

Abstract
Background
Rapid advancements in high-throughput sequencing technologies allow for detailed and accurate measurement of omics features within their biological context. The integration of different omics types creates heterogeneous datasets, presenting challenges in analysis due to variations in measurement units, sample numbers, and features. Currently, there is a lack of generalized guidelines for making decisions in multi-omics study design (MOSD), such as selecting an appropriate number of samples and features, type of preprocessing and integration for robust analysis results. We propose a suggestive guideline for MOSD, involving nine important factors: sample size, feature selection, preprocessing strategy, noise characterization, class balance, number of classes, cancer subtype combination, omics combination, and clinical features.
Results
To assess the effectiveness of our proposed MOSD guidelines, we designed and conducted seven benchmark tests using 10 clustering methods on various TCGA cancer datasets with an objective of clustering cancer subtypes. The results indicated robust performance in terms of cancer subtype discrimination when adhering to the following criteria: 26 or more samples per class, selecting less than 10% of omics features, maintaining a sample balance under a 3:1 ratio, and keeping the noise level below 30%. Feature selection was particularly important, improving clustering performance by 34%.
Conclusion
These findings provide evidence-based recommendations for MOSD, enabling researchers to optimize analytical approaches and enhance the reliability of results across cancer datasets. The proposed MOSD framework offers a suggestive guideline addressing both computational and biological factors for multi-omics data integration.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12864-025-11925-y.
Keywords: Multi-omics, Integration, Study design, Machine learning
Introduction
Recent advances in high-throughput sequencing and omics technologies have enabled the generation of extensive multi-omics data, proving valuable in identifying causal relationships for deeper understanding of biological functions [1–3]. Multi-omics integration (MOI) involves the simultaneous analysis of genomics, transcriptomics, epigenomics, proteomics, and metabolomics and has substantially advanced investigations into the molecular underpinnings of complex diseases such as cancer [4–8].
Various MOI tools have been developed for specific combinations of omics data, including IGC [9], PLRS [10], Oncodrive-CIS [11], MethylMix [12], MMIA [13], MethGET [14], and linkedOmics [15]. Tools, such as, MOGSA [16], ActivePathways [17], multiGSEA [18], and iPanda [19] have also emerged to facilitate the interpretation of complex multi-omics results. Large-scale multi-omics archives, such as, The Cancer Genome Atlas (TCGA) [20], International Cancer Genome Consortium (ICGC) [21], Cancer Cell Line Encyclopedia (CCLE) [22], and Clinical Proteomic Tumor Analysis Consortium (CPTAC) [23] provide extensive multi-omics data with annotated clinical labels across different cancer types. However, leveraging this integration effectively poses numerous analytical and computational challenges. From a computational perspective, harmonizing distinct data types requires specialized approaches, as different omics data types can have varying distributions and sources of noise [24]. For instance, transcript expression follows a binomial distribution, while CpG islands associated with methylation display a bimodal distribution [25]. Biologically, different omics layers may produce complementary but occasionally conflicting signals, as demonstrated in studies of colorectal carcinomas where methylation profiles were linked to genetic lineages defined by copy number alterations (CNAs), but transcriptional programs showed inconsistent connections to subclonal genetic identities [26].
Previous research has identified several critical challenges in multi-omics integration, including the curse of dimensionality, data heterogeneity, missing data, class imbalances, and scalability [27]. Various studies have investigated specific aspects of these challenges - from sample size and noise variation effects on clustering performance [28] to the importance of feature selection [29]. Although these studies offer valuable insights into components of multi-omics analysis, none provide a unified framework that systematically addresses both computational and biological complexities across a broad range of factors [24, 28–31]. The extension of MOI analysis to new multi-omics datasets with smaller size or different omics types presents additional challenges. Questions persist about optimal feature selection, such as determining the number of genes or omics features to use, and establishing minimum sample size requirements for qualitative analysis.
To address these gaps, we propose a structured suggestive guideline for Multi-omics Study Design (MOSD). This guideline synthesizes and extends previous work to identify nine key factors across computational and biological domains. Through comprehensive benchmarking across multiple TCGA datasets using ten clustering-based analytical methods, we evaluate how these factors affect MOI analysis across 10 cancer types. Our goal is to provide clear, evidence-based recommendations for researchers engaged in multi-omics research, offering minimal guidelines for designing multi-omics experiments and performing downstream analysis.
Materials and methods
Definition of MOSD factors
Through comprehensive literature review and systematic analysis, nine critical factors were identified that fundamentally influence multi-omics integration outcomes. This framework originated from Mirza et al. [27], who initially identified five key challenges in MOI analysis: the curse of dimensionality, data heterogeneity, missing data, class imbalances, and scalability. Building upon these foundations and incorporating insights from subsequent benchmark studies, these factors were organized into computational and biological aspects to provide a more complete analytical framework.
The computational factors begin with sample size, which addresses both the curse of dimensionality and scalability challenges. Chauvel et al. [28] demonstrated through both simulation and TCGA breast cancer data how clustering performance changes according to sample numbers, evaluating two clustering methods using the adjusted rand index (ARI) across mRNA, miRNA, methylation and protein omics data from 348 samples. This finding was further supported by Tini et al. [24], who evaluated five unsupervised algorithms using the F-measure method across multiple datasets including simulation data, murine liver data (BXD), and breast cancer gene expression data. Feature selection is a process where genes or omics features that are likely related to some biological trait of interest are carefully chosen by some algorithm, which is a standard procedure in most omics analysis. Pierre-Jean et al. [29] evaluated 13 unsupervised MOI methods for measuring the impact of feature selection. The study used three distinct simulated datasets (Gaussian, binary, and beta-like omics representations) to examine how the proportion of relevant variables impacts clustering performance. Tini et al. [24] also investigated feature selection’s impact, though their filtering method based on coefficient of variation showed limitations in determining the optimal number of meaningful features for class distinction. Preprocessing and noise characterization of multi-omics data represent vital computational factors. Duan et al. [31] demonstrated this in their benchmark of 10 MOI methods across nine TCGA cancer datasets, where they evaluated robustness by adding Gaussian noise at five different variance levels. Their work emphasized the importance of measuring both noise intensity and its distribution across the dataset. Sample balance across classes, initially identified by Mirza [27], was thoroughly examined by Rappoport et al. [30] in their benchmark of nine MOI methods across 10 TCGA datasets, where they evaluated clustering performance using both survival differences and clinical label correlations.
The biological factors incorporate cancer subtype combinations and multi-omics layer integration. Rappoport et al. [30] evaluated this through two measures: scoring survival differences of divided clusters as log ranks and analyzing 41 clinical labels using chi-square and Kruskal–Wallis tests. Duan et al. [31] further explored this aspect by testing 11 different combinations from four omics types to identify optimal configurations for individual and overall cancer subtyping, while also evaluating clustering accuracy, clinical significance, and computational efficiency. The four omics types were gene expression (GE), miRNA (MI), methylation (ME) and copy number variation (CNV). The framework also emphasizes clinical feature correlation, incorporating molecular subtypes, gender, pathological stage, and age. This aspect was particularly highlighted in Pierre-Jean et al.’s work [29] with real datasets (BXD, liver cancer, obesity), though their study noted limitations in applying simulation-derived factors to real clinical data. The framework addresses several limitations identified in previous studies. While Chauvel et al. [28] only performed single tests on TCGA-BRCA data, and Rappoport et al. [30] couldn’t fully grasp the conditions needed to improve MOI method performance, this approach provides a more comprehensive evaluation. Similarly, it addresses the limitations noted in Tini et al.’s work [24] regarding noise application and feature selection, and expands upon Duan et al.’s analysis [31] by incorporating additional data conditions such as size and class balance.
Based on the comprehensive review of previous studies (Table 1), nine factors were identified and categorized into computational and biological factors. The computational factors include: (1) sample size, (2) feature selection, (3) preprocessing strategy, (4) noise characterization, (5) class balance and (6) number of classes. The biological factors comprise: (7) cancer subtype combinations, (8) omics combinations, and (9) clinical feature correlation. These factors provide a structured framework for evaluating and improving multi-omics data integration methods. By consolidating the efforts of previous research and addressing their limitations, we aimed to provide a more complete analytical approach that enhances both the precision and reproducibility of multi-omics analyses. The validation process developed specific benchmarks for each factor and applied them across various TCGA cancer datasets, allowing for comprehensive assessment of their impact on multi-omics integration outcomes.
Table 1.
Related study analysis
| Study | Computational factors | Biological factors | Datasets | Omics | Method | Estimation method | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample size | Feature selection | Preprocessing | Noise | Balance | Number of classes | Cancer subtype combination | Omics combination | Clinical feature | |||||
| Chauvel et al. [28] | S | S | X | S | X | S | X | X | X | BRCA (348) | GE | BCC | ARI |
| ME | MDI | ||||||||||||
| MI | iCluster | ||||||||||||
| Protein | moCluster | ||||||||||||
| JIVE | |||||||||||||
| iNMF | |||||||||||||
| Rappoport et al. [30] | X | X | X | X | X | O | X | X | O | AML (170) | GE | k-means | Survival analysis (logrank) |
| BIC (621) | ME | Spectral | Clinical distribution | ||||||||||
| COAD (220) | MI | LRAcluster | |||||||||||
| GBM (274) | PINS | ||||||||||||
| KIRC (183) | SNF | ||||||||||||
| LIHC (367) | rMKL-LPP | ||||||||||||
| LUSC (341) | MCCA | ||||||||||||
| SKCM (448) | MultiNMF | ||||||||||||
| OV (287) | iClusterBayes | ||||||||||||
| SARC (257) | |||||||||||||
| Tini et al. [24] | S | O | X | O | X | S | X | O | O | BXD (66) | GE | MCCA | F-measure |
| Platelet (12) | ME | JIVE | |||||||||||
| BRCA (491) | MI | MCIA | |||||||||||
| MFA | |||||||||||||
| SNF | |||||||||||||
| Pierre-Jean et al. [29] | S | S | S | O | S | S | X | O | O | BXD (64) | GE | RGCCA | F-measure |
| Obesity (13) | ME | intNMF | ARI | ||||||||||
| Liver cancer (360) | CNV | SNF | Survival analysis | ||||||||||
| Mutation | LRACluster | ||||||||||||
| PINSPlus | |||||||||||||
| CC | |||||||||||||
| MCIA | |||||||||||||
| mixKernel | |||||||||||||
| SGCCA | |||||||||||||
| iClusterPlus | |||||||||||||
| MoCluster | |||||||||||||
| CIMLR | |||||||||||||
| MOFA | |||||||||||||
| Duan et al. [31] | X | O | X | O | X | O | X | O | X | ACC (77) | GE | SNF | Precision |
| BRCA (760) | ME | NEMO | NMI | ||||||||||
| COAD (291) | MI | CIMLR | ARI | ||||||||||
| KIRP (273) | CNV | MultiNMF | F-measure | ||||||||||
| KIRC (314) | PFA | ||||||||||||
| LIHC (364) | LRAcluster | ||||||||||||
| LUAD (453) | moCluster | ||||||||||||
| LUSC (363) | iClusterBayes | ||||||||||||
| THYM (119) | PINS | ||||||||||||
| Subtype-GAN | |||||||||||||
Data acquisition and assembly
A comprehensive analysis of data sourced from TCGA [20] was used to provide the suggestive MOSD guidelines. This study utilized multi-omics data from TCGA repository, focusing on 3,988 patients across ten cancer types: bladder urothelial carcinoma (BLCA, n=400), breast invasive carcinoma (BRCA, n=592), colon adenocarcinoma (COAD, n=249), head and neck squamous cell carcinoma (HNSC, n=471), kidney renal papillary cell carcinoma (KIRP, n=268), liver hepatocellular carcinoma (LIHC, n=357), lung adenocarcinoma (LUAD, n=442), skin cutaneous melanoma (SKCM, n=350), stomach adenocarcinoma (STAD, n=365), and thyroid carcinoma (THCA, n=494). These cancer types were selected based on the availability of patient-matched omics and clinical data, with a minimum threshold of 100 patients per cancer type to ensure robust statistical analysis. Detailed breakdown of subtype and sample counts is listed in Supplementary Table S1.
As shown in Table 2, the multi-omics layers incorporated in this study comprised gene expression (GE), miRNA (MI), mutation data, copy number variation (CNV), and methylation (ME), representing the commonly utilized data types in multi-omics research [32]. The genomic features varied across cancer types, with GE features ranging from 2,097 to 2,229, while LIHC demonstrated the most extensive profile with 17,390 GE features and 21,933 CNV features. Notably, mutation data was particularly comprehensive for LIHC, encompassing 396,065 features. Figure 1 shows the structure of the multi-omics data, which were divided to omics and clinical feature data types for the analysis.
Table 2.
Statistics of the TCGA dataset
| Cancer | Number of omics features | Number of samples for each clinical feature | ||||||
|---|---|---|---|---|---|---|---|---|
| GE | ME | MI | CNV | Subtype | Pathologic-stage | Gender | Age | |
| BLCA | 17390 | 396065 | 2195 | 21933 | 1 (41) | StageII (128) | MALE (296) | Middle (124) |
| 2 (41) | StageIII (136) | FEMALE (104) | Old (244) | |||||
| 3 (29) | StageIV (132) | Total (400) | Total (368) | |||||
| 4 (15) | Total (396) | |||||||
| Total (126) | ||||||||
| BRCA | 2222 | LumA (323) | StageI (96) | - | Young (16) | |||
| LumB (118) | StageII (345) | Middle (221) | ||||||
| Basal (102) | StageIII (151) | Old (81) | ||||||
| Her2 (39) | Total (592) | Total (318) | ||||||
| Total (582) | ||||||||
| COAD | 2097 | CMS1 (34) | StageI (36) | MALE (137) | Middle (99) | |||
| CMS2 (67) | StageII (83) | FEMALE (112) | Old (130) | |||||
| CMS3 (35) | StageIII (67) | Total (249) | Total (229) | |||||
| CMS4 (63) | StageIV (29) | |||||||
| Total (199) | Total (215) | |||||||
| HNSC | 2229 | Basal (74) | StageI (26) | MALE (347) | Middle (261) | |||
| Mesenchymal (65) | StageII (71) | FEMALE (124) | Old (168) | |||||
| Atypical (59) | StageIII (74) | Total (471) | Total (429) | |||||
| Classical (44) | StageIV (234) | |||||||
| Total (242) | Total (405) | |||||||
| KIRP | 2099 | C1 (84) | StageI (164) | MALE (200) | Middle (141) | |||
| C2a (34) | StageII (18) | FEMALE (68) | Old (91) | |||||
| C2b (21) | StageIII (49) | Total (268) | Total (232) | |||||
| Total (139) | StageIV (14) | |||||||
| Total (245) | ||||||||
| LIHC | 2157 | 1 (65) | StageI (166) | MALE (243) | Middle (171) | |||
| 2 (55) | StageII (84) | FEMALE (114) | Old (129) | |||||
| 3 (63) | StageIII (61) | Total (357) | Total (300) | |||||
| Total (183) | Total (311) | |||||||
| LUAD | 2211 | 1 (17) | StageI (233) | MALE (205) | Middle (174) | |||
| 2 (22) | StageII (108) | FEMALE (237) | Old (212) | |||||
| 3 (38) | StageIII (62) | Total (442) | Total (386) | |||||
| 4 (26) | StageIV (19) | |||||||
| 5 (43) | Total (422) | |||||||
| 6 (33) | ||||||||
| Total (179) | ||||||||
| SKCM | 2203 | BRAF (116) | StageI (72) | MALE (220) | Middle (175) | |||
| RAS (78) | StageII (67) | FEMALE (130) | Old (103) | |||||
| TWT (37) | StageIII (137) | Total (350) | Young (29) | |||||
| NF1 (23) | StageIV (20) | Total (307) | ||||||
| Total (254) | Total (296) | |||||||
| STAD | 2161 | CIN (202) | StageI (46) | MALE (240) | Old (177) | |||
| MSI (59) | StageII (116) | FEMALE (125) | Middle (133) | |||||
| GS (46) | StageIII (163) | Total (365) | Total (310) | |||||
| EBV (29) | StageIV (28) | |||||||
| Total (336) | Total (353) | |||||||
| THCA | 2201 | 1 (131) | StageI (279) | MALE (133) | Middle (237) | |||
| 2 (67) | StageII (50) | FEMALE (361) | Young (166) | |||||
| 3 (82) | StageIII (109) | Total (494) | Old (45) | |||||
| 4 (105) | StageIV (46) | Total (448) | ||||||
| 5 (90) | Total (484) | |||||||
| Total (475) | ||||||||
Fig. 1.

Structure and organization of multi-omics data integration. Multi-omics datasets are structured as a collection of two-dimensional matrices, where each matrix represents a distinct omics layer (e.g., genomics, transcriptomics, proteomics). Rows consistently represent biological samples across all matrices, while columns contain features specific to each omics type (e.g., genes, proteins, methylation probes). A complementary clinical matrix maintains the same sample-wise organization (rows) but contains relevant clinical variables (columns) such as age, sex, and disease status. This standardized structure enables integrated analysis across multiple biological layers while maintaining sample correspondence
Clinical annotations were systematically integrated, including molecular subtypes, gender distribution, pathological staging, and age demographics. The molecular subtypes for each cancer type were extracted from TCGA clinical data and varied by cancer type, with some cancers showing distinct classification system. For instance, for BRCA, we applied PAM50-based subtypes [33] (Luminal A, Luminal B, Basal, HER2), and for COAD, Consensus Molecular Subtype 1-4 (CMS1–4) were classified based on Guinney et al. [34]. For other cancer types, including LUAD, STAD, SKCM, KIRP, and HNSC, we utilized the ‘Subtype’ column provided in the TCGA clinical matrices, which included both categorical descriptors and numeric classifications (ranging 1-6 depending on cancer type). These subtype labels were used as-is for benchmarking purposes without reclassification or biological reinterpretation. Age stratification into Young, Middle, and Old categories demonstrated a predominance of middle-aged and elderly patients, with specific age-related patterns varying by cancer type. The University of California Santa Cruz (UCSC) Xena browser [35] was used for collecting the multi-omics data. Patient barcodes were verified through TCGAbiolinks [36] to ensure data matching.
Data preprocessing
The examination of preprocessing effects on integration performance involved the creation of three distinct dataset types, each representing different levels of data refinement. The first category comprises Raw Datasets (RAW), which maintain data in its most basic processed form through log2 transformation of the original values. This approach preserves the inherent structure of the data while enabling basic computational analysis. The second category consists of Normalized Datasets (NORM), which underwent quantile normalization followed by 0-1 scaling. This process establishes uniform distributions across various omics layers, facilitating more standardized comparisons.
The third category encompasses Gene Level Datasets (GL), representing an advanced preprocessing approach where normalized data aligns with gene expression characteristics. To enable integrative analysis across heterogeneous omics layers, we converted all omics data into a gene-level format, following a gene-centric integration strategy as described in Jung et al. [37] For methylation, we selected probes located within a 2 kilobase upstream region from the promoter region of each gene. The average beta value across these probes was computed per gene. miRNA expression values were assigned to their corresponding target genes based on experimentally validated miRNA–gene interactions, and the geometric mean of all miRNAs targeting a gene was used as the representative value. For CNV, segmented values overlapping gene bodies were averaged per gene. After alignment across omics layers, each matrix was min–max scaled to a 0–1 range to ensure comparability and avoid dominance from any single data type during integration. This process ensures that all omics matrices share the same gene and sample dimensions, enabling structured gene-level multi-omics representation. The complete preprocessing methodology and associated code reside in the project’s GitHub repository, ensuring transparency and reproducibility.
The preprocessing strategy specifically addressed the challenge of missing data, which previous research has identified as detrimental to multi-omics analyses. To ensure consistency across omics layers and avoid the influence of missing values, we adopted a conservative approach by strictly excluding any omics features that contained missing entries. In addition, sample groups with fewer than ten individuals were removed to maintain sufficient representation for statistical analysis. No imputation was applied during preprocessing Such measures maintain robust representation across categories while minimizing potential biases stemming from data sparsity.
The processing of clinical data involved careful categorization to enable meaningful correlation with multi-omics findings. The framework retained categorical features including molecular subtype, gender, and pathological stage as natural labels for subsequent analyses. Age data underwent transformation from its continuous form into three discrete categories: young (0-39 years), middle aged (40-64 years), and older (65 years and above), following protocols in cancer research literature [38]. This age categorization facilitates the investigation of age specific molecular patterns within each cancer type [39].
MOI analysis methods and implementation
We evaluated ten MOI methods that employ various computational approaches including clustering, matrix factorization, and network integration across ten TCGA cancer datasets to determine optimal conditions for cancer data analysis. These methods were selected based on three criteria: ease of implementation in R or Python, prevalence in academic research, and effectiveness with default parameters. Rather than focusing solely on comparison of method performance, our analysis examined how each method’s characteristics interacted with the nine factors through seven benchmark tests. Through a series of controlled benchmark tests, we identified parameter conditions that consistently improved analysis quality under each method, which we refer to as analytically favorable or recommended setups for multi-omics study.
Similarity network fusion
Similarity Network Fusion (SNF) [40] constructs similarity networks for each omics type using metrics such as Pearson correlation or chi-square and then fuses these networks into a unified representation. Its advantage in MOSD lies in integrating disparate omics layers, handling clinical features of both discrete and continuous types, and tolerating small sample sizes.
Spectrum
Spectrum [41] is a density-aware spectral clustering method that leverages eigenvectors from the multi-omics sample-graph matrix to mitigate noise and illuminate biologically relevant structures. It uses a k-nearest neighbors kernel, obviating the need for extensive parameter tuning across varied datasets. Spectrum’s advantages for MOSD include scalability to large datasets and the ability to capture subtle density variations, aided by a Gaussian Mixture Model (GMM) rather than traditional k-means.
PINSPlus
PINSPlus (PP) [42], an extension of PINS [43], integrates multi-omics data by building connectivity graphs for each omics layer and fusing them to identify stable clusters. PP showed to be able to find both known and unknown subtypes with significant clinical implications.
Neighborhood-based multi-omics clustering
Neighborhood-based Multi-omics Clustering (NEMO) [44] is a flexible algorithm that operates through three main steps: 1) calculation of pairwise distances between samples within each omics layer using specified distance metrics, 2) construction of k-nearest neighbor graphs for each sample based on local neighborhood patterns, and 3) clustering of samples using graph-based clustering algorithms applied to these neighborhood representations. NEMO supports various feature types including numerical, discrete, ordinal, and imaging data, and excels with partial datasets by eliminating the need for missing data imputation while maintaining faster execution compared to other multi-omics clustering methods.
Cluster of cluster assignments
Cluster of Cluster Assignments (COCA) [45] converts each omics dataset into a one-hot encoded cluster matrix and then applies consensus clustering, typically using Pearson’s correlation to form a similarity matrix. The consensus clustering provides stability and helps identify cross-omics correlations without strict normalization requirements.
Low-rank approximation clustering
Low-Rank Approximation Clustering (LRAcluster) [46] adopts a probabilistic model that treats omics data as Gaussian-distributed variables under a low-rank constraint. It compresses high-dimensional multi-omics data into a more interpretable, low-dimensional representation. LRAcluster accurately detected shared covariance structures across omics types and highlighted critical regulatory signatures and cancer subtypes.
Consensus clustering
Consensus Clustering (CC) [47] repeatedly applies clustering algorithms to resampled versions of the dataset and aggregates these results into a consensus solution, mitigating random fluctuations. This is particularly valuable in MOSD when handling high-dimensional and heterogeneous data. By quantifying cluster assignment frequencies, CC not only consolidates robust groupings but also reveals the stability of potential cluster solutions across multiple resampling iterations.
Multi-omics factor analysis
Multi-Omics Factor Analysis (MOFA) [48] decomposes multi-omics data into latent factors that capture the main source of variation across different layers. By fitting a linear factor model, it effectively integrates incomplete data, highlights outliers, and identifies molecular signatures driving heterogeneity. MOFA’s interpretability and capacity for imputing missing values offer considerable benefits within MOSD, allowing deeper insight into latent structures that span multiple data types.
iClusterPlus
iClusterPlus [49] is an advanced version of iCluster [50] that integrates multi-omics data using a probabilistic joint latent-variable model. The method incorporates different omics data types (binary, categorical, and continuous) into a low-dimensional latent representation through estimation of latent variables W and H that capture relationships between samples and features. Model optimization employs a lasso penalty (L1-norm) on H to obtain sparse solutions, enabling dimension reduction by removing uninformative features. This approach improves clustering performance while increasing interpretability and computational efficiency through comprehensive multi-omics integration.
Integrative non-negative matrix factorization
Integrative Non-negative Matrix Factorization (IntNMF) [51] extends classic NMF [52] by sharing a common basis matrix W across multiple two-dimensional omics datasets, while allowing each data type to have a distinct coefficient matrix H. It enhances the detection of subtypes and leverages resampling-based cross-validation for selecting the optimal number of clusters. IntNMF’s strength lies in its ability to handle large-scale datasets without relying on strict distributional assumptions. Collectively, the ten methods offer a wide range of strategies for multi-omics integration and clustering, each with unique advantages for different data structures and MOSD factors. By systematically benchmarking their performance across TCGA cancer datasets, we aimed to pinpoint favorable conditions, such as sample size requirements, handling of missing values, or optimal omics combinations, that maximize analytical precision and biological relevance under the MOSD framework.
Benchmark design
To address the heterogeneous nature of multi-omics data and evaluate parameter influence on analysis outcomes, a series of comprehensive benchmarks aligned with the previously defined computational and biological factors in MOSD has been developed. Each benchmark test isolated a single study design factor while maintaining all other variables at fixed default settings. This allowed fair comparison of individual factor impacts on clustering performance. The full test design, including parameter ranges, evaluation objectives, target values, and default settings for each benchmark, is summarized in Table S2. This systematic isolation of variables allowed for precise measurement of each factor’s unique contribution to analysis outcomes. The evaluation process employed multiple performance metrics, including Normalized Mutual Information (NMI), Adjusted Rand Index (ARI), and the F-measure. To ensure statistical robustness, each benchmark underwent ten iterations with random sample selection.
Determining the minimal required sample size
A key objective in MOSD is to identify the minimal sample size required for reliable multi-omics clustering. In this test, we randomly selected subsets of increasing size beginning with as few as five samples to track when performance metrics plateaued. As illustrated in Fig. 2, this plateau reflects the threshold beyond which additional samples provide diminishing returns for accurate cancer subtyping, thus indicating the minimum cohort size needed for robust analysis.
Fig. 2.

Benchmarking designs of key factors. Systematic evaluation of six critical factors in multi-omics integration: a Sample size - Performance metrics with increasing sample sizes showing minimum cohort requirements. b Feature Selection - Impact of varying feature numbers selected via statistical testing across omics types. c Preprocessing test - comparison of RAW (Log2 transformed), NORM (Quantile normalization with 0-1 scaling), and GL (Gene level dataset) approaches on clustering performance. d Class balance - effects of classes with imbalanced sample on method accuracy. e Noise tolerance - Method resilience to incremental Gaussian noise (10-100%). f Cancer subtype combination - Performance analysis across different cancer subtype combinations. Each panel demonstrates the parameter’s influence on MOI performance
Optimizing feature selection
Determining and adequate balance between feature quantity and quality is important in MOSD. To assess the effect of feature quantity on MOI performance, we incrementally selected features based on their relevance to sample group labels (i.e., cancer subtypes). Chi-square tests were applied for GE, MI, and ME omics types, while ANOVA was applied to CNV data due to its inclusion of negative values. Features were ranked by p-value, and top-ranked subsets were selected at increasing proportions—1% steps for early iterations, then 10%—up to 100% of features. This strategy was chosen over variance-based or classifier-specific ranking to avoid bias from model dependencies and to provide a group-informed yet algorithm-agnostic selection method. The exact number of features per omics layer in each benchmark iteration is provided in the GitHub repository. The proportion of selected features in each scenario is shown in Fig. 2.
Measuring the impact of different preprocessing methods
Preprocessing can significantly affect the outcomes of multi-omics analysis. We compared three approaches: RAW, NORM, and GL. RAW data underwent only log2 transformation, while NORM underwent quantile normalization and min-max scaling. GL integration averaged omics features at the gene level, as recommended by Jung et al. [47, 53] potentially enhancing gene-centric signals yet discarding non-gene-aligned features. By applying these variants to the same sets of MOI methods, we examined whether specific preprocessing strategies yield consistently better clustering performance (Fig. 2).
Evaluating class balance
Class imbalance, where certain cancer subtypes or clinical categories contain fewer samples, can bias clustering and reduce the detection of smaller classes [54]. In this benchmark, we progressively introduced imbalance in sample sizes to gauge how well different MOI methods cope with skewed distributions (Fig. 2).
Assessing noise tolerance
Multi-omics datasets often include noise, or errors, from technical and biological reasons [29]. To evaluate its influence, Gaussian noise was introduced in 10% increments up to 100% in each omics layer (Fig. 2). The objective was to observe the maximum tolerable noise threshold where clustering performance started declining significantly.
Cancer subtype combinations
Different subtypes within the same cancer type frequently display unique molecular signatures. We explored whether varying combinations of these subtypes influence clustering performance, focusing on how distinguishable they are (Fig. 2). All clustering evaluations were performed on datasets containing at least two distinct subtype labels, ensuring meaningful calculation of performance metrics. The benchmark also examined the number of subtypes assessed—ranging from a low count (2–3) to more diverse sets—to identify at which point distinguishing multiple subtypes becomes challenging or more susceptible to errors.
Omics combination
Finally, we investigated how specific omics-layer combinations affect clustering for four clinical features (molecular subtype, gender, pathological stage, age). Data configurations ranged from single-omics (GE alone) to multi-omics (e.g., GE-ME-CNV or GE-ME-MI-CNV). Although combining more omics can reveal intricate biological insights, it can also amplify heterogeneity, aligning with findings by Duan et al. [31] and Reel et al. [55]. Hence, determining which combinations yield the most accurate results for each clinical feature is a key focus in MOSD, and our test delineates situations where additional omics layers either enhance or degrade performance. Through these targeted benchmarks, we provide a systematic exploration of how each factor contributed to multi-omics analysis results. This approach offers actionable insights into the design, execution, and interpretation of multi-omics studies.
Performance evaluation for MOI methods
Evaluating the performance of clustering-based MOI methods is an essential component of the MOSD framework. Three metrics were used to evaluate the clustering performance, ARI, NMI, and F-measure [56]. ARI is calculated from the Rand Index (RI), which measures clustering accuracy:
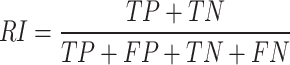 |
Here, TP represents the sample pairs correctly grouped within the same cluster, while TN represents the pairs correctly grouped in different clusters. FP indicates the pairs incorrectly grouped in the same cluster, and FN represents pairs incorrectly grouped in different clusters. ARI adjusts the RI by accounting for chance:
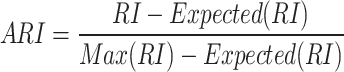 |
The Expected(RI) represents the expected RI value by random chance, while Max(RI) indicates the maximum possible RI value. NMI measures the mutual information between the true labels (T) and clustering results (C). The probability distributions are defined as:
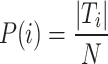 |
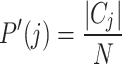 |
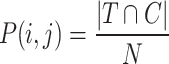 |
Here, N represents the total number of elements, while  indicates the number of elements in the true class i, and
indicates the number of elements in the true class i, and 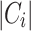 represents the number of elements in the predicted cluster j. The intersection
represents the number of elements in the predicted cluster j. The intersection  denotes the number of elements shared between the true class and predicted cluster. The mutual information is then calculated as:
denotes the number of elements shared between the true class and predicted cluster. The mutual information is then calculated as:
 |
To normalize MI, we calculate the entropy of both true labels and clustering results. Entropy measures the uncertainty or randomness in the clustering assignments. The entropy for true labels and clustering results are calculated as:
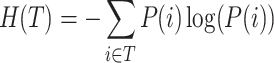 |
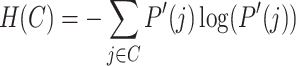 |
where P(i) is the probability of an element belonging to the true class i, and 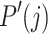 is the probability of an element being assigned to cluster j. Using these entropy values, we normalized MI to obtain NMI:
is the probability of an element being assigned to cluster j. Using these entropy values, we normalized MI to obtain NMI:
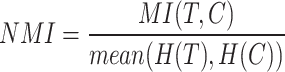 |
This ensures the NMI value falls between 0 and 1, where 1 indicates perfect clustering alignment with true labels, and 0 indicates completely random clustering. The average of the entropies in the denominator acts as a normalization factor, making NMI more comparable across different clustering scenarios. The F-measure computation involves three set operations:
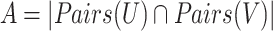 |
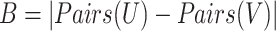 |
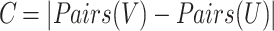 |
The sets U and V represent the predicted clusters and true clusters respectively, where Pairs(X) generates all possible pairs of elements within set X. The F-measure is then calculated as:
 |
To obtain an overall performance metric, we compute a cluster-score as the arithmetic mean of all three metrics:
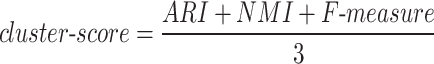 |
Although ARI theoretically ranges from –1 to 1, all observed values in our benchmark were non-negative, eliminating the need for normalization before averaging with NMI and F-measure. This combined cluster-score serves as our primary evaluation metric, providing balanced assessment of clustering performance across all benchmark tests. Individual metrics are examined when detailed analysis is required. We note that future studies encountering negative ARI values should consider normalization to a 0–1 scale to ensure balanced integration into composite scoring.
Results
Optimal and suboptimal configurations in benchmark tests
Identifying the most and least effective parameter configurations were searched to characterize the importance of each of the nine key factors. Regarding sample size and class balance, smaller but balanced classes often performed better than larger but imbalanced ones, as exemplified in BLCA where 11 samples per class achieved the highest cluster-score (0.55), while very small classes (i.e., 5 samples) showed poor performance. Feature selection demonstrated that using fewer features generally yielded better results than using all features, which consistently produced the lowest scores across cancer types. The class balance benchmark showed varying optimal configurations across cancer types. Some cancers performed best with equal class sizes (e.g., BRCA with 40 samples per class), while others showed resilience to moderate imbalance. The noise tolerance test revealed that increasing noise levels universally degraded performance, with 100% noise consistently producing the lowest scores (often below 0.10) across all cancer types.
For biological factors, certain subtype combinations demonstrated notably stronger signals. For instance, Luminal B-Basal subtypes in BRCA and CIN-EBV in STAD showed particularly high discrimination (score >0.90). For omics combinations, GE-ME pairings frequently emerged as the most effective omics pairs across multiple cancer types, while combinations involving CNV often showed poorer performance. Notably, some tests, such as gender analysis in BRCA, were excluded due to extreme data imbalance, highlighting the importance of considering data distribution in study design.
These findings across different parameter configurations provide insights for optimizing multi-omics analysis approaches and aid in parameter selection. The complete detailed results are available in Supplementary Tables S3 and S4.
Impact of sample size
The relationship between sample size and clustering performance was examined across all cancer types (Fig. 3a). The analysis revealed that increasing sample size from 5 to 38 samples per class had a subtle but positive effect on average cluster scores, as indicated by the gradually rising trend line (red). However, the most notable impact of larger sample sizes was on score consistency rather than absolute performance - the violin plot distributions become narrower as sample size increases, particularly after 26 samples, indicating reduced variance in clustering performance. While the average cluster score shows only modest improvement (i.e., ranging approximately from 0.5 to 0.55), the minimum scores become notably higher with increased sample size, suggesting that larger sample sizes help prevent poor clustering outcomes. Detailed cancer-type specific trends are provided in Supplementary Figure S1.
Fig. 3.

Impact of 4 factors on Multi-omics clustering performance. Violin plots showing the distribution of clustering performance scores across different parameters: a Sample size effect - Performance trends with increasing sample sizes (5-38 samples per class), showing modest improvement in mean scores (red trend line) but notably decreased variance with larger samples. b Feature selection impact - The relationship between feature proportion and clustering performance, demonstrating optimal performance at 1-10% feature selection, decline at 20-50%, and consistent poor performance at 60-100%. c Class balance analysis - comparison of clustering performance between nearly balanced (<3:1 ratio) and imbalanced (>3:1 ratio) classes, showing significantly higher performance (****p < 0.0001) in relatively balanced conditions. d Noise tolerance - distinct impact of performance response to increasing noise levels: stable (0-50%), decline (50-80%), and severe degradation (80-100%). Red lines indicate overall trends, and violin plot widths represent score distribution density at each parameter level
Effect of feature selection
Feature selection appears to influence clustering accuracy, with certain tendencies observed in the relationship between feature proportion and performance (Fig. 3b). In the analyzed dataset, lower feature proportion (1–10% of features) tended to yield higher cluster scores, averaging around 0.55, albeit with a relatively wide distribution. As the feature proportion increased (20–50%), a gradual decline in performance was observed, followed by a further reduction at higher feature proportion (60–100%), where mean cluster scores approached 0.4. Notably, the distribution patterns change markedly across these phases. The lower feature proportion (1-10%) showed wider distributions, indicating greater variability in outcomes but also higher potential for optimal performance. The transition phase showed gradually narrowing distributions, suggesting more consistent but declining performance. The higher feature proportion (particularly 90-100%) showed both decreased performance and narrower distributions, indicating consistently poorer outcomes when using most or all features.
Such pattern suggests that selective feature usage, particularly under 20%, is recommended for optimal clustering performance. The sharp decline in performance with larger feature sets clearly indicates that selecting fewer but biologically relevant ones is beneficial. Detailed cancer-type specific trends are available in Supplementary Figure S1.
Preprocessing influence
The comparative analysis of three preprocessing approaches—RAW, NORM, and GL—revealed nuanced patterns across different cancer types and analytical contexts. While NORM datasets showed a slight overall advantage, the performance differences between methods were generally modest, typically below 0.1, suggesting robustness across the different preprocessing approaches. NORM showed strength in gender-based analyses, with consistently higher performance improvements, ranging from +0.20 to +0.27, across multiple cancer types including BLCA, COAD, HNSC, and LUAD. This suggests that normalization may be especially beneficial when analyzing gender-specific molecular patterns. Cancer-type specific patterns were observed where some types showed consistent preferences for particular preprocessing methods. For instance, LUAD demonstrated better performance with GL preprocessing across multiple test categories, while STAD consistently favored NORM preprocessing, indicating that optimal preprocessing strategies may be cancer-type dependent.
Raw data preprocessing showed advantages in noise tolerance tests for several cancer types (notably BLCA, BRCA, and KIRP), potentially indicating that normalization may sometimes weaken the signals relevant to certain biological features. These findings suggest that while the choice of preprocessing can impact analysis outcomes, the effect is often context-dependent, varying by cancer type and analytical objective. The complete comparative analysis is available in Supplementary Table S5.
Influence of class balance
The analysis of sample balance showed distinct difference in clustering performance between nearly balanced and imbalanced classes (Fig. 3c). Datasets with nearly balanced classes (imbalance ratio < 3:1) yielded significantly higher and more consistent cluster scores compared to imbalanced sample classes (imbalance ratio >3:1), as indicated by the statistically significant difference (adj. p < 0.0001). Classes with nearly balanced samples showed not only higher cluster scores (i.e., 0.58) but also exhibited a wider range of potential high-performance outcomes, as illustrated by the broader upper portion of the violin plot. In contrast, classes with imbalanced samples demonstrated lower performance (0.52) and a distinctive bimodal distribution, suggesting two distinct performance patterns in imbalanced datasets. The narrower distribution in the imbalanced classes indicates more predictable, though generally lower, performance outcomes. Collectively, the overall reliability of clustering results is likely to decline when the sample ratio exceeds 3:1. Detailed cancer-type specific trends are available in Supplementary Figure S1.
Noise-induced effects on clustering outcomes
The analysis of noise tolerance in multi-omics integration showed distinct performance drop after a certain level of noise (Fig. 3d). The violin plots demonstrate a clear response to increased noise levels. The performance was stable with 0-50% noise where cluster scores maintained around 0.5 with consistent performance. The cluster score started to decline with 50% noise showing gradual decline. Interestingly, the variance in performance decreased substantially with increasing noise, suggesting that high noise levels not only degrade performance but also make outcomes more predictably poor. The sharp decline after 80% noise indicates a critical threshold beyond which reliable clustering becomes nearly impossible, regardless of the MOI method used. Detailed cancer-type specific responses to noise are available in Supplementary Figure S1.
Cancer subtype combination effect
Analysis of cancer subtype combinations across ten TCGA cancer types revealed distinct patterns in clustering performance (Fig. 4). Cancer-type-specific molecular characteristics significantly influence multi-omics clustering performance, with cluster-score varying across both cancer types and specific subtype combinations. In BRCA, Luminal A vs. Luminal B classification consistently showed lower performance due to their shared estrogen receptor positivity and overlapping expression profiles, differing primarily in proliferation-related genes [33, 57]. Similarly, COAD clustering of CMS2 and CMS3 subtypes yielded lower accuracy than more distinct pairs like CMS1 and CMS4, reflecting their shared epithelial characteristics and overlapping genomic features [34, 58]. These results demonstrate that clustering difficulty correlates with underlying molecular similarity between subtypes, emphasizing the importance of selecting biologically distinct subtypes for reliable integrative clustering outcomes. Method-specific performance patterns emerged across cancer types. SNF, CC, and Spectrum methods consistently maintained higher performance levels across most cancer types, while iClusterPlus showed consistently lower performance when handling multiple subtype distinctions. This pattern was particularly evident in cancers with complex molecular landscapes such as LUAD, which exhibited the most diverse combination patterns and generally lower clustering scores across all methods.
Fig. 4.

Method performance across cancer subtype combinations in ten cancer types. Analysis of clustering performance across different cancer subtype combinations in ten TCGA (The Cancer Genome Atlas) cancer types (BLCA, BRCA, COAD, HNSC, KIRP, LHC, LUAD, SKCM, STAD, and THCA). The x-axis represents different subtype combinations, and the y-axis shows cluster scores. The mean performance (red dashed line) indicates overall trends in clustering capability as subtype complexity increases
Cancer types demonstrated varying levels of clustering stability. The KIRP showed relatively stable performance across fewer subtype combinations, while THCA and HNSC displayed more variable performance patterns depending on the specific subtype combinations analyzed. The general trend across all cancer types indicated declining performance as the number of subtypes in the combination increased, particularly when including molecularly similar subtypes. These observations show the importance of carefully considering both cancer type-specific molecular characteristics and method selection when designing multi-omics studies involving subtype analysis. The results suggest that optimal study design should prioritize clearly distinct molecular subtypes when possible, while acknowledging that performance may vary significantly based on the specific cancer type and analysis method chosen.
Optimizing omics combinations for clinical features
Evaluating different omics-layer combinations for Subtype, and Pathological Stage (Fig. 5) revealed that integrating GE–ME often led to superior clustering performance, especially for Subtype (Fig. 5a). While GE–MI was also beneficial for Subtype, it did not perform as well for the other test. In contrast, the Pathological Stage (Fig. 5b) benchmarks generally exhibited lower cluster-scores, suggesting that this clinical feature may be more challenging for MOI methods to distinguish. This pattern is reflected in the narrower regions seen in the corresponding panel (Fig. 5b), regardless of the method employed. A closer examination of the combination test showed notable method-specific differences. For instance, SNF, COCA, and NEMO show consistently strong performance when classifying Gender using GE–ME, whereas other approaches (e.g., iCluster, MOFA) may excel in different contexts or for different clinical variables. These findings suggest the importance of matching the method to the data type and clinical interest: the interplay between specific omics layers (e.g., GE–ME vs. GE–MI) and particular clinical variables (e.g., Subtype vs. Gender) can substantially influence clustering success. Consequently, researchers need to evaluate a range of MOI approaches and omics-layer combinations to optimize performance for their specific study goals. The additional results for Gender and Age are available in Supplementary Figure S2.
Fig. 5.

Performance analysis of multi-omics combinations across clinical features. Radar chart visualization of clustering performance across different omics combinations and clinical features, presented in two sections: summary charts (top) showing mean performance values and detailed method/cancer-specific charts (bottom). a Subtype Analysis - Superior clustering performance observed with combinations including GE-ME-MI (Gene Expression-Methylation-miRNA), demonstrating the value of integrating multiple omics layers for subtype identification. b Pathological Stage Classification - Generally lower clustering performance across omics combinations, indicating challenges in stage distinction through multi-omics integration
Comparative performance of ten MOI methods
The comparative analysis of ten MOI methods across different test scenarios revealed distinct patterns of performance strengths (Table 3). SNF demonstrated consistently strong performance in structural tests, leading in sample size (5/10), feature selection (4/10), and subtype combination analyses (5/10). NEMO showed particular strength in clinical feature analysis, achieving top performance in stage-based omics combinations (5/10) and maintaining strong performance across multiple test categories. Method performance varied notably by analytical context. CC showed specific strength in gender-based analyses (4/9), suggesting utility for sex-specific molecular patterns. PP demonstrated robust performance under noise conditions (3/10), while also maintaining competitive performance in various other categories. Other methods showed more specialized performance profiles. LRAcluster and IntNMF performed well in specific clinical contexts, particularly in gender and age-related analyses, while COCA and Spectrum showed moderate but consistent performance across various test scenarios. This performance heterogeneity emphasizes that method selection should be guided by specific analytical objectives rather than general performance metrics alone. Notably, the balanced performance across top methods in several categories (e.g., three methods sharing top performance in balance analysis) suggests that multiple approaches may be viable depending on specific research contexts. These findings highlight the importance of matching MOI method selection to specific analytical goals and data characteristics in the MOSD framework.
Table 3.
Performance ranking of MOI methods across different test scenarios
| Test case | Top method | Runner-up | Notable performer |
|---|---|---|---|
| Sample size | SNF (5/10) | NEMO (2/10) | PP (2/10) |
| Feature selection | SNF (4/10) | NEMO (2/10) | PP, CC (2/10 each) |
| Balance | SNF, NEMO (3/10 each) | PP (2/10) | CC (1/10) |
| Noise tolerance | PP (3/10) | SNF, NEMO, CC (2/10 each) | - |
| Subtype combination | SNF (5/10) | NEMO (2/10) | MOFA, CC (1/10 each) |
| Omics (Subtype) | NEMO (4/10) | SNF, Spectrum (2/10 each) | CC, IntNMF (1/10 each) |
| Omics (Gender) | CC (4/9) | LRAcluster (2/9) | SNF, COCA, IntNMF (1/9 each) |
| Omics (Stage) | NEMO (5/10) | PP, CC (2/10 each) | COCA (1/10) |
| Omics (Age) | NEMO (3/10) | IntNMF, CC, LRAcluster (2/10 each) | Spectrum (1/10) |
A comparison of ten multi-omics integration methods across various test scenarios. Numbers in parentheses indicate the frequency of achieving the highest cluster score out of total cases (e.g., 5/10 means the method achieved the highest score in 5 out of 10 cancer types). Top method indicates the best performing method in each test scenario, followed by the second-best performer (Runner-up) and other methods showing notable performance (Notable Performer). Test cases include fundamental computational assessments (sample size, feature selection, balance, noise tolerance) and biological analyses (subtype and omics combinations with various clinical features). The gender analysis includes only 9 cases due to the exclusion of one cancer type with extreme gender imbalance
Discussion
MOI has expanded the understanding of cancer biology [3], while practical challenges persist. Factors such as sample size, feature selection, and omics combination selection influence whether analyses yield reliable or misleading insights [59]. Despite advancements in machine learning algorithms, a unifying framework to address variability in data quality, noise, and imbalances has been lacking [60]. The proposed MOSD framework encompasses both computational and biological factors, contributing to standardization of MOI approaches and bridging the gap between theory and practice.
Feature selection plays a significant role in clustering performance—an aspect sometimes overshadowed by focus on algorithms [61]. The findings suggest that feature curation can enhance interpretation and reduce computational overhead, even with modest sample sizes or inherent imbalances. This indicates the importance of considering both quantity and biological relevance of selected features. Benchmark tests revealed that using fewer features (1-10% of the total) generally yielded higher clustering performance, with improvements of up to 34% compared to using all features. This finding aligns with Pierre-Jean et al. [29]’s observation that feature selection impacts clustering quality, while providing more specific guidance on selection ratios. Sample size analysis demonstrated that 26 samples per class supports robust clustering performance, particularly for reducing variance in results. While absolute performance showed modest improvement with larger sample sizes, the consistency of results improved substantially, indicating that larger cohorts help prevent poor clustering outcomes rather than dramatically improving average performance.
Class balance emerged as another important factor, with results showing that imbalance ratios exceeding 3:1 degraded clustering performance. This finding expands on Mirza [27]’s identification of class imbalance as a challenge in MOI analysis and offers a specific threshold for multi-omics study design. The comparison of raw, normalized, and gene-level preprocessing approaches demonstrated that preprocessing method selection affects clustering outcomes. The gene-level approach recommended by Jung et al. [47] showed advantages in certain contexts, particularly when analyzing molecular subtypes, though effectiveness varied by cancer type and analysis method. For biological factors, certain cancer subtype combinations (e.g., Luminal B-Basal in BRCA) showed high discrimination potential, while others were more challenging to distinguish. This pattern was consistent across different MOI methods, highlighting the relevance of considering molecular distinctiveness of subtypes when designing studies.
Additionally, combining numerous data types does not automatically improve results—particularly when clinical features are difficult to discriminate based on non-linear relationships [62]. The GE-ME pairing frequently emerged as an effective omics combination across multiple cancer types, suggesting that focused integration of complementary omics types may be more effective than comprehensive multi-omics approaches. Method selection also influenced outcomes, with different algorithms showing distinct strengths across various test scenarios. SNF demonstrated consistent performance in structural tests, while NEMO performed well in clinical feature analysis. This method-specific performance variation indicates the importance of matching analytical approaches to specific research questions.
Conclusion
The MOSD framework provides a guide to prioritizing factors such as sample size, feature selection, and omics integration strategy for reliable and interpretable MOI results. The framework incorporates insights from benchmarking of ten MOI methods across ten TCGA cancer datasets, offering evidence-based recommendations for multi-omics analysis. Guidelines emerging from this study include: maintaining approximately 26 samples per class for reliable results, selecting less than 10% of features to enhance performance, ensuring sample balance under a 3:1 ratio between classes, and keeping noise levels below 30%. For biological factors, distinct molecular subtypes should be prioritized, and omics combinations selected based on the specific clinical features of interest, with GE-ME often providing effective results for subtype identification.
These findings have implications for the design, execution, and interpretation of multi-omics studies, potentially improving efficiency and reproducibility of cancer research. Future research could explore framework adaptation to emerging data modalities and additional factors—such as temporal sampling or single-cell resolution—that may further refine multi-omics analyses. Continued refinement of these guidelines may support more efficient translation of complex molecular data into meaningful clinical and biological insights.
Supplementary Information
Acknowledgements
The authors of this paper thank the Division of Healthcare and Artificial Intelligence group within the Korea National Institute of Health for constructing the valuable multi-omics COVID-19 cohort dataset.
Authors’ contributions
I.J and H.K. conceived the study and designed the experiments. H.K. developed the multi-omics integration methods and performed the data analysis. H.K., E.H., and I.J contributed to the experimental design and interpretation of the results. H.K., E.H., and I.J wrote the manuscript. I.J supported the funding and supervised the study. All authors reviewed and approved the final version of the manuscript.
Funding
This work was supported by the Korea National Institute of Health (KNIH) research project (project no. 2024-ER-0801-01) and by the National Research Foundation of Korea (NRF) funded by the Korean Government (MSIT) (RS-2024-00440285).
Data availability
The data and associated code supporting the findings of this study are publicly available in our GitHub repository at https://github.com/cobi-git/MOPARAM. The repository includes all necessary materials to reproduce the results presented, along with detailed documentation and instructions for use.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Eonyong Han and Hwijun Kwon contributed equally to this work.
References
- 1.Karczewski KJ, Snyder MP. Integrative omics for health and disease. Nat Rev Genet. 2018;19(5):299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ebrahim A, Brunk E, Tan J, O’brien EJ, Kim D, Szubin R, et al. Multi-omic data integration enables discovery of hidden biological regularities. Nat Commun. 2016;7(1):13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hasin Y, Seldin M, Lusis A. Multi-omics approaches to disease. Genome Biol. 2017;18:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Misra BB, Langefeld C, Olivier M, Cox LA. Integrated omics: tools, advances and future approaches. J Mol Endocrinol. 2019;62(1):R21-45. [DOI] [PubMed] [Google Scholar]
- 5.Nicora G, Vitali F, Dagliati A, Geifman N, Bellazzi R. Integrated multi-omics analyses in oncology: a review of machine learning methods and tools. Front Oncol. 2020;10:1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwon MS, Kim Y, Lee S, Namkung J, Yun T, Yi SG, et al. Integrative analysis of multi-omics data for identifying multi-markers for diagnosing pancreatic cancer. BMC Genomics. 2015;16:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chai H, Zhou X, Zhang Z, Rao J, Zhao H, Yang Y. Integrating multi-omics data through deep learning for accurate cancer prognosis prediction. Comput Biol Med. 2021;134: 104481. [DOI] [PubMed] [Google Scholar]
- 8.Sammut SJ, Crispin-Ortuzar M, Chin SF, Provenzano E, Bardwell HA, Ma W, et al. Multi-omic machine learning predictor of breast cancer therapy response. Nature. 2022;601(7894):623–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lai YP, Wang LB, Wang WA, Lai LC, Tsai MH, Lu TP, et al. iGC–an integrated analysis package of gene expression and copy number alteration. BMC Bioinformatics. 2017;18:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leday GG, van de Wiel MA. PLRS: a flexible tool for the joint analysis of DNA copy number and mRNA expression data. Bioinformatics. 2013;29(8):1081–2. [DOI] [PubMed] [Google Scholar]
- 11.Tamborero D, Lopez-Bigas N, Gonzalez-Perez A. Oncodrive-CIS: a method to reveal likely driver genes based on the impact of their copy number changes on expression. PLoS One. 2013;8(2):e55489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gevaert O. Methylmix: an R package for identifying DNA methylation-driven genes. Bioinformatics. 2015;31(11):1839–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chae H, Rhee S, Nephew KP, Kim S. BioVLAB-MMIA-NGS: microRNA-mRNA integrated analysis using high-throughput sequencing data. Bioinformatics. 2015;31(2):265–7. [DOI] [PubMed] [Google Scholar]
- 14.Teng CS, Wu BH, Yen MR, Chen PY. MethGET: web-based bioinformatics software for correlating genome-wide DNA methylation and gene expression. BMC Genomics. 2020;21:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vasaikar SV, Straub P, Wang J, Zhang B. Linkedomics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018;46(D1):D956-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meng C, Basunia A, Peters B, Gholami AM, Kuster B, Culhane AC. MOGSA: integrative single sample gene-set analysis of multiple omics data. Mol Cell Proteomics. 2019;18(8):S153-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paczkowska M, Barenboim J, Sintupisut N, Fox NS, Zhu H, Abd-Rabbo D, et al. Integrative pathway enrichment analysis of multivariate omics data. Nat Commun. 2020;11(1):735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Canzler S, Hackermüller J. multiGSEA: a GSEA-based pathway enrichment analysis for multi-omics data. BMC Bioinformatics. 2020;21:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ozerov IV, Lezhnina KV, Izumchenko E, Artemov AV, Medintsev S, Vanhaelen Q, et al. In silico pathway activation network decomposition analysis (iPANDA) as a method for biomarker development. Nat Commun. 2016;7(1):13427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, et al. The cancer genome atlas pan-cancer analysis project. Nat Genet. 2013;45(10):1113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Consortium ICG, et al. International network of cancer genome projects. Nature. 2010;464(7291):993. [DOI] [PMC free article] [PubMed]
- 22.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edwards NJ, Oberti M, Thangudu RR, Cai S, McGarvey PB, Jacob S, et al. The CPTAC data portal: a resource for cancer proteomics research. J Proteome Res. 2015;14(6):2707–13. [DOI] [PubMed] [Google Scholar]
- 24.Tini G, Marchetti L, Priami C, Scott-Boyer MP. Multi-omics integration–a comparison of unsupervised clustering methodologies. Brief Bioinform. 2019;20(4):1269–79. [DOI] [PubMed] [Google Scholar]
- 25.Branciamore S, Chen ZX, Riggs AD, Rodin SN. CpG island clusters and pro-epigenetic selection for CpGs in protein-coding exons of HOX and other transcription factors. Proc Natl Acad Sci U S A. 2010;107(35):15485–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nam AS, Chaligne R, Landau DA. Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat Rev Genet. 2021;22(1):3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mirza B, Wang W, Wang J, Choi H, Chung NC, Ping P. Machine learning and integrative analysis of biomedical big data. Genes. 2019;10(2):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chauvel C, Novoloaca A, Veyre P, Reynier F, Becker J. Evaluation of integrative clustering methods for the analysis of multi-omics data. Brief Bioinform. 2020;21(2):541–52. [DOI] [PubMed] [Google Scholar]
- 29.Pierre-Jean M, Deleuze JF, Le Floch E, Mauger F. Clustering and variable selection evaluation of 13 unsupervised methods for multi-omics data integration. Brief Bioinform. 2020;21(6):2011–30. [DOI] [PubMed] [Google Scholar]
- 30.Rappoport N, Shamir R. Multi-omic and multi-view clustering algorithms: review and cancer benchmark. Nucleic Acids Res. 2018;46(20):10546–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duan R, Gao L, Gao Y, Hu Y, Xu H, Huang M, et al. Evaluation and comparison of multi-omics data integration methods for cancer subtyping. PLoS Comput Biol. 2021;17(8):e1009224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clark K, Vendt B, Smith K, Freymann J, Kirby J, Koppel P, et al. The cancer imaging archive (TCIA): maintaining and operating a public information repository. J Digit Imaging. 2013;26:1045–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guinney J, Dienstmann R, Wang X, De Reynies A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21(11):1350–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goldman MJ, Craft B, Hastie M, Repečka K, McDade F, Kamath A, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol. 2020;38(6):675–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silva TC, Colaprico A, Olsen C, D’Angelo F, Bontempi G, Ceccarelli M, et al. TCGA Workflow: Analyze cancer genomics and epigenomics data using Bioconductor packages. F1000Research. 2016;5:1542. [DOI] [PMC free article] [PubMed]
- 37.Jung I, Kim M, Rhee S, Lim S, Kim S. MONTI: a multi-omics non-negative tensor decomposition framework for gene-level integrative analysis. Front Genet. 2021. 10.3389/fgene.2021.682841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andersen NH, Christiansen JA, la Cour K, Aagesen M, Tang LH, Joergensen DS, et al. Differences in functioning between young adults with cancer and older age groups: a cross-sectional study. Eur J Cancer Care. 2022;31(6): e13660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu Y, Xu Y, Mao L, Lei W, Xiang J, Gao L, et al. Gene expression analysis reveals age and ethnicity signatures between young and old adults in human PBMC. Front Aging. 2022. 10.3389/fragi.2021.797040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang B, Mezlini AM, Demir F, Fiume M, Tu Z, Brudno M, et al. Similarity network fusion for aggregating data types on a genomic scale. Nat Methods. 2014;11(3):333–7. [DOI] [PubMed] [Google Scholar]
- 41.Yan D, Huang L, Jordan MI. Fast approximate spectral clustering. In: Proceedings of the 15th ACM SIGKDD international conference on Knowledge discovery and data mining. New York (NY): Association for Computing Machinery; 2009. p. 907–16. 10.1145/1557019.1557118.
- 42.Nguyen H, Shrestha S, Draghici S, Nguyen T. PINSplus: a tool for tumor subtype discovery in integrated genomic data. Bioinformatics. 2019;35(16):2843–6. [DOI] [PubMed] [Google Scholar]
- 43.Nguyen T, Tagett R, Diaz D, Draghici S. A novel approach for data integration and disease subtyping. Genome Res. 2017;27(12):2025–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rappoport N, Shamir R. NEMO: cancer subtyping by integration of partial multi-omic data. Bioinformatics. 2019;35(18):3348–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158(4):929–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu D, Wang D, Zhang MQ, Gu J. Fast dimension reduction and integrative clustering of multi-omics data using low-rank approximation: application to cancer molecular classification. BMC Genomics. 2015;16:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Monti S, Tamayo P, Mesirov J, Golub T. Consensus clustering: a resampling-based method for class discovery and visualization of gene expression microarray data. Mach Learn. 2003;52:91–118. [Google Scholar]
- 48.Argelaguet R, Velten B, Arnol D, Dietrich S, Zenz T, Marioni JC, et al. Multi-Omics factor analysis–a framework for unsupervised integration of multi-omics data sets. Mol Syst Biol. 2018;14(6): e8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mo Q, Wang S, Seshan VE, Olshen AB, Schultz N, Sander C, et al. Pattern discovery and cancer gene identification in integrated cancer genomic data. Proc Natl Acad Sci U S A. 2013;110(11):4245–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shen R, Olshen AB, Ladanyi M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics. 2009;25(22):2906–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chalise P, Fridley BL. Integrative clustering of multi-level ‘omic data based on non-negative matrix factorization algorithm. PLoS One. 2017;12(5):e0176278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee D, Seung HS. Algorithms for non-negative matrix factorization. Adv Neural Inf Process Syst. 2000;13:556–562.
- 53.Jeon J, Han EY, Jung I. MOPA: an integrative multi-omics pathway analysis method for measuring omics activity. PLoS One. 2023;18(3):e0278272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xuan L, Zhigang C, Fan Y. Exploring of clustering algorithm on class-imbalanced data. In: 2013 8th International Conference on Computer Science & Education. Piscataway, NJ, USA: IEEE; 2013. p. 89–93.
- 55.Reel PS, Reel S, Pearson E, Trucco E, Jefferson E. Using machine learning approaches for multi-omics data analysis: a review. Biotechnol Adv. 2021;49: 107739. [DOI] [PubMed] [Google Scholar]
- 56.Pfitzner D, Leibbrandt R, Powers D. Characterization and evaluation of similarity measures for pairs of clusterings. Knowl Inf Syst. 2009;19:361–94. [Google Scholar]
- 57.Wirapati P, Sotiriou C, Kunkel S, Farmer P, Pradervand S, Haibe-Kains B, et al. Meta-analysis of gene expression profiles in breast cancer: toward a unified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res. 2008;10:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Linnekamp JF, Hooff SRV, Prasetyanti PR, Kandimalla R, Buikhuisen JY, Fessler E, et al. Consensus molecular subtypes of colorectal cancer are recapitulated in in vitro and in vivo models. Cell Death Differ. 2018;25(3):616–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ritchie MD, Holzinger ER, Li R, Pendergrass SA, Kim D. Methods of integrating data to uncover genotype-phenotype interactions. Nat Rev Genet. 2015;16(2):85–97. [DOI] [PubMed] [Google Scholar]
- 60.Leek JT, Scharpf RB, Bravo HC, Simcha D, Langmead B, Johnson WE, et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet. 2010;11(10):733–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saeys Y, Inza I, Larranaga P. A review of feature selection techniques in bioinformatics. Bioinformatics. 2007;23(19):2507–17. [DOI] [PubMed] [Google Scholar]
- 62.Huang S, Chaudhary K, Garmire LX. More is better: recent progress in multi-omics data integration methods. Front Genet. 2017;8:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data and associated code supporting the findings of this study are publicly available in our GitHub repository at https://github.com/cobi-git/MOPARAM. The repository includes all necessary materials to reproduce the results presented, along with detailed documentation and instructions for use.