

Abstract
SP-A (surfactant protein A) is a lipid-binding collectin primarily involved in innate lung immunity. SP-A interacts with the bacterial rough LPS (lipopolysaccharide) Re-LPS (Re595 mutant of LPS from Salmonella minnesota), but not with smooth LPS. In the present study, we first examined the characteristics of the interaction of human SP-A with Re-LPS. Fluorescence intensity and anisotropy measurements of FITC-labelled Re-LPS in the presence and absence of SP-A indicated that SP-A bound to Re-LPS in solution in a Ca2+-independent manner, with a dissociation constant of 2.8×10−8 M. In the presence of calcium, a high-mobility complex of SP-A and [3H]Rb-LPS (Rb mutant of LPS from Escherichia coli strain LCD 25) micelles was formed, as detected by sucrose density gradients. Re-LPS aggregation induced by SP-A was further characterized by light scattering. On the other hand, human SP-A inhibited TNF-α (tumour necrosis factor-α) secretion by human macrophage-like U937 cells stimulated with either Re-LPS or smooth LPS. We further examined the effects of human SP-A on the binding of Re-LPS to LBP (LPS-binding protein) and CD14. SP-A decreased the binding of Re-LPS to CD14, but not to LBP, as detected by cross-linking experiments with 125I-ASD-Re-LPS [125I-labelled sulphosuccinimidyl-2-(p-azidosalicylamido)-1,3-dithiopropionate derivative of Re-LPS] and fluorescence analysis with FITC-Re-LPS. When SP-A, LBP and CD14 were incubated together, SP-A reduced the ability of LBP to transfer 125I-ASD-Re-LPS to CD14. These SP-A effects were not due to the ability of SP-A to aggregate Re-LPS in the presence of calcium, since they were observed in both the absence and the presence of calcium. These studies suggest that SP-A could contribute to modulate Re-LPS responses by altering the competence of the LBP–CD14 receptor complex.
Keywords: CD14, inflammation, lipopolysaccharide-binding protein (LBP), lung, rough lipopolysaccharide, surfactant protein A (SP-A)
Abbreviations: bis-ANS, 1,1′-bis-(4-anilino)naphthalenesulphonic acid; BPI, bactericidal permeability-increasing protein; CMC, critical micellar concentration; FBS, fetal bovine serum; KDO, 2-keto-3-deoxyoctulosonic acid; LPS, lipopolysaccharide; Re-LPS, Re595 mutant of LPS from Salmonella minnesota; 125I-ASD-Re-LPS, 125I-labelled sulphosuccinimidyl-2-(p-azidosalicylamido)-1,3-dithiopropionate derivative of Re-LPS; LBP, LPS-binding protein; Rb-LPS, Rb mutant of LPS from Escherichia coli strain LCD 25; SP-A, surfactant protein A; TLR4, Toll-like receptor 4; TNF-α, tumour necrosis factor-α
INTRODUCTION
Bacterial LPS (lipopolysaccharide) is the major component of the outer leaflet of the outer membrane of Gram-negative bacteria. LPS is the major inducer of host responses to Gram-negative bacteria. Such bacteria express either smooth (S-form or wild-type) LPS or rough (R-form) LPS mutants. Bacteria with R-LPS phenotypes are more common among pathogens that colonize the upper aerodigestive tract [1]. All rough LPS mutants (Ra, Rb, Rc, Rd and Re) lack O-antigen, but possess lipid A and progressively shorter core oligosaccharides [1].
Myeloid cells use at least four proteins to mount a sensitive cellular response to LPS: LBP (LPS-binding protein), CD14, TLR4 (Toll-like receptor 4) and MD2 [2–6]. LBP is a 60 kDa glycoprotein found in the serum [2] and also in the alveolar fluid [7]. In the lung, LBP is produced by alveolar epithelial type II cells [7]. LBP catalyses the transfer of LPS to a binding site of CD14, increasing the binding affinity of CD14 to LPS [2]. CD14 is a 55 kDa glycoprotein, glycosylphosphatidylinositol-linked to the surface of mature myeloid cells (mCD14) or present as a soluble form (sCD14) in serum [3] or in the alveolar fluid [8]. CD14 is required to bring LPS into close proximity to the TLR4–MD2 complexes present at the cell surface [4–6]. The transmembrane TLR4 serves as the primary mediator of LPS signalling [6].
Due to inhalation of airborne particles containing bacteria and LPS, the thin alveolar epithelium is continuously exposed to this potent pro-inflammatory molecule. When LPS molecules enter the host via airways, they interact with alveolar macrophages in a fluid environment characterized by the presence of surfactant membranes, which are involved in reducing the surface tension of the fluid lining the alveoli and in host defence. Several components present in this lipid-rich alveolar fluid are involved in the down-regulation of LPS responses that promote excessive inflammation and compromise gas exchange. First, given the lipophilic nature of LPS, surfactant membranes might incorporate this molecule, making LPS unavailable for signalling. Secondly, SP-A (surfactant protein-A), SP-C and SP-D interact with LPS and/or inhibit LPS responses [9,10]. The alveolar fluid also contains other proteins and peptides with capability of binding to LPS that have direct antimicrobial activity (e.g. α-defensin or cathelicidin) [11]. In animal models, LPS can induce lung injury and acute respiratory distress syndrome. In humans, acute inhalation of LPS induces systemic and airway inflammation [12]. Host defence in the alveolar interface is extremely demanding, since even moderate degrees of inflammation and exudation compromise the primary function of the lung.
SP-A belongs to the structurally homologous family of innate immune defence proteins known as collectins for their collagen-like and lectin domains. SP-A is involved in innate host-defence and inflammatory immunomodulator processes of the lung, as demonstrated by skilful experiments with mice deficient in SP-A. These SP-A knockout mice show increased susceptibility to bacterial and viral infections and enhanced inflammatory responses in the lung to a variety of stimuli [9,10].
SP-A binds to the lipid A and rough LPS but not to smooth LPS [13–16] and induces aggregation of these molecules in the presence of calcium [13,16–18]. On the other hand, several studies have shown that SP-A can modulate smooth LPS responses in vitro and in vivo. Human SP-A inhibits TNF-α (tumour necrosis factor-α) release from human alveolar macrophages [19,20] or human macrophage-like cell line U937 [15,16] stimulated with smooth LPS. In vivo experiments show that SP-A-deficient mice, intratracheally challenged with smooth LPS, produce more nitric oxide and pro-inflammatory cytokines than do wild-type mice [9]. These results are consistent with the anti-inflammatory action of SP-A in the alveolar lavage, opposite in direction to the effect of LBP. Previous studies indicate that SP-A may inhibit smooth LPS response through a direct interaction with CD14 [15,21]. While there seems to be an agreement on the role of SP-A in the suppression of stimulation of smooth LPS, to which SP-A hardly binds, there is a controversy over whether SP-A enhances or suppresses the host inflammatory response to rough LPS, to which SP-A binds [15,16,21–23].
The first objective of the present study was to investigate the affinity and characteristics of the binding of SP-A to Re-LPS (Re595 mutant of LPS from Salmonella minnesota), which have not yet been studied in detail. The second objective was to determine whether SP-A stimulates or decreases the inflammatory response to rough LPS and subsequently investigate the effect of SP-A on the binding of rough LPS to either LBP alone or CD14 in the absence and presence of LBP. In the present study, we have used 125I-labelled Re-595-LPS with covalently attached UV-activated phenylazide that is capable of cross-linking to nearby proteins in order to check the effect of SP-A on the interaction between Re-LPS and LBP and/or CD14. Our results show that SP-A does not affect the binding of LBP to rough LPS but inhibits the binding of CD14 to rough LPS in the presence or absence of LBP.
EXPERIMENTAL
Materials
Rough LPS from S. minnesota (serotype Re 595), smooth LPS from Escherichia coli (serotype 055:B5) and PMA were purchased from Sigma (St. Louis, MO, U.S.A.). Tritium-labelled LPS from the LCD 25 strain of E. coli K12 (Rb mutant) was obtained from List Biological Laboratories (Campbell, CA, U.S.A.). Fluorescein, FITC isomer I and bis-ANS [1,1′-bis-(4-anilino)naphthalenesulphonic acid] were purchased from Molecular Probes (Eugene, OR, U.S.A.). The organic solvents (methanol and chloroform) used to dissolve rough LPS were HPLC-grade (Scharlau Chemie, Barcelona, Spain). RPMI 1640 medium, heat-inactivated FBS (fetal bovine serum) and the Limulus amebocyte lysate kit were obtained from BioWhittaker (Walkersville, MD, U.S.A.). ELISA kit for TNF-α immunoassays was obtained from BD PharMingen (San Diego, CA, U.S.A.). Macrophage-like cell line U937 cells were supplied by the A.T.C.C. (Manassas, VA, U.S.A.). Lavages from alveolar proteinosis patients were donated by Dr K. B. M. Reid (Oxford University, Oxford, U.K.).
SP-A, CD14 and LBP
Broncho-alveolar lavages from alveolar proteinosis patients were used as a source of human SP-A. The structural properties and lipid-related functions of human SP-A from alveolar proteinosis patients are similar to those of human SP-A from healthy subjects [17]. SP-A was purified from isolated surfactant using sequential butanol and octyl glucoside extractions as described previously [16,17,24]. Quantification of SP-A was carried out by amino acid analysis in a Beckman System 6300 High Performance analyser as described elsewhere [24]. The purity of SP-A was checked by one-dimensional SDS/PAGE in 12% polyacrylamide gel under reducing conditions. Endotoxin content of human SP-A isolated by this method was approx. 0.3 pg of endotoxin/μg of SP-A, as determined by Limulus amebocyte lysate assay.
For the analysis of thermal stability of the collagen-like domain of human SP-A, melting curves were obtained on a Jasco J-715 spectropolarimeter fitted with a 150 W xenon lamp as described previously [16,17]. Molar ellipticities were monitored at 207 nm while the sample temperature was raised from 20 to 70 °C and dropped from 70 to 20 °C with an average heating rate of 20 °C/h. SP-A concentrations were 120 μg/ml and quartz cells with a 1 mm path length were used. The folded fraction (F) was calculated as described elsewhere [16,17]. The temperature at which the collagen triple helix of SP-A was 50% unfolded (F=0.5) was taken as the melting temperature (Tm).
Recombinant human soluble CD14 and LBP were expressed in a baculovirus system and purified as described previously [25,26].
Preparation of LPS
For experiments in which concentrations of Re-LPS (molecular mass 2.5 kDa) were in the range 10 to 10−7 M, Re-LPS was prepared from a stock solution of the LPS in chloroform/methanol/water (17:7:1, by vol.). The required amounts of Re-LPS were taken and the solvent evaporated to dryness under a gentle stream of nitrogen. Traces of solvent were subsequently removed by evacuation under reduced pressure overnight. Re-LPS (or lipid A, molecular mass 2 kDa) films were hydrated for 1 h at 41 °C in 5 mM Tris/HCl buffer (pH 7.4) containing 0.1 mM EDTA. After vortex-mixing, the LPS solution was sonicated for 2 min. For experiments in which LPS concentrations were higher than 10−6 M (LPS aggregation experiments), the required amounts of LPS were directly dissolved in 5 mM Tris/HCl buffer (pH 7.4) containing 150 mM NaCl and 0.1 mM EDTA. LPS supramolecular aggregates were allowed to swell for 1 h at a temperature above that for the gel-to-liquid crystalline (β↔α) phase transition of the acyl chains of the corresponding LPS [27]. After intense vortex-mixing, the LPS solution was sonicated for 2 min.
The CMCa (where CMC is critical micellar concentration) of Re-LPS, defined as a transition value from aggregate to supra-aggregate structures, was determined using fluorescence spectroscopy. Bis-ANS (0.5×10−6 M) was incubated in the absence and presence of increasing amounts of Re-LPS (0–34 μM) in 20 mM Tris/HCl (pH 7.4), 150 mM NaCl and 0.2 mM EDTA for 15 min at 37 °C. The samples (either Re-LPS alone or bis-ANS with and without Re-LPS) were excited at 395 nm and fluorescence emission recorded from 460 to 600 nm in an SLM-Aminco AB-2 spectrofluorimeter with a thermostated cuvette holder (±0.1 °C), using 5×5 mm path-length quartz cuvettes. As the bis-ANS molecule partitioned into the hydrophobic compartment of the Re-LPS supra-aggregates, its emission peak shifted from 525 to 500 nm and the quantum yield increased. This is consistent with a decrease of the microenvironment polarity around the probe upon binding to Re-LPS supra-aggregate structures. The CMCa was determined at the point where the two slopes of the curve intersected.
A fluorescent Re-LPS derivative (FITC-Re-LPS), in which the phosphoethanolamine groups of Re-LPS were bound to FITC, was prepared as described in [2]. The Re-LPS concentration of the fluorescent derivative was determined by quantification of the KDO (2-keto-3-deoxyoctulosonic acid), and the content of fluorescein in FITC-Re LPS was determined spectrophotometrically at 493 nm, using a molar absorption coefficient of 77000 M−1·cm−1 (pH 9) [2]. Typically, the fluorescent Re-LPS derivative had substitution ratios of 15–20 mol% fluorescein. In addition, 125I-ASD-Re-LPS [125I-labelled sulphosuccinimidyl-2-(p-azidosalicylamido)-1,3-dithiopropionate derivative of Re-LPS] was prepared as described in [28]. These derivative-formation procedures did not alter the biological activity of the unsubstituted molecule [2].
LPS-binding assays
Fluorescence assays of FITC-Re-LPS
Fluorescence measurements were carried out using an SLM-Aminco AB-2 spectrofluorimeter with a thermostated cuvette holder (±0.1 °C), using 5×5 mm path-length quartz cuvettes. Fluorescence emission spectra of FITC-Re-LPS (4×10−7 M) were measured in the absence and presence of one among SP-A, LBP and CD14 in a buffer containing 150 mM NaCl, 1 mM EDTA and 5 mM Tris/HCl (pH 8) at 25 or 37 °C. The blanks (protein alone) and FITC-Re-LPS samples (with and without protein) were excited at 470 nm and emission spectra recorded from 500 to 650 nm. The apparent dissociation constant (KD) for FITC-Re-LPS–SP-A complexes was obtained by analysing the time dependence of the fluorescence change when 2×10−8 M FITC-Re-LPS reacted with various concentrations of SP-A (0–120 μg/ml) at 37 °C. Fluorescence emission was monitored at 520 nm for 30 min. These experiments were performed exactly as described previously for KD determination for Re-LPS–LBP and Re-LPS–CD14 complexes [2]. On the other hand, kinetics of FITC-Re-LPS binding to either LBP or CD14 was studied in the absence and presence of SP-A to determine whether SP-A hindered the binding of LBP and CD14 to FITC-Re-LPS.
Fluorescence emission anisotropy measurements of FITC-Re-LPS (4×10−7 M) in the absence and presence of one among SP-A, LBP and CD14 were obtained with Glan Prism polarizers. Excitation wavelength λx and emission wavelength λm were set at 470 and 520 nm respectively. For each sample, fluorescence emission intensity data in parallel and perpendicular orientations with respect to the exciting beam were collected ten times each and then averaged. Anisotropy r was calculated as
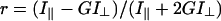 |
(1) |
where I∥ and I⊥ are the parallel and perpendicular polarized intensities measured with the vertically polarized excitation light and G is the monochromator grating correction factor.
125I-ASD-Re-LPS cross-linking assays
SP-A, LBP, CD14 and combinations were preincubated for 15 min at 37 °C in darkness with 2×10−7 M 125I-ASD-Re-LPS in a buffer (150 mM NaCl and 25 mM Tris/HCl, pH 7.4) containing either 2.5 mM EDTA or 0.5 mM Ca2+. Afterwards, samples were irradiated with UV-light for 5 min at 4 °C for photolysis as described elsewhere [2,28]. The proteins were then separated by one-dimensional SDS/PAGE (12% acrylamide separating gel) under reducing conditions (5% 2-mercaptoethanol). The 125I-labelled proteins were detected by autoradiography after Coomassie Blue staining to detect protein molecular-mass standards.
Sedimentation analysis of [3H]Rb-LPS (Rb mutant of LPS from E. coli strain LCD 25)
[3H]Rb-LPS (10−7 M) and human SP-A (0–0.3 μM) were incubated in a buffer (150 mM NaCl and 25 mM Tris/HCl, pH 7.4) containing either 2 mM Ca2+ or 2 mM EDTA, for 45 min at 37 °C. The LPS–SP-A complexes were chilled to 4 °C before 0.2 ml were layered on the top of a linear 5–50% sucrose gradient (4 ml) in a buffer (150 mM NaCl and 25 mM Tris/HCl, pH 7.4) containing either 2 mM Ca2+ or 2 mM EDTA, layered on to 0.25 ml of 70% sucrose. The gradients were centrifuged for 1 h 45 min at 4 °C in a prechilled TV-865 rotor (DuPont, Burbank, CA, U.S.A.) at 50000 rev./min, to improve resolution of small LPS aggregates formed in the presence of EDTA, or at 30000 rev./min for 30 min, to improve the resolution of large LPS aggregates formed in the presence of CaCl2. Fractions of 350 μl were collected and the amount of [3H]Rb-LPS was determined by liquid-scintillation counting. In addition, aliquots of the samples were removed before centrifugation to determine the total radioactive material for calculation of recovery. Typically, recovery of radioactive material from the gradient was 65–90% as described previously [2].
LPS aggregation assays
LPS aggregation induced by SP-A was studied at 37 °C by measuring the change in turbidity at 400 nm in a Beckman DU-640 spectrophotometer as described elsewhere [16–18]. The potential contribution of SP-A self-association to the light absorption at 400 nm was routinely checked under the experimental conditions in which Re-LPS aggregation assays were performed [16–18]. The calcium requirement for Re-LPS aggregation induced by SP-A was studied by titration experiments [29,30] in which increased amounts of a concentrated solution of CaCl2 were added to the protein solution in the presence of Re-LPS. The assay buffer consisted of 5 mM Tris/HCl, 150 mM NaCl and 150 μM EDTA (pH 7.4). The free-Ca2+ concentration at each point of the titration experiment was estimated by a computer program as described elsewhere [29]. The turbidity at 400 nm was registered once stabilized after each change of Ca2+ concentration.
Cell assays
Human monocyte-like U937 cells were grown in RPMI 1640 supplemented with 10% (v/v) heat-inactivated FBS, 2 mM glutamine and penicillin G sodium (100 units/ml)/streptomycin sulphate (100 μg/ml), and 0.25 μg/ml amphotericin B under a 95% air/5% CO2 humidified atmosphere at 37 °C. U937 cells were dispensed into 24-well plates at 1×106 cells/ml and differentiated into macrophages by incubation with 10 nM PMA for 24 h at 37 °C in a 5% CO2 humidified atmosphere. Then, the adhered cells were washed with the medium and, 24 h later, washed and pretreated with increasing concentrations (1–20 μg/ml) of SP-A for 10 min before 4 h Re-LPS (4×10−8 M) or smooth LPS (5×10−8 M) stimulation in the presence of 5% heat-inactivated FBS at 37 °C. Cell viability was confirmed by Trypan Blue exclusion.
For measurement of TNF-α production, cell-free culture supernatants were collected and assayed for TNF-α with an enzyme-linked immunoassay kit. Three different cell cultures were used (n=3). The assays from each U937 cell culture were performed in triplicate, the triplicate values were averaged, and their mean treated as a single point. The results are presented as the means (±S.E.M.) obtained by combining the results from each cell preparation. Results were expressed as a percentage of the level of TNF-α production by cells stimulated with LPS in the absence of SP-A. For statistical analysis, mean comparison between groups was done by one-way ANOVA followed by Bonferroni post hoc analysis; a confidence level of 95% or greater (P<0.05) was considered significant.
RESULTS
LPS and lipid A, as amphiphilic molecules, form aggregates in aqueous environments above a critical concentration (CMC). Below this value, LPS molecules are present only as monomers. Above CMC, monomers are in equilibrium with micelles (‘micelles’ as synonym for amphiphilic aggregates). At higher concentrations of LPS, the amphiphilic aggregates form nearly spherical supramolecular aggregate structures, which can be multilamellar or non-lamellar depending on the physicochemical environment [27].
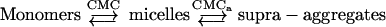 |
The CMC for Re-LPS, determined experimentally by applying equilibrium dialysis, is 3.3×10−8 M at 22 °C and 2.8×10−8 M at 37 °C [31]. CMCa can be defined as a transition value from aggregate to supra-aggregate structures. Using fluorescence spectroscopy of bis-ANS, we found that the CMCa value for Re-LPS from S. minnesota was 3.2×10−6 M (8 μg/ml). This is in accordance with the CMCa value of 14 μg/ml for LPS from E. coli (serotype 026:B6) obtained by dynamic light-scattering spectroscopy [32]. The molecular mass of the supramolecular aggregates above CMCa is approximately three times larger than those LPS aggregates below CMCa [32]. Bivalent cations stabilize LPS aggregates by forming bridges among phosphate groups located in the 1 and 4′ positions of the diglucosamine backbone and in the first KDO of LPS [27].
SP-A binds to Re-LPS monomers or Re-LPS micelles in a Ca2+-independent manner
The binding of SP-A to rough LPS in solution was followed by changes in FITC-Re-LPS fluorescent properties, such as intensity and anisotropy, in comparison with those changes produced by LBP and CD14 as representative LPS-binding proteins (Figure 1). FITC-Re-LPS has been widely used to study the interaction of Re-LPS with LBP, CD14 and MD2 [2,25,26,33].
Figure 1. Ca2+-independent binding of SP-A to Re-LPS monitored by changes in FITC-Re-LPS fluorescence.

(A, B) Fluorescence excitation (λm=520 nm) and emission (λx=470 nm) spectra of 4×10−7 M FITC-Re-LPS in the absence (a) and presence (b) of human SP-A (76.9 nM) in a buffer containing EDTA. (C) Human SP-A (76.9 nM) (b) (black bar), CD14 (72.7 nM) (c) (striped bar) and LBP (23.3 nM) (d) (open bar) induce a significant increase in fluorescence anisotropy upon binding to 4×10−7 M FITC-Re-LPS (a) (shaded bar) in a buffer containing either 1 mM EDTA or 1 mM Ca2+. All data points are the means±S.D. for experiments performed in triplicate. *P<0.01 versus control.
Figures 1(A) and 1(B) show the fluorescence excitation and emission spectra respectively of 4×10−7 M FITC-Re-LPS in the absence and presence of SP-A. Addition of SP-A in the presence of 1 mM EDTA produced an increase of total fluorescence excitation and emission intensities (Figures 1A and 1B) and a red-shift of the emission maximum of FITC-Re-LPS from 516 to 518 nm (Figure 1B). CD14 and LBP also produced an increase of fluorescence intensity as reported previously [2,25,26] (results not shown). These results indicated that the binding of any of these proteins to FITC-Re-LPS generated a different environment of the dye relative to that in the absence of bound protein. In order to confirm that the changes observed in FITC-Re-LPS fluorescence properties in the presence of SP-A, CD14 and LBP were due to the binding of these proteins to the Re-LPS moiety, but not due to the fluorescent moiety of the labelled LPS, we studied the potential interaction between these proteins and fluorescein. The addition of SP-A, CD14 or LBP to fluorescein did not affect the fluorescence spectrum of the dye (results not shown).
We also examined the effect of SP-A, CD14 and LBP on the fluorescence anisotropy of FITC-Re-LPS and free fluorescein (Figure 1C). FITC-Re-LPS (4×10−7 M) possessed an emission anisotropy r of approx. 0.067±0.003 in the presence of 1 mM EDTA and 0.084±0.006 in the presence of 1 mM Ca2+ in a buffer containing 5 mM Tris/HCl and 100 mM NaCl (pH 8), at 25 °C (λx=470 nm; λm=520 nm). The formation of complexes between FITC-Re-LPS and one among SP-A, CD14 and LBP led to an increase in fluorescence emission anisotropy, indicating that the binding of these proteins to Re-LPS caused mechanical restrictions in the rotational mobility of the dye. Control experiments with free fluorescein were performed to determine whether the protein-induced changes in FITC-Re-LPS emission anisotropy were due to the specific binding of these proteins to the LPS moiety or due to the fluorescent probe. The fluorescence emission anisotropy of free fluorescein was very low and was not affected by addition of 3-fold excess of any of these proteins (results not shown).
We then measured the time dependence of the fluorescence change when 2×10−8 M FITC-Re-LPS reacted with increasing amounts of SP-A in 150 mM NaCl, 2 mM EDTA and 25 mM Tris/HCl buffer (pH 7.4) at 37 °C. The magnitude of the fluorescence change increased as a function of SP-A concentration and was saturable (results not shown). The apparent equilibrium dissociation constant (KD) for FITC-Re-LPS–SP-A complexes, calculated from the saturation curve fitted to a rectangular hyperbola, was (2.8±0.6)×10−8 M (n=3) considering a molecular mass of 650 kDa for octadecameric human SP-A. Using the same methodology and the same ionic conditions, KD values for FITC-Re-LPS–LBP and FITC-Re-LPS–sCD14 complexes are 3.5×10−9 and 2.9×10−8 M respectively [2]. These data indicate that octadecameric human SP-A binds to Re-LPS as well as sCD14 and approx. 10-fold less tightly than LBP.
SP-A enhances the sedimentation velocity of [3H]Rb-LPS micelles
We used sucrose density gradients to analyse the state of Re-LPS upon binding to human SP-A. Figure 2 shows the sedimentation measurements of [3H]Rb-LPS (10−7 M) in the absence and presence of SP-A. The binding of SP-A to [3H]Rb-LPS did not have any effect on the sedimentation velocity of [3H]Rb-LPS in the presence of EDTA. The presence of calcium would enhance and stabilize [3H]Rb-LPS micelles. Figure 2 shows that, in the presence of calcium, Rb-LPS micelles floated to low density and that SP-A greatly increased the sedimentation velocity of [3H]Rb-LPS micelles. This indicates the formation of Rb-LPS–SP-A complexes with high size and density. Results indicated that, at SP-A concentrations higher than 10 μg/ml, SP-A induced an increase in the sedimentation velocity of [3H]Rb-LPS micelles in a concentration-dependent manner (results not shown). This is consistent with the Ca2+-dependent activity of SP-A to induce aggregation of rough LPS micelles [13,16–18].
Figure 2. SP-A enhances the sedimentation velocity of [3H]Rb-LPS micelles in the presence of calcium.

[3H]Rb-LPS (10−7 M) was preincubated with or without SP-A (195 μg/ml) for 45 min at 37 °C in the presence of either 2 mM Ca2+ or 2 mM EDTA. Afterwards, linear (5–50%) sucrose density gradients were performed as described in the Experimental section. Sedimentation direction is from right to left. The radioactivity of each fraction is presented as percentage of total recovered radioactivity.
Characteristics of Re-LPS aggregation induced by SP-A
The process of Re-LPS aggregation induced by SP-A in the presence of calcium was further analysed by measuring changes in turbidity at 400 nm. These experiments were carried out under the same ionic conditions as the sedimentation studies, except that concentrations of Re-LPS 50–100 times higher were needed to produce detectable light absorption at 400 nm. In these experiments, turbidity at 400 nm is continuously monitored as a function of time to measure changes in particle size as a consequence of SP-A-induced Re-LPS aggregation. Given that SP-A self-associates in the presence of calcium [30], control experiments to determine the potential contribution of SP-A self-association to the turbidity change at 400 nm were routinely performed.
Figure 3(A) shows that the minimal concentration of Re-LPS to produce detectable aggregation by this method was 5 μg/ml (2×10−6 M), showing a saturation effect at 20 μg/ml (8×10−6 M) at an SP-A concentration of 10 μg/ml. At this SP-A concentration, Ca2+-dependent SP-A self-association is hardly detected by measuring changes in turbidity at 400 nm. Figure 3(B) shows that SP-A triggered off aggregation of Re-LPS micelles in a dose-dependent manner. The concentration of SP-A required for half-maximal Re-LPS aggregation, calculated from the saturation curve fitted by non-linear regression, was in the nanomolar range (9.2 nM). This suggests that calcium might increase the binding affinity of SP-A to Re-LPS. Figure 3(C) shows that the Ca2+ concentration required for half-maximal Re-LPS aggregation at physiological ionic strength was approx. 150±60 μM (n=3). In the absence of NaCl, the threshold concentration of Ca2+ required to induce Re-LPS aggregation was 0.5 mM (results not shown). The calcium requirement for this process is similar to that calculated for human SP-A self-association in the presence and absence of salts [30]. Both processes, SP-A-induced Re-LPS aggregation and Ca2+-dependent SP-A self-association, were completely reversed by the addition of EDTA [16–18,30].
Figure 3. Characteristics of Re-LPS aggregation induced by SP-A.

(A) Extent of SP-A-induced Re-LPS aggregation as a function of Re-LPS concentration. Different kinetic assays were performed in which increasing amounts of Re-LPS were added to both the sample and reference cuvette in 5 mM Tris/HCl buffer (pH 7.4), containing 150 mM NaCl and 0.2 mM EDTA at 37 °C. After 10 min equilibration at 37 °C, 10 μg/ml SP-A was added to the sample cuvette in each experiment. Re-LPS aggregation induced by SP-A started after the addition of 2.5 mM calcium to both the sample and reference cuvette. The contribution of self-association of SP-A (10 μg/ml) to the turbidity change at 400 nm was negligible. (B) Extent of SP-A-induced Re-LPS aggregation as a function of SP-A concentration. Different kinetic assays were performed as described in (A), but increasing amounts of SP-A were added to both the sample and reference cuvettes to eliminate the contribution of self-association of SP-A to the light absorption change at 400 nm. Re-LPS aggregation induced by SP-A started after the addition of 4×10−6 M Re-LPS (20 μg/ml) to the sample cuvette and Ca2+ (2.5 mM) to both the sample and reference cuvettes. (C) Ca2+ dependence of SP-A-induced Re-LPS aggregation. Titration experiments were performed at 37 °C as described in the Experimental section. (A–C) Results of one representative experiment from three experiments performed are shown.
Re-LPS aggregation induced by SP-A was inhibited by polymyxin-B but not by mannan or by acidic pH (results not shown). At acidic pH, the lectin activity of SP-A and other collectins such as MBP or conglutinin, is abrogated [34]. These results indicate that the lectin activity of the protein is not involved in the aggregation of Re-LPS micelles in the presence of calcium.
A structurally intact collagen-like domain is needed for Ca2+-dependent SP-A self-association [30]. That the folding of the collagen domain might also play an important role in Re-LPS aggregation was inferred from the finding that Re-LPS aggregation induced by recombinant human SP-A2 and co-expressed SP-A1/SP-A2, produced in insect cells, was inhibited at 37 °C by unfolding of the collagen-like domain of these recombinant proteins [18]. These insect cell-derived recombinant human SP-A lack proline hydroxylation, showing reduced thermal stability of the collagen domain, with Tm values around 33 °C [18]. The requirement of a folded collagen-like domain for Re-LPS aggregation induced by human SP-A was also analysed here. Figure 4, left-hand panel, shows kinetics of Re-LPS aggregation at 37 °C, in which human SP-A was previously heated at the indicated temperature for 10 min and then cooled to 37 °C before assessment of Re-LPS aggregation. The process was partially inhibited when SP-A was preheated over 47 °C. Figure 4, right-hand panel, shows the thermal denaturation of the collagen triple helix of human SP-A, monitored by CD spectroscopy and the renaturation profile when the temperature was cooling down from 70 to 20 °C. This experiment indicates that partial renaturation occurred after cooling to 37 °C, which might explain why SP-A maintained some of its ability to induce Re-LPS aggregation after preheating at 65 °C and cooling down to 37 °C. On the other hand, the extent of rough LPS aggregation induced by SP-A also depended on the rigidification of the acyl chains of Re-LPS, as observed for SP-A-induced phospholipid vesicle aggregation [24]. The extent of rough LPS aggregation was higher below the (β↔α) phase-transition temperature for the acyl chains of Re-LPS from S. minnesota (around 33 °C) [27] than above it (results not shown).
Figure 4. Effects of partial denaturation of the SP-A collagen-like domain on the ability of SP-A to induce Re-LPS aggregation.

In the left panel, SP-A-induced Re-LPS aggregation is shown as described in the text, but SP-A was preheated at the indicated temperatures before being added to the sample cuvette (10 μg/ml). After a 10 min equilibration at 37 °C, Ca2+ (2.5 mM) was added to the sample and reference cuvettes, and the change in absorbance at 400 nm was monitored. In the right panel, unfolding and refolding curves of human SP-A are shown. F was calculated as described in the Experimental section. The results shown are from a representative experiment of three experiments performed.
SP-A inhibits TNF-α production by macrophages stimulated with either rough or smooth LPS
To investigate whether human SP-A stimulates or decreases the inflammatory response to LPS, increasing amounts (0–10 μg/ml) of SP-A were preincubated with human macrophage-like cells 10 min before 4 h activation with an optimal concentration of either rough LPS (4×10−8 M) or smooth LPS (5×10−8 M) (Figure 5). SP-A alone (without LPS) had no effect on TNF-α production by resting differentiated U937 cells for 4 h after SP-A addition (results not shown), which is consistent with several studies [15,16,22]. Human SP-A showed a clear inhibitory effect upon rough or smooth LPS-induced TNF-α secretion, which was dependent upon the SP-A concentration. This is in line with previous studies that demonstrate that pretreatment of alveolar macrophages with recombinant human SP-A1 expressed in mammalian cells [16] or natural human SP-A [23] causes a marked inhibition of LPS-induced responses independent of the LPS chemotype. Given that SP-A hardly binds and aggregates smooth LPS, the anti-inflammatory effect of SP-A must be mediated by blocking the binding of LPS to its receptor complex and/or by direct SP-A modulation of signal pathways that decrease pro-inflammatory cytokine production.
Figure 5. SP-A inhibits TNF-α production by macrophages stimulated with either rough or smooth LPS.

Differentiated macrophage-like U937 cells (1×106 cells/ml) were preincubated in the absence or presence of various concentrations of human SP-A for 10 min before 4 h activation with either 4×10−8 M rough LPS (black bars) or 5×10−8 M smooth LPS (open bars) in the presence of 5% FBS at 37 °C. Cell-free supernatants were collected at 4 h after LPS treatment and the levels of TNF-α were measured by ELISA. The results are presented as the means (±S.E.M.) from three different cell cultures. Results were expressed as percentage of LPS-induced TNF-α levels. *P<0.05 compared with response elicited by LPS in the absence of SP-A.
SP-A does not inhibit the binding of LBP to Re-LPS
To investigate whether SP-A interferes in the binding of LBP to Re-LPS, we used a radioactive photoactivatable derivative of Re-595 LPS (125I-ASD-Re-LPS). LBPs are detected by covalent attachment of the radiolabelled group to the protein after photolysis [2,33]. Figure 6(A) shows the results of a photoaffinity labelling experiment in which photoactivatable LPS was incubated with LBP in the absence and presence of various concentrations of SP-A at 37 °C. SP-A did not inhibit the cross-linking of photoactivatable rough LPS with LBP, at the molar ratios tested, in the presence of either 0.5 mM Ca2+ or 2 mM EDTA. In addition, preincubation of SP-A with 125I-ASD-Re-LPS in the presence of calcium had no effect on the subsequent binding of LBP to the labelled LPS (results not shown).
Figure 6. SP-A does not inhibit the binding of LBP to Re-LPS.

(A) LBP (0.7 μg) was preincubated with increasing amounts of SP-A (0–30 μg) in a buffer containing 2 mM EDTA at 37 °C. Afterwards, proteins were incubated with 20 ng of 125I-ASD-Re-LPS for 15 min at 37 °C in 50 μl of buffer and photolysed. The labelled proteins were then separated by SDS/PAGE (12% acrylamide separating gel) under reducing conditions and detected by autoradiography after Coomassie Blue staining. (B) Time dependence of the fluorescence change when 2×10−8 M FITC-Re-LPS (0.05 μg/ml) reacts with either 0.08 μM LBP alone (black line) or first with 0.09 μM human SP-A and then with 0.08 μM LBP (grey line) at 37 °C. Fluorescence emission intensity was monitored at 520 nm. The net change in fluorescence emission intensity after the addition of LBP to FITC-Re-LPS preincubated in the absence or presence of SP-A is indicated. Similar experiments were performed with different SP-A:Re-LPS molar ratios (from 11.5:1 to 0.25:1) with similar results.
To corroborate these results, we next studied whether the preincubation of SP-A with FITC-Re-LPS affected the subsequent binding of LBP to FITC-Re-LPS (Figure 6B). To this end, the fluorescence emission of FITC-Re-LPS was monitored both after preincubation of Re-LPS with different amounts of SP-A for 25 min and after the subsequent addition of LBP. Figure 6(B) clearly indicates that LBP bound to the complexes formed from Re-LPS and SP-A. Moreover, addition of LBP produced a similar increase in fluorescence when FITC-Re-LPS had been preincubated in the absence or presence of SP-A. Together, these results indicate that SP-A did not inhibit the binding of LBP to Re-LPS.
SP-A decreases the binding of CD14 to Re-LPS
We next examine whether SP-A affects the interaction of CD14 with Re-LPS. Figure 7(A) shows a photoaffinity labelling experiment in which photoactivatable LPS was incubated with CD14 in the absence and presence of various concentrations of SP-A at 37 °C. The cross-linking of CD14 with photoactivatable rough LPS decreased when the concentration of SP-A increased. Identical results were found in the presence of either 0.5 mM Ca2+ or 2 mM EDTA.
Figure 7. SP-A decreases the binding of CD14 to Re-LPS.

(A) CD14 (2 μg) was preincubated with increasing amounts of SP-A (0–40 μg) in a buffer containing 2 mM EDTA at 37 °C. Afterwards, proteins were incubated with 20 ng of 125I-ASD-Re-LPS for 15 min at 37 °C in 50 μl of buffer and photolysed. The labelled proteins were then separated as described in Figure 6. When the buffer contained 0.5 mM Ca2+ instead of EDTA, similar results were obtained. (B) Time dependence of the fluorescence change when 2×10−7 M FITC-Re-LPS was incubated in the absence (line E) or presence of 0.075 μM CD14 alone (line A), 0.075 μM CD14+0.03 μM SP-A (line B), 0.075 μM CD14+1 μg/ml of an anti-CD14 antibody (28C5) (line C) and 0.03 μM SP-A alone (line D) in a buffer containing 1 mM EDTA at 37 °C.
To confirm that SP-A decreased the binding of CD14 to Re-LPS in solution, we examined the time dependence of the fluorescence change when FITC-Re-LPS was incubated in the absence (Figure 7B, line E) or presence of CD14 alone (line A), CD14+SP-A (line B), CD14+1 μg/ml 28C5, an anti-CD14 antibody that has been shown to block Re-LPS binding to CD14 [25] (line C) and SP-A alone (line D) at 37 °C. Figure 7(B) shows that CD14 preincubated with SP-A binds to FITC-Re-LPS to a significantly lesser extent than CD14 alone.
Our findings are quite different from the results of Sano et al. [15], who observed that the apparent binding of CD14 to Re-LPS augmented in the presence of SP-A. The differences between the two studies may depend on the different experimental approaches used to determine binding of Re-LPS to CD14. In Sano et al. [15], solid phase-binding assays were used. The effect of SP-A on the binding of CD14 to Re-LPS coated on to microtitre wells was detected by an anti-CD14 polyclonal antibody. It is possible that CD14-SP-A complexes could bind to immobilized LPS via SP-A rather than via CD-14, given that SP-A binds to Re-LPS by the globular domain [21] and to CD14 through the neck domain [21]. The different structural domain topology of the binding of SP-A to its ligands (either CD14 or Re-LPS) enables the protein to interact with both ligands. We do not know which CD14 domain SP-A recognizes, nor whether this domain is the same as that recognized by Re-LPS. However, our LPS-binding studies in fluid phase suggest that SP-A blocks the LPS-binding site on CD14.
SP-A reduces the ability of LBP to transfer LPS to CD14
Figure 8 shows experiments with radioactive photoactivatable Re-LPS in the presence of combinations of LBP, CD14 and SP-A. Lanes 1, 2 and 7 show the cross-linking of Re-LPS with one among CD14, SP-A and LBP respectively when these proteins were incubated alone. When SP-A and LPB were incubated together, SP-A did not affect the binding of LBP to Re-LPS, but the labelling of SP-A was enhanced by LBP (comparison of lanes 1, 6 and 7). As reported above, SP-A decreased the cross-linking of Re-LPS with CD14 (lanes 2 and 3) and LBP did the opposite, i.e. catalysed the binding of Re-LPS to CD14 (lanes 2 and 5). When the three proteins were incubated together, SP-A reduced the catalysing activity of LBP to transfer LPS to CD14 (lanes 4 and 5). Photoaffinity labelling experiments were performed in the absence and presence of Ca2+ with identical results. Together, these results indicate that SP-A decreases the binding of CD14 to Re-LPS in the absence or presence of LBP.
Figure 8. SP-A reduces the ability of LBP to transfer LPS to CD14.

SP-A (20 μg), LBP (0.7 μg), CD14 (2 μg) and combinations were preincubated for 15 min at 37 °C with 11 ng of 125I-ASD-Re-LPS in 25 μl of a buffer containing 2 mM EDTA. Afterwards, samples were irradiated with UV-light for 5 min at 4 °C for photolysis. The labelled proteins were then separated by SDS/PAGE (12% acrylamide separating gel) and identified by autoradiography. When the buffer contained Ca2+ instead of EDTA, similar results were obtained. Numbers on the right denote molecular-mass standards. SP-Ad and SP-Am denote dimers and monomers of SP-A respectively.
DISCUSSION
Pathogen-derived components such as LPS, peptidoglycan or zymosan are potent stimulators of inflammation. SP-A does not bind to smooth LPS, peptidoglycan or zymosan [10]. In contrast, SP-A binds to rough LPS, which contains fewer sugar residues than smooth LPS because of the absence of the O domain [13,14]. The affinity of rough LPS–SP-A interaction is still unknown. In addition, there is no consensus on whether rough LPS–SP-A binding is calcium-dependent [13,14,21]. The present study documents that SP-A was able to bind to FITC-Re-LPS monomers in solution, in a calcium-independent manner, with a KD of 2.8×10−8 M. Comparison with KD values for FITC-Re-LPS–LBP and FITC-Re-LPS–CD14 complexes, determined under the same conditions and methodology [2], indicates that the affinity of octadecameric human SP-A for Re-LPS appears to be similar to that of soluble CD14, but approx. 10-fold less than LBP.
The fact that SP-A–Re-LPS binding is Ca2+-independent, and that mannan has no inhibitory effect on SP-A binding to Re-LPS in the presence of calcium [13], strongly supports the concept that the phosphorylated diglucosamine backbone of Re-LPS or lipid A interacts with an SP-A site different from the carbohydrate-binding domain. The recent determination of the crystal structure of the trimeric carbohydrate recognition domain and neck domain of SP-A [35] has provided new insights in understanding the potential mode of SP-A interaction with lipids such as DPPC or Re-LPS. These two SP-A ligands are structurally related. It is conceivable that some of the acyl chains of Re-LPS or DPPC could fit in the apolar pocket formed by four tyrosine side chains (Tyr161, Tyr164, Tyr208 and Tyr192) that belong to a discontinuous beta sheet structure in the globular domain of SP-A. In addition, numerous electrostatic interactions can be formed between the polar moieties of Re-LPS or DPPC and a cluster of basic or amine side chains on SP-A created by Arg216, Arg222 and Gln220 [35]. BPI (bactericidal permeability-increasing protein), another LPS-binding protein produced by neutrophils that is a powerful bactericidal for Gram-negative bacteria, also contains apolar pockets that bind phosphatidylcholine or the lipid A region of LPS [36]. It is believed that the phospholipid molecule binds to BPI with its acyl chain buried in the BPI apolar pocket whereas the head group lies at the opening of the pocket and is exposed to the solvent. The interaction of SP-A with Gram-negative bacteria that express rough LPS phenotype also causes increased bacterial permeability [37], presumably by a direct effect of SP-A on the physical properties of the microbial cell membrane. The effect of SP-A interaction on the structure and physical properties of Re-LPS membranes remains to be determined.
In the presence of Ca2+, SP-A induced Re-LPS aggregation in a dose-dependent manner. This process was inhibited by polymyxin-B but not by mannan or low pH, indicating that the lectin activity of the protein is not implicated. The mechanism involved in the Re-LPS aggregation phenomenon is poorly understood. We hypothesize that SP-A-induced Re-LPS aggregation can be mediated by Ca2+-self-association of the protein. This hypothesis is based on several observations: (i) both processes were reversed by the addition of EDTA; (ii) the calcium activation constant (KaCa2+) for SP-A-induced LPS aggregation (Figure 3C) was similar to that for human SP-A self-association [30]; (iii) both processes were inhibited by the unfolding of SP-A collagen-like domain (Figure 4 of this paper and [17,30] for SP-A self-association); and (iv) the supratrimeric assembly of human SP-A is essential for both protein self-association and SP-A-induction of Re-LPS aggregation [16]. Thus trimers of SP-A cannot either self-associate or induce aggregation of Re-LPS. These results strongly suggest that the ability of human SP-A to induce Re-LPS correlates with the ability of SP-A to self-associate in the presence of calcium.
Calcium causes conformational changes in the globular domain of SP-A [16–18,30,38]. Protein association might occur among adjacent globular heads and nearby regions provided that a supratrimeric assembly and a structurally intact collagen domain ensure the correct grouping and orientation of globular heads in the oligomer [16]. Self-associated SP-A could bind to the surface of Re-LPS aggregates or bacteria by its globular heads. This would result in bacterial aggregation while the SP-A collagen tails of these aggregates would be free to interact with receptors on the surface of macrophages initiating phagocytosis. It is thought that significant self-association of SP-A is required for the optimal stimulation of macropinocytosis induced by interaction of SP-A collagen tails with the calreticulin/CD91 system on the surface of phagocytic cells [20]. The α2 macroglobulin receptor CD91 interacts with the cell-surface calreticulin, which is known as a receptor for the collagen-like domain of C1q, which is structurally similar to SP-A [9,10].
The large excess of SP-A in the lung air-spaces probably minimizes the biological effects of LPS or microbes that enter the alveolus. Aggregation of rough LPS could be important to reduce LPS toxicity [31] and facilitate phagocytosis of large aggregates, preventing adherence of endotoxins to the alveolar epithelium. SP-A-induced aggregation of Gram-negative bacteria that express the rough LPS phenotype can also aid phagocytosis of bacteria [9,10], affect microbial physiology by inducing bacteria to down-regulate microbial synthetic functions, and favour or potentiate SP-A bactericidal activity [37].
SP-A is also involved in inflammatory immunomodulator processes of the lung. Consistent with previous results [16,23] we found that human SP-A inhibited activation of human macrophage-like U937 cells by either Re-LPS or smooth LPS in a dose-dependent manner. Because SP-A does not bind to smooth LPS, the fact that SP-A inhibits smooth or rough LPS inflammatory response suggests a direct anti-inflammatory effect of SP-A by binding to cell receptors or to soluble accessory molecules present in serum or in the alveolar fluid. In the present study, we showed that SP-A inhibited the binding of Re-LPS to CD14, but not to LBP. The cross-linking of CD14 with photoactivatable 125I-ASD-Re-LPS decreased when the concentration of SP-A increased. Because the hydrophobic region of the SP-A neck domain interacts with CD14 [21], these results suggest that the binding of SP-A to CD14 inhibits the subsequent binding of CD14 to Re-LPS. Alternatively, these results might be explained by the ability of SP-A to aggregate Re-LPS in the presence of calcium, which could block the subsequent binding of CD14 to Re-LPS–SP-A aggregates. However, this possibility must be ruled out because the SP-A-mediated inhibition of the cross-linking between CD14 and 125I-ASD-Re-LPS was observed both in the absence and presence of calcium. While the interaction of SP-A with CD14 does not require calcium [21], Re-LPS aggregation induced by SP-A does.
The present study also shows that when SP-A, CD14 and LBP were incubated together with photoactivatable 125I-ASD-Re-LPS, SP-A reduced the catalysing activity of LBP to transfer LPS to CD14 in a calcium-independent manner. These results suggest that SP-A might partly inhibit Re-LPS inflammatory response by altering the competence of the LBP–CD14 receptor complex. This mechanism, which has already been suggested to explain the inhibitory effect of SP-A on smooth LPS inflammatory response [15,21], could also explicate the inhibitory effect of SP-A on rough LPS responses. SP-A-mediated alteration of the coupling of the LBP–CD14 receptor complex might be one of the protective mechanisms that help to maintain the resting lung in a quiet, non-inflamed state. SP-A also inhibits inflammatory cytokine production in a CD14-independent manner [20]. A mechanism independent of the LPS signalling pathway involves SP-A stimulation of SIRPα (signal-inhibitory regulatory protein α) on alveolar macrophages through their globular heads. SIRPα activation by SP-A might suppress the inflammatory response induced by LPS and other pro-inflammatory stimuli present in the oxygen-rich environment of this highly exposed organ.
In conclusion, the present study provides evidence that SP-A binds Re-LPS in a Ca2+-independent manner and promotes Re-LPS aggregation in the presence of Ca2+ by a mechanism independent of SP-A lectin activity. SP-A-induced Re-LPS aggregation seems to be mediated by Ca2+-dependent self-association of the protein. On the other hand, SP-A might partly inhibit Re-LPS inflammatory response by altering the competence of the LBP–CD14 receptor complex, since SP-A decreases the binding of CD14 to Re-LPS and reduces LBP activity to transfer LPS to CD14. These SP-A effects are independent of the ability of SP-A to aggregate Re-LPS.
Acknowledgments
This work was supported by the European Community grant QLK2-CT-2000-00325, Fondo de Investigaciones Sanitarias (Instituto de Salud Carlos III, Ministerio de Sanidad y Consumo, Madrid, Spain) grant PI03/0137 and National Institutes of Health (Bethesda, MD, U.S.A.) grant GM066119.
References
- 1.Alexander C., Rietschel E. T. Bacterial lipopolysaccharides and innate immunity. J. Endotoxin Res. 2001;7:167–202. [PubMed] [Google Scholar]
- 2.Tobias P. S., Soldau K., Gegner J. A., Mintz D., Ulevitch R. J. Lipopolysaccharide binding protein-mediated complexation of lipopolysaccharide with soluble CD14. J. Biol. Chem. 1995;270:10482–10488. doi: 10.1074/jbc.270.18.10482. [DOI] [PubMed] [Google Scholar]
- 3.Wright S. D., Ramos R. A., Tobias P. S., Ulevitch R. J., Mathison J. C. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science. 1990;249:1431–1433. doi: 10.1126/science.1698311. [DOI] [PubMed] [Google Scholar]
- 4.Ulevitch R. J., Tobias P. S. Recognition of Gram-negative bacteria and endotoxin by the innate immune system. Curr. Opin. Immunol. 1999;11:19–22. doi: 10.1016/s0952-7915(99)80004-1. [DOI] [PubMed] [Google Scholar]
- 5.Shimazu R., Akashi S., Ogata H., Nagai Y., Fukudome K., Miyake K., Kimoto M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 1999;189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beutler B. TLR4: central component of the sole mammalian LPS sensor. Curr. Opin. Immunol. 2000;12:20–26. doi: 10.1016/s0952-7915(99)00046-1. [DOI] [PubMed] [Google Scholar]
- 7.Dentener M. A., Vreugdenhil A. C., Hoet P. H., Vernooy J. H., Nieman F. H., Heumann D., Janssen Y. M., Buurman W. A., Wouters E. F. Production of the acute-phase protein lipopolysaccharide-binding protein by respiratory type II epithelial cells: implications for local defense to bacterial endotoxins. Am. J. Respir. Cell Mol. Biol. 2000;23:146–153. doi: 10.1165/ajrcmb.23.2.3855. [DOI] [PubMed] [Google Scholar]
- 8.Dubin W., Martin T. R., Swoveland P., Leturcq D. J., Moriarty A. M., Tobias P. S., Bleecker E. R., Goldblum S. E., Hasday J. D. Asthma and endotoxin: lipopolysaccharide-binding protein and soluble CD14 in bronchoalveolar compartment. Am. J. Physiol. 1996;270:L736–L744. doi: 10.1152/ajplung.1996.270.5.L736. [DOI] [PubMed] [Google Scholar]
- 9.Wright J. R. Immunomodulatory functions of surfactant proteins. Nat. Rev. Immunol. 2005;5:58–68. doi: 10.1038/nri1528. [DOI] [PubMed] [Google Scholar]
- 10.Sano H., Kuroki Y. The lung collectins, SP-A and SP-D, modulate pulmonary innate immunity. Mol. Immunol. 2005;42:279–287. doi: 10.1016/j.molimm.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 11.Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 2003;3:710–720. doi: 10.1038/nri1180. [DOI] [PubMed] [Google Scholar]
- 12.Nightingale J. A., Rogers D. F., Hart L. A., Kharitonov S. A., Chung K. F., Barnes P. J. Effect of inhaled endotoxin on induced sputum in normal, atopic, and atopic asthmatic subjects. Thorax. 1998;53:563–571. doi: 10.1136/thx.53.7.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Iwaarden J. F., Pikaar J. C., Storm J., Brouwer E., Verhoef J., Oosting R. S., van Golde L. M., van Strijp J. A. Binding of surfactant protein A to the lipid A moiety of bacterial lipopolysaccharides. Biochem. J. 1994;303:407–411. doi: 10.1042/bj3030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalina M., Blau H., Riklis S., Kravtsov V. Interaction of surfactant protein A with bacterial lipopolysaccharide may affect some biological functions. Am. J. Physiol. 1995;268:L144–L151. doi: 10.1152/ajplung.1995.268.1.L144. [DOI] [PubMed] [Google Scholar]
- 15.Sano H., Sohma H., Muta T., Nomura S., Voelker D. R., Kuroki Y. Pulmonary surfactant protein A modulates the cellular response to smooth and rough lipopolysaccharides by interaction with CD14. J. Immunol. 1999;163:387–395. [PubMed] [Google Scholar]
- 16.Sanchez-Barbero F., Strassner J., Garcia-Canero R., Steinhilber W., Casals C. Role of the degree of oligomerization in the structure and function of human surfactant protein A. J. Biol. Chem. 2005;280:7659–7670. doi: 10.1074/jbc.M410266200. [DOI] [PubMed] [Google Scholar]
- 17.Garcia-Verdugo I., Sanchez-Barbero F., Bosch F. U., Steinhilber W., Casals C. Effect of hydroxylation and N187-linked glycosylation on molecular and functional properties of recombinant human surfactant protein A. Biochemistry. 2003;42:9532–9542. doi: 10.1021/bi0347196. [DOI] [PubMed] [Google Scholar]
- 18.Garcia-Verdugo I., Wang G., Floros J., Casals C. Structural analysis and lipid-binding properties of recombinant human surfactant protein A derived from one or both genes. Biochemistry. 2002;41:14041–14053. doi: 10.1021/bi026540l. [DOI] [PubMed] [Google Scholar]
- 19.Arias-Diaz J., Garcia-Verdugo I., Casals C., Sanchez-Rico N., Vara E., Balibrea J. L. Effect of surfactant protein A (SP-A) on the production of cytokines by human pulmonary macrophages. Shock. 2000;14:300–306. doi: 10.1097/00024382-200014030-00010. [DOI] [PubMed] [Google Scholar]
- 20.Gardai S. J., Xiao Y. Q., Dickinson M., Nick J. A., Voelker D. R., Greene K. E., Henson P. M. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell (Cambridge, Mass.) 2003;115:13–23. doi: 10.1016/s0092-8674(03)00758-x. [DOI] [PubMed] [Google Scholar]
- 21.Sano H., Chiba H., Iwaki D., Sohma H., Voelker D. R., Kuroki Y. Surfactant proteins A and D bind CD14 by different mechanisms. J. Biol. Chem. 2000;275:22442–22451. doi: 10.1074/jbc.M001107200. [DOI] [PubMed] [Google Scholar]
- 22.Stamme C., Muller M., Hamann L., Gutsmann T., Seydel U. Surfactant protein A inhibits lipopolysaccharide-induced immune cell activation by preventing the interaction of lipopolysaccharide with lipopolysaccharide-binding protein. Am. J. Respir. Cell Mol. Biol. 2002;27:353–360. doi: 10.1165/rcmb.4812. [DOI] [PubMed] [Google Scholar]
- 23.Wu Y., Adam S., Hamann L., Heine H., Ulmer A. J., Buwitt-Beckmann U., Stamme C. Accumulation of inhibitory κB-α as a mechanism contributing to the anti-inflammatory effects of surfactant protein-A. Am. J. Respir. Cell Mol. Biol. 2004;31:587–594. doi: 10.1165/rcmb.2004-0003OC. [DOI] [PubMed] [Google Scholar]
- 24.Casals C., Miguel E., Perez-Gil J. Tryptophan fluorescence study on the interaction of pulmonary surfactant protein A with phospholipid vesicles. Biochem. J. 1993;296:585–593. doi: 10.1042/bj2960585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Viriyakosol S., Mathison J. C., Tobias P. S., Kirkland T. N. Structure-function analysis of CD14 as a soluble receptor for lipopolysaccharide. J. Biol. Chem. 2000;275:3144–3149. doi: 10.1074/jbc.275.5.3144. [DOI] [PubMed] [Google Scholar]
- 26.Tobias P. S., Soldau K., Iovine N. M., Elsbach P., Weiss J. Lipopolysaccharide (LPS)-binding proteins BPI and LBP form different types of complexes with LPS. J. Biol. Chem. 1997;272:18682–18685. doi: 10.1074/jbc.272.30.18682. [DOI] [PubMed] [Google Scholar]
- 27.Brandenburg K., Andra J., Muller M., Koch M. H., Garidel P. Physicochemical properties of bacterial glycopolymers in relation to bioactivity. Carbohydr. Res. 2003;338:2477–2489. doi: 10.1016/j.carres.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 28.Wollenweber H. W., Morrison D. C. Synthesis and biochemical characterization of a photoactivatable, iodinatable, cleavable bacterial lipopolysaccharide derivative. J. Biol. Chem. 1985;260:15068–15074. [PubMed] [Google Scholar]
- 29.Ruano M. L., Miguel E., Perez-Gil J., Casals C. Comparison of lipid aggregation and self-aggregation activities of pulmonary surfactant-associated protein A. Biochem. J. 1996;313:683–689. doi: 10.1042/bj3130683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruano M. L., Garcia-Verdugo I., Miguel E., Perez-Gil J., Casals C. Self-aggregation of surfactant protein A. Biochemistry. 2000;39:6529–6537. doi: 10.1021/bi000188z. [DOI] [PubMed] [Google Scholar]
- 31.Takayama K., Mitchell D. H., Din Z. Z., Mukerjee P., Li C., Coleman D. L. Monomeric Re lipopolysaccharide from Escherichia coli is more active than the aggregated form in the Limulus amebocyte lysate assay and in inducing Egr-1 mRNA in murine peritoneal macrophages. J. Biol. Chem. 1994;269:2241–2244. [PubMed] [Google Scholar]
- 32.Santos N. C., Silva A. C., Castanho M. A., Martins-Silva J., Saldanha C. Evaluation of lipopolysaccharide aggregation by light scattering spectroscopy. ChemBioChem. 2003;4:96–100. doi: 10.1002/cbic.200390020. [DOI] [PubMed] [Google Scholar]
- 33.Viriyakosol S., Tobias P. S., Kitchens R. L., Kirkland T. N. MD-2 binds to bacterial lipopolysaccharide. J. Biol. Chem. 2001;276:38044–38051. doi: 10.1074/jbc.M105228200. [DOI] [PubMed] [Google Scholar]
- 34.Haurum J. S., Thiel S., Haagsman H. P., Laursen S. B., Larsen B., Jensenius J. C. Studies on the carbohydrate-binding characteristics of human pulmonary surfactant-associated protein A and comparison with two other collectins: mannan-binding protein and conglutinin. Biochem. J. 1993;293:873–878. doi: 10.1042/bj2930873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Head J. F., Mealy T. R., McCormack F. X., Seaton B. A. Crystal structure of trimeric carbohydrate recognition and neck domains of surfactant protein A. J. Biol. Chem. 2003;278:43254–43260. doi: 10.1074/jbc.M305628200. [DOI] [PubMed] [Google Scholar]
- 36.Beamer L. J., Carroll S. F., Eisenberg D. Crystal structure of human BPI and two bound phospholipids at 2.4 angstrom resolution. Science. 1997;276:1861–1864. doi: 10.1126/science.276.5320.1861. [DOI] [PubMed] [Google Scholar]
- 37.Wu H., Kuzmenko A., Wan S., Schaffer L., Weiss A., Fisher J. H., Kim K. S., McCormack F. X. Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. J. Clin. Invest. 2003;111:1589–1602. doi: 10.1172/JCI16889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ridsdale R. A., Palaniyar N., Holterman C. E., Inchley K., Possmayer F., Harauz G. Cation-mediated conformational variants of surfactant protein A. Biochim. Biophys. Acta. 1999;1453:23–34. doi: 10.1016/s0925-4439(98)00057-x. [DOI] [PubMed] [Google Scholar]