin and calreticulin form part of the quality-control system for glycoproteins in the endoplasmic reticulum [51,52], which bind monoglucosylated N-glycans. They bind to terminal glucose residues on N-linked oligosaccharides and retain misfolded glycoproteins in the endoplasmic reticulum [51,52]. They act as chaperones for N-linked glycoproteins so as to keep the folding intermediates in a folding-competent state. Calnexin is a transmembrane protein and calreticulin is a soluble protein retained in the lumen by a C-terminal retention signal. The luminal N-terminal portion of calnexin is very similar to calreticulin, although one of the repeated segments of calnexin is absent from calreticulin. They share the same jelly-roll tertiary structure as the legume lectins. However, they remain monomeric and do not form higher oligomers. Similar to calnexin/calreticulin, the Vp4 sialic-acid-binding domain also has the lectin-like jelly-roll tertiary structure, but remains a monomer. The sequence alignments of calnexin, calreticulin and the Vp4 sialic-acid-binding domain with the legume lectins are shown in Figure 3. When these alignments were analysed for the presence of the signature sequences of the legume lectin interface types, we found that although these proteins have some of the residues required for a few of the interface types, they lack most of the residues required for all of the legume lectin oligomerization types (II, X1, X2, X3 and X4), indicating why they remain monomeric.
NETWORK MODEL OF LECTIN INTERFACES
Apart from the amino-acid-cluster analysis at the lectin interfaces, we have also analysed the protein structure graphs of the lectin oligomers from the network perspective. This includes the size of the largest cluster in these proteins at different Imin values and the residue hubs obtained at their interfaces. These concepts have been presented for the first time in the present review. One representative from each legume lectin interface type, galectin interface type and pentraxin interface type has been chosen for this analysis (Table 3).
Table 3. Interface hubs in various lectin interface types from lectin structure graphs.
Abbreviations: DGL, Dioclea grandiflora (mucana) lectin; PDB, Protein Data Bank.
| Lectin* | PDB code | Interface types in quaternary structure† | Interface hubs (Imin in %)‡ | Imin at which interface cluster is the largest (%) |
|---|---|---|---|---|
| ConA | 2cna | II (canonical)+X2 tetramer | W88A, W88B, Q137A, Q137B (4%) | 6 |
| GS4 | 1gsl | X4 (back-to-back) dimer | R194A, R194B, Y72A, Y72B (4%) | 6 |
| ECoRL | 1axy | X3 (handshake) dimer | I178A, I178B, H180A, H180B (2%) | 4 |
| DBL | 1bjq | II+X1 tetramer | H51A, V57A, R60A, V64A, K116A, N55B, R60B (2%) | 0 |
| DGL | 1dgl | II+X2 tetramer | I187A, I187B (2%) | 0 |
| GS1 | 1hql | X4+unusual tetramer | W10A, W10B (4%) | 0 |
| PNA | 2pel | II+X4+unusual tetramer | N29C, Q33C, E72C, K74C, L219C, R221C, N29D, L219D, R221D (2%) | 0 |
| Galectin-1 | 1a78 | Galectin-1 dimer | L121A, I131A, F90B, F128B (2%) | 0 |
| Galectin-2 | 1uld | Galectin-2 dimer | H96A, Q101A, H96B, Q101B (4%) | 6 |
| Galectin-7 | 2gal | Galectin-7 dimer | R20A, R22A, K98A, R20B, R22B (2%) | 0 |
| Pentraxin (CRP) | 1b09 | Pentraxin pentamer | F199A, K201A, L204A, R118B (2%) | 4 |
| Pentraxin(SAP) | 1gyk | Pentraxin pentamer | K116A (4%) | 4 |
* Galectin-3 (1a3k), Charcot–Leyden protein (1lcl), calreticulin (1gv9), calnexin (1jhn) and arcelin-5 (1ioa) are monomeric types and are not presented here; only one representative from the other dimeric interface types are presented; congerin (1c1f), which has an interface similar to that of galectin-1, is also not presented.
† All the interfaces contributing to the quaternary structure of the lectin are given; the specific interfaces whose hubs are given in the Table are highlighted in bold.
‡ Interface hubs are given at Imin=4%; if no interface hubs were obtained at Imin=4%, those obtained at Imin=2% are given; the one-letter notation for amino acids is used.
Interface hubs
In network terminology, hubs are highly connected nodes in the network. In the protein structure graphs, hubs are those residues that make more than four contacts with other residues and the interface hubs are those which make more than four contacts, with at least one residue belonging to the chain other than its own. Table 3 gives the interface hubs obtained in the ten selected lectins at Imin=4% or 2% (when there are no interface hubs at Imin=4%). Most of these interface hubs are also present in the signature motifs of their respective interface types (Figures 3 and 6). It can be observed from Table 3 that there are nine aromatic hubs, eleven hydrophobic hubs, nine negatively charged hubs and 21 positively charged hubs. Out of the positively charged ones, 11 are arginine residues, five are histidine residues and five are lysine residues. We see a predominance of charged hubs, especially arginine, in these interfaces. Except X2, unusual GS1 and galectin-1 interface types, all the others have at least one charged hub in their interfaces. The contribution of charged hubs in these interfaces is about 60%. The hydrophobic and aromatic contributions are relatively lesser. This is significantly different from the hub preferences seen in the monomeric protein cores, where there was no clear domination of the charged residues at any Imin value, except a meagre excess of arginine at Imin=4% (K. V. Brinda and S. Vishveshwara, unpublished work). This dominance of the polar and charged residue hubs at interfaces could be due the fact that these residues can be stabilized by interactions with water when they are exposed on the surface of the monomer and can be neutralized by oppositely charged residues when they get buried at the interface during oligomerization. However, in the monomeric proteins, the charged residues are preferentially exposed on the surface and hence the probability of their forming hubs is generally less. The preference of arginine residues at protein/protein and protein/DNA interfaces have been elucidated previously by a few groups [4,43,44]. The dominance of charged residues as hubs in the interfaces is only an observation based on the present set of lectins, and a detailed analysis of a complete non-redundant set of protein oligomers holds the conclusive results for the hub preferences at protein interfaces.
The interface hubs obtained in the canonical (type II) interface of ConA and SAP pentraxin dimeric interface are shown in Figure 8. The Figure clearly shows that the hubs play a major role in integrating the two monomers structurally through a series of non-covalent interactions with residues belonging to both monomers. Hence, these hubs contribute significantly to the stability of the interface and so a mutation of the hub can severely destabilize the interface. The hubs also provide robustness to the interface interaction network, because a random mutation of a non-hub residue is unlikely to affect the stability of the interface interaction network, whereas the mutation of a hub can have drastic effects on the oligomerization and hence the function of the protein.
Figure 8. Interface hubs in (a) ConA (type II) interface and (b) SAP pentraxin (dimeric) interface at Imin=4%.

The proteins are shown in cartoon representation, and each monomer in the dimer is coloured differently. The interface hubs and the residues with which they interact are represented as van der Waals spheres with each residue being coloured differently. The residue name and number are indicated in the Figure, and the chain to which each residue belongs is given within parentheses. Using the one-letter amino acid notation, W88 (A) and Q137 (A) in ConA and K116 (A) in SAP petraxin form the interface hubs. It can be seen from the Figure that these hubs interact with different types of residues from both the chains, thus stabilizing the dimeric interfaces.
Size of the largest cluster as a function of interaction cut-off (Imin)
The normalized size-of-the-largest-cluster-versus-Imin plots for the selected lectins (one representative from each interface type) is shown in Figure 9. As the Figure shows, the plots are sigmoidal with a transition (where there is a drastic decrease in size) at about Imin=3–4%. The largest clusters at different Imin values were obtained for each of these lectins. Whether the largest cluster belongs to the interface or not was examined in each of these lectins at Imin values varying from below the transition (0%) to above the transition (6%). Clearly, at Imin=0%, the oligomer exists as one big cluster in all the lectins and hence the largest cluster includes the interface in all the proteins at Imin=0%. An example is shown in Figure 10(a), where, at Imin=0%, the oligomer exists as one big cluster that includes the interface. However, as the Imin is increased, the size of the largest cluster reduces and after the transition it splits into smaller distinct clusters. Hence, the largest cluster at and above the transition (4 and 6% respectively) may or may not belong to the interface, depending upon the strength of interface of the particular lectin. An example of a lectin with the largest cluster still at the interface at Imin=6% is shown in Figure 10(b). In the present set we find that the type II and X4 interfaces of the legume lectins and the galectin-2 type interface contribute to the largest cluster, even at Imin=6%, the X3 interface of legume lectins and the dimeric interface of the pentraxins contribute to the largest cluster even at Imin=4%, whereas the largest clusters of X1 and X2 interfaces of legume lectins and that of galectins 1 and 7 do not involve their oligomeric interfaces beyond Imin=0%. This shows that the type II, X4, galectin-2, X3 and pentraxin interfaces are as strong as their hydrophobic cores and these are considerably stronger than the X1, X2 and the galectins 1 and 7 interfaces.
Figure 9. Plot of the size of the largest cluster (normalized with respect to the total number of residues in the protein structure, N) versus Imin for selected lectins.

The plot clearly shows that the curve is sigmoidal in nature.
Figure 10. Largest cluster at (a) Imin=0% and (b) Imin=6% in ConA (2cna) type II dimer.

The protein is shown in cartoon representation with the monomers coloured differently. The cluster-forming residues are shown in a grey ball-and-stick representation. At Imin=0%, the whole dimer forms a single large cluster, as expected. However, at Imin=6%, the single large cluster splits into smaller ones. In this case the largest cluster at Imin=6% is an interface cluster, as can be seen in the Figure.
Thus the Imin value used in the present analysis is a good measure of the strength of the interaction of the interacting residues, which can be used to estimate the strength of the interacting interfaces too. It is noteworthy that, in several cases, the oligomerization has formed by a strong interface that is almost as strong as their monomeric protein cores. Moreover, the identification of interface hubs can be a useful tool for predicting hot spots at protein interfaces, which can be mutated to considerably destabilize the interface. Hence, the interface hub analysis can aid in designing mutants in order to understand protein quaternary association and protein–protein interactions in general.
CONCLUSIONS
The present review provides a comprehensive analysis of the nature and types of quaternary associations found in plant and animal lectins. Specifically, the graph-spectral algorithm has provided the signature sequence motifs characterizing the oligomeric interfaces of legume lectins, galectins and pentraxins, thus giving insights into the factors determining the nature and type of quaternary association in each one of them. It has clearly elucidated why each one of them prefers a specific type of association and why the other known types are excluded in these lectins. The prediction of the oligomerization modes of lectins with unknown structures using the signature motifs established using the present method is also very rational and successful.
The interface hubs identified using the protein structure graphs provide a simple yet novel tool for identifying hot spots at protein interfaces. The graph representation of protein structures based on the strength of the non-covalent interactions among amino acid residues also aids in estimating the strengths of protein interfaces. Thus the graph theoretical algorithm presented in combination with traditional sequence-alignment methods is found to be extremely successful in identifying the signature motifs for a given type of quaternary association, has a very high predictive value and can be used in designing mutants that can destabilize the oligomeric interface. The present study has also aided in assessing the relative strengths of interactions at different types of lectin interfaces.
The representation of protein structures as a network of non-covalently interacting amino acid residues is an interesting way to analyse both monomeric and oligomeric protein structures, which gives valuable insights into the tertiary and quaternary structures of proteins. This can be a useful tool to understand protein–protein interactions and various other aspects of protein structure.
Acknowledgments
A. S. thanks the Department of Biotechnology (DBT), India, for funding this project. S. V. acknowledges the computational genomics initiative at the Indian Institute of Science, funded by the Department of Biotechnology, India, for support. K. V. B. thanks the Council of Scientific and Industrial Research, India, for the award of the fellowship.
References
- 1.Fernandez A., Scheraga H. A. Insufficiently dehydrated hydrogen bonds as determinants of protein interactions. Proc. Natl. Acad. Sci. U.S.A. 2003;100:113–118. doi: 10.1073/pnas.0136888100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu D., Tsai C. J., Nussinov R. Hydrogen bonds and salt bridges across protein–protein interfaces. Protein Eng. 1997;10:999–1012. doi: 10.1093/protein/10.9.999. [DOI] [PubMed] [Google Scholar]
- 3.Chakrabarti P., Janin J. Dissecting protein–protein recognition sites. Proteins. 2002;47:334–343. doi: 10.1002/prot.10085. [DOI] [PubMed] [Google Scholar]
- 4.Bahadur R. P., Chakrabarti P., Rodier F., Janin J. Dissecting subunit interfaces in homodimeric proteins. Proteins. 2003;53:708–719. doi: 10.1002/prot.10461. [DOI] [PubMed] [Google Scholar]
- 5.Janin J., Wodak S. J. Protein modules and protein–protein interaction. Introduction. Adv. Protein Chem. 2002;61:1–8. doi: 10.1016/s0065-3233(02)61000-9. [DOI] [PubMed] [Google Scholar]
- 6.Valdar W. S., Thornton J. M. Protein–protein interfaces: analysis of amino acid conservation in homodimers. Proteins Struct. Funct. Genet. 2001;42:108–124. [PubMed] [Google Scholar]
- 7.Ma B., Elkayam T., Wolfson H., Nussinov R. Protein–protein interactions: structurally conserved residues distinguish between binding sites and exposed protein surfaces. Proc. Natl. Acad. Sci. U.S.A. 2003;100:5772–5777. doi: 10.1073/pnas.1030237100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones S., Thornton J. M. Analysis of protein-protein interaction sites using surface patches. J. Mol. Biol. 1997;272:121–132. doi: 10.1006/jmbi.1997.1234. [DOI] [PubMed] [Google Scholar]
- 9.Smith G. R., Sternberg M. J. Prediction of protein–protein interactions by docking methods. Curr. Opin. Struct. Biol. 2002;12:28–35. doi: 10.1016/s0959-440x(02)00285-3. [DOI] [PubMed] [Google Scholar]
- 10.Camacho C. J., Vajda S. Protein–protein association kinetics and protein docking. Curr. Opin. Struct. Biol. 2002;12:36–40. doi: 10.1016/s0959-440x(02)00286-5. [DOI] [PubMed] [Google Scholar]
- 11.Valencia A., Pazos F. Computational methods for the prediction of protein interactions. Curr. Opin. Struct. Biol. 2002;12:368–373. doi: 10.1016/s0959-440x(02)00333-0. [DOI] [PubMed] [Google Scholar]
- 12.Kortemme T., Baker D. Computational design of protein–protein interactions. Curr. Opin. Chem. Biol. 2004;8:91–97. doi: 10.1016/j.cbpa.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 13.Cole C., Warwicker J. Side-chain conformational entropy at protein–protein interfaces. Protein Sci. 2002;11:2860–2870. doi: 10.1110/ps.0222702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wodak S. J., Mendez R. Prediction of protein–protein interactions: the CAPRI experiment, its evaluation and implications. Curr. Opin. Struct. Biol. 2004;14:242–249. doi: 10.1016/j.sbi.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 15.Liener I. E., Sharon N., Goldstein I. J. Functions and Applications in Biology and Medicine. New York: Academic Press; 1986. The Lectins: Properties. [Google Scholar]
- 16.Varki A., Cummings R., Esko J., Freeze H., Hart G., Marth J., editors. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2002. Essentials in Glycobiology. [PubMed] [Google Scholar]
- 17.Murzin A. G., Brenner S. E., Hubbard T., Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 18.Loris R., Hamelryck T., Boukaert J., Wyns L. Legume lectin structure. Biochim. Biophys. Acta. 1998;1383:9–36. doi: 10.1016/s0167-4838(97)00182-9. [DOI] [PubMed] [Google Scholar]
- 19.Vijayan M., Chandra N. Lectins. Curr. Opin. Struct. Biol. 1999;9:707–714. doi: 10.1016/s0959-440x(99)00034-2. [DOI] [PubMed] [Google Scholar]
- 20.Elgavish S., Shaanan B. Chemical characteristics of dimer interfaces in the legume lectin family. Protein Sci. 2001;10:753–761. doi: 10.1110/ps.44001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manoj N., Suguna K. Signature of quaternary structure in the sequences of legume lectins. Protein Eng. 2001;10:735–745. doi: 10.1093/protein/14.10.735. [DOI] [PubMed] [Google Scholar]
- 22.Srinivas V. R., Reddy G. B., Ahmad N., Swaminathan C. P., Mitra N., Surolia A. Legume lectin family, the ‘natural mutants of the quaternary state’, provide insights into the relationship between protein stability and oligomerization. Biochim. Biophys. Acta. 2001;1527:102–111. doi: 10.1016/s0304-4165(01)00153-2. [DOI] [PubMed] [Google Scholar]
- 23.Brinda K. V., Mitra N., Surolia A., Vishveshwara S. Determinants of quaternary association in legume lectins. Protein Sci. 2004;13:1735–1749. doi: 10.1110/ps.04651004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barabasi A. L. Cambridge, MA: Perseus Publishing; 2002. Linked: the new science of networks. [Google Scholar]
- 25.Wuchty S., Ravasz E., Barabási A.-L. The Architecture of Biological Networks. In: Deisboeck T. S., Yasha Kresh J., Kepler T. B., editors. Complex Systems in Biomedicine. New York: Kluwer Academic Publishing; 2003. [Google Scholar]
- 26.Dokholyan N. V., Shakhnovich B., Shakhnovich E. I. Expanding protein universe and its origin from the biological Big Bang. Proc. Natl. Acad. Sci. U.S.A. 2002;99:14132–14136. doi: 10.1073/pnas.202497999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grindley H. M., Artymiuk P. J., Rice D. W., Willett P. Identification of tertiary structure resemblance in proteins using a maximal common subgraph isomorphism algorithm. J. Mol. Biol. 1993;229:707–721. doi: 10.1006/jmbi.1993.1074. [DOI] [PubMed] [Google Scholar]
- 28.Przytycka T., Srinivasan R., Rose G. D. Recursive domains in proteins. Protein Sci. 2002;11:409–417. doi: 10.1110/ps.24701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Atilgan A. R., Akan P., Baysal C. Small-world communication of residues and significance for protein dynamics. Biophys. J. 2004;86:85–91. doi: 10.1016/S0006-3495(04)74086-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greene L. H., Higman V. A. Uncovering network systems within protein structures. J. Mol. Biol. 2003;334:781–791. doi: 10.1016/j.jmb.2003.08.061. [DOI] [PubMed] [Google Scholar]
- 31.Vendruscolo M., Paci E., Dobson C. M., Karplus M. Three key residues form a critical contact network in a protein folding transition state. Nature (London) 2001;409:641–645. doi: 10.1038/35054591. [DOI] [PubMed] [Google Scholar]
- 32.Vendruscolo M., Dokholyan N. V., Paci E., Karplus M. Small-world view of the amino acids that play a key role in protein folding. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2002;65:061910. doi: 10.1103/PhysRevE.65.061910. [DOI] [PubMed] [Google Scholar]
- 33.Dokholyan N. V., Li L., Ding F., Shakhnovich E. I. Topological determinants of protein folding. Proc. Natl. Acad. Sci. U.S.A. 2002;99:8637–8641. doi: 10.1073/pnas.122076099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bahar I., Atilgan A. R., Erman B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold. Des. 1997;2:173–181. doi: 10.1016/S1359-0278(97)00024-2. [DOI] [PubMed] [Google Scholar]
- 35.Bahar I., Atilgan A. R., Demirel M. C., Erman B. Vibrational dynamics of folded proteins: significance of slow and fast motions in relation to function and stability. Phys. Rev. Lett. 1998;80:2733–2736. [Google Scholar]
- 36.Samudrala R., Moult J. Handling context-sensitivity in protein structures using graph theory: bona fide prediction. Proteins: Struct. Funct. Genet. 1997;1:43–49. doi: 10.1002/(sici)1097-0134(1997)1+<43::aid-prot7>3.3.co;2-z. [DOI] [PubMed] [Google Scholar]
- 37.Samudrala R., Moult J. A graph-theoretic algorithm for comparative modeling of protein structure. J. Mol. Biol. 1998;279:287–302. doi: 10.1006/jmbi.1998.1689. [DOI] [PubMed] [Google Scholar]
- 38.Sistla R. K., Brinda K. V., Vishveshwara S. Identification of domains and domain interface residues in multidomain proteins from graph spectral method. Proteins. 2005;59:616–626. doi: 10.1002/prot.20444. [DOI] [PubMed] [Google Scholar]
- 39.Reichmann D., Rahat O., Albeck S., Meged R., Dym O., Schreiber G. The modular architecture of protein–protein binding interfaces. Proc. Natl. Acad. Sci. U.S.A. 2005;102:57–62. doi: 10.1073/pnas.0407280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kannan N., Vishveshwara S. Identification of side-chain clusters in protein structures by a graph spectral method. J. Mol. Biol. 1999;292:441–464. doi: 10.1006/jmbi.1999.3058. [DOI] [PubMed] [Google Scholar]
- 41.Vishveshwara S., Brinda K. V., Kannan N. Protein structure: insights from graph theory. J. Theor. Comp. Chem. 2002;1:187–211. [Google Scholar]
- 42.Kannan N., Vishveshwara S. Aromatic clusters: a determinant of thermal stability of thermophilic proteins. Protein Eng. 2000;13:753–761. doi: 10.1093/protein/13.11.753. [DOI] [PubMed] [Google Scholar]
- 43.Brinda K. V., Kannan N., Vishveshwara S. Analysis of homodimeric protein interfaces by graph-spectral methods. Protein. Eng. 2002;4:265–277. doi: 10.1093/protein/15.4.265. [DOI] [PubMed] [Google Scholar]
- 44.Sathyapriya R., Vishveshwara S. Interaction of DNA with clusters of amino acids in proteins. Nucleic Acids Res. 2004;32:4109–4118. doi: 10.1093/nar/gkh733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kannan N., Chander P., Ghosh P., Vishveshwara S., Chatterji D. Stabilizing interactions in the dimer interface of α-subunit in Escherichia coli RNA polymerase: a graph spectral and point mutation study. Protein Sci. 2001;10:46–54. doi: 10.1110/ps.26201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Reference deleted.
- 48.Thompson J. D., Higgins D. G., Gibson T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walser P. J., Haebel P. W., Kunzler M., Sargent D., Kues U., Aebi M., Ban N. Structure and functional analysis of the fungal galectin CGL2. Structure. 2004;12:689–702. doi: 10.1016/j.str.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 50.Kilpatrick J. M., Volanakis J. E. Molecular genetics, structure, and function of C-reactive protein. Immunol. Res. 1991;10:43–53. doi: 10.1007/BF02918166. [DOI] [PubMed] [Google Scholar]
- 51.Trombetta E. S., Helenius A. Lectins as chaperones in glycoprotein folding. Curr. Opin. Struct. Biol. 1998;8:587–592. doi: 10.1016/s0959-440x(98)80148-6. [DOI] [PubMed] [Google Scholar]
- 52.Parodi A. J. Protein glucosylation and its role in protein folding. Annu. Rev. Biochem. 2000;69:69–93. doi: 10.1146/annurev.biochem.69.1.69. [DOI] [PubMed] [Google Scholar]