

Abstract
Type 1 diabetes (T1D) is a chronic autoimmune disease that leads to progressive destruction of pancreatic beta cells. Compared to healthy controls, a characteristic feature of patients with T1D is the presence of self-reactive T cells with a memory phenotype. These autoreactive memory T cells in both the CD4+ and CD8+ compartments are likely to be long-lived, strongly responsive to antigenic stimulation with less dependence on costimulation for activation and clonal expansion, and comparatively resistant to suppression by regulatory T cells (Tregs) or downregulation by immune-modulating agents. Persistence of autoreactive memory T cells likely contributes to the difficulty in preventing disease progression in new-onset T1D and maintaining allogeneic islet transplants by regular immunosuppressive regimens. The majority of immune interventions that have demonstrated some success in preserving beta cell function in the new-onset period have been shown to deplete or modulate memory T cells. Based on these and other considerations, preservation of residual beta cells early after diagnosis or restoration of beta cell mass by use of stem cell or transplantation technology will require a successful strategy to control the autoreactive memory T cell compartment, which could include depletion, inhibition of homeostatic cytokines, induction of hyporesponsiveness, or a combination of these approaches.
Keywords: Autoimmunity, Effector memory T cells, Central memory T cells, Costimulation blockade, Homeostatic cytokines, CD2, Alefacept, CD3
Introduction
Type 1 diabetes (T1D) results from destruction of insulin-producing beta cells by autoreactive T cells that have escaped central and peripheral immune tolerance [1]. T1D is an organ-specific autoimmune disease that occurs in the context of disease-specific genetic backgrounds and epigenetic influences in those exposed to one or more environmental triggers, but the precise etiology remains elusive [1]. Insulin therapy is lifesaving but does not entirely avert serious complications, including microvascular and macrovascular disease and death [2•]. Higher levels of endogenous insulin secretion correlate with lower rates of complications, and hence, there is a strong interest in interventions that can preserve or restore even modest levels of beta-cell function [3]. Maintenance of endogenous insulin secretion—whether by naturally occurring beta cells, insulin-producing cells derived from stem cells, or transplanted allogeneic islets—will necessitate, at least in part, an immune intervention that halts diabetes autoimmunity and, ideally, restores immunologic tolerance.
The Immunopathology of T1D
T1D results from an autoimmune process with a strong genetic predisposition and likely environmental triggers. The evidence suggests that there are defects in both central and peripheral immune tolerance resulting in the emergence, activation, and persistence of autoreactive effector and memory T cells (both CD4+ and CD8+) that damage and eventually destroy most of the insulin-producing beta cells in the pancreatic islets. The strongest genetic influence comes from polymorphisms in HLA class I and II alleles, followed by 40 or more other loci, the vast majority of which are immune response genes, reinforcing the notion that T1D is a disease of immune dysregulation [4]. HLA susceptibility alleles may lead to alterations in binding affinities of the MHC-peptide complex to cognate T cell receptors (TCRs) on both CD4+ and CD8+ T cells, enabling thymic escape of autoreactive T cells (failure of central tolerance).
Release and processing of beta-cell antigens and subsequent presentation by antigen-presenting cells (APCs) are thought to occur during times of increased rates of physiologic beta-cell turnover [5], during periods of increased beta-cell stress, or following infections with beta-cell trophic pathogens which lead to upregulation of MHC class I molecules and a pro-inflammatory milieu [6]. These events, combined with presumed genetic defects in peripheral tolerance checkpoints, lead to the activation of autoreactive T cells and the initiation and propagation of an islet-specific immune attack. A defect in peripheral tolerance could potentially result from various genetic abnormalities, including T1D-associated polymorphisms in the genes encoding IL-2 and the IL-2 receptor α subunit, cytotoxic T-lymphocyte-associated protein 4 (CTLA4), and the FoxP3 transcription factor [1, 4], all of which are required for the development and maintenance of regulatory T cells (Tregs). At the same time, diabetogenic T cells in T1D appear to be unusually resistant to suppression by Tregs [7], a process that may involve pro-inflammatory mediators or intrinsic properties of memory and effector T cells.
A key observation made during the past decade was that autoreactive T cells are also found in the peripheral blood of healthy controls, indicating that the immunopathology of T1D is more subtle than previously thought. Both CD4+ and CD8+ T cells reactive to a variety of T1D autoantigens occur in the peripheral blood of healthy controls at frequencies similar to those found in patients with T1D. However, a key difference between the two populations is that the autoreactive T cells in healthy subjects tend to have a naïve phenotype whereas those in T1D patients are memory T cells [8, 9, 10•, 11••]. Moreover, the autoreactive T cells in T1D patients show hallmarks of antigen-driven expansion, including telomere shortening, terminal differentiation and memory status, and skewed oligoclonal T cell receptor repertoires [9, 11••].
These findings suggest that there is a critical break-down of peripheral tolerance in T1D consisting of the activation, expansion, and differentiation of circulating naïve beta-cell reactive T cells into highly pathogenic, armed, effector, and memory T cells which, because they are long-lived and do not require costimulation for further proliferation, result in an intractable autoreactive state with progressive destruction of beta cells. The key role of memory T cells in T1D pathogenesis is further highlighted by the phenomenon of autoimmune recurrence in the context of islet and pancreas transplantation. This was first observed in T1D patients who had received pancreatic grafts from HLA-identical twins or siblings [12–14] but was later also seen in recipients of cadaveric pancreatic grafts [15] and in recipients of islet allografts [16]. Biopsies of failed grafts revealed selective destruction of beta cells with no evidence of alloimmune rejection [13–15]. Recurrence of T1D autoimmunity was strongly suggested by the identification of autoreactive, antigen-specific CD8+ and CD4+ T cells after islet and pancreas transplantation [17–20]. These autoreactive T cells have a memory phenotype, which is known to be difficult to eradicate with most immunosuppressive therapies, and further may preferentially expand following lympho-depleting regimens due to homeostatic proliferation [18–20]. Taken together, these various lines of evidence suggest that persistent, autoreactive memory T cells are a critical component of T1D autoimmunity. Further, it is likely that therapeutic modulation of these memory T cells will be an essential component of any successful immune intervention strategy to preserve or restore beta-cell function in T1D.
Memory T Cell Subsets in Autoimmunity
Our understanding of the differentiation and lineage commitment of T cell subsets is largely based on studies of infection models. The classical description of T cell memory posits that antigen-specific naïve T cells are activated following engagement of the T cell receptor and appropriate costimulatory signals and then proliferate and differentiate into effector T cells (Teff). Subsequently, effector T cells undergo a contraction phase resulting in either apoptosis or differentiation into long-lived memory T cells. In the original description, memory T cells were divided into two types: central memory (Tcm) and effector memory (Tem) cells [21]. Tcm cells are long-lived memory T cells that retain expression of CCR7 and CD62L, homing receptors that allow these cells to migrate to secondary lymphoid organs. In contrast, Tem cells have lost constitutive expression of CCR7 and most CD62L and instead display chemokine and adhesion receptors required for homing to inflammed tissues; further, Tem cells are characterized by rapid effector function. In a typical acute infection, the Tem compartment declines with time after resolution of the infection, but a stable quiescent Tcm population can persist for decades, giving rise to new Teff cells upon reencountering the antigen. Compared to naïve T cells, both Tcm and Tem have high sensitivity to antigenic stimulation, are less dependent on costimulation, and show strong proliferative responses (mainly Tcm) to homeostatic cytokines such as IL-7 and IL-15 [21].
Figure 1 illustrates the relationships and identification of the major T cell lineages, including CD4+ Tn, Tcm, Tem, and Treg cells and CD8+ Tn, Tcm, and Tem cells. Although this is a useful framework, the neat distinction between Tcm and Tem may not be entirely accurate, however, and there is evidence for considerable heterogeneity in terms of surface receptor expression, effector function, location, and trafficking. Thus, there is a subset of Tem that retains CD45RA expression (a marker of naïve T cells), called Temra cells, and more recently, two additional types of memory T cells have been proposed: resident memory T cells (Trm) and stem memory T cells (Tsm) [22•]. Adding further complexity is evidence for interconversion between memory subsets. Perhaps most important is that while some clarity is emerging around T cell memory in the context of infection, much less is known about the nature and dynamics of self-reactive memory T cells in autoimmunity. As noted earlier, there is no doubt that memory T cells are a cardinal feature of chronic autoimmunity, but comparatively little is known about the origin, identity, and dynamics of autoreactive memory T cells. It has been proposed that the constant presence of self-antigens in autoimmunity may preclude the development of Tcm and Trm cells, and thus, Tem may be the dominant type of memory T cell in autoimmune conditions [23•].
Fig. 1.
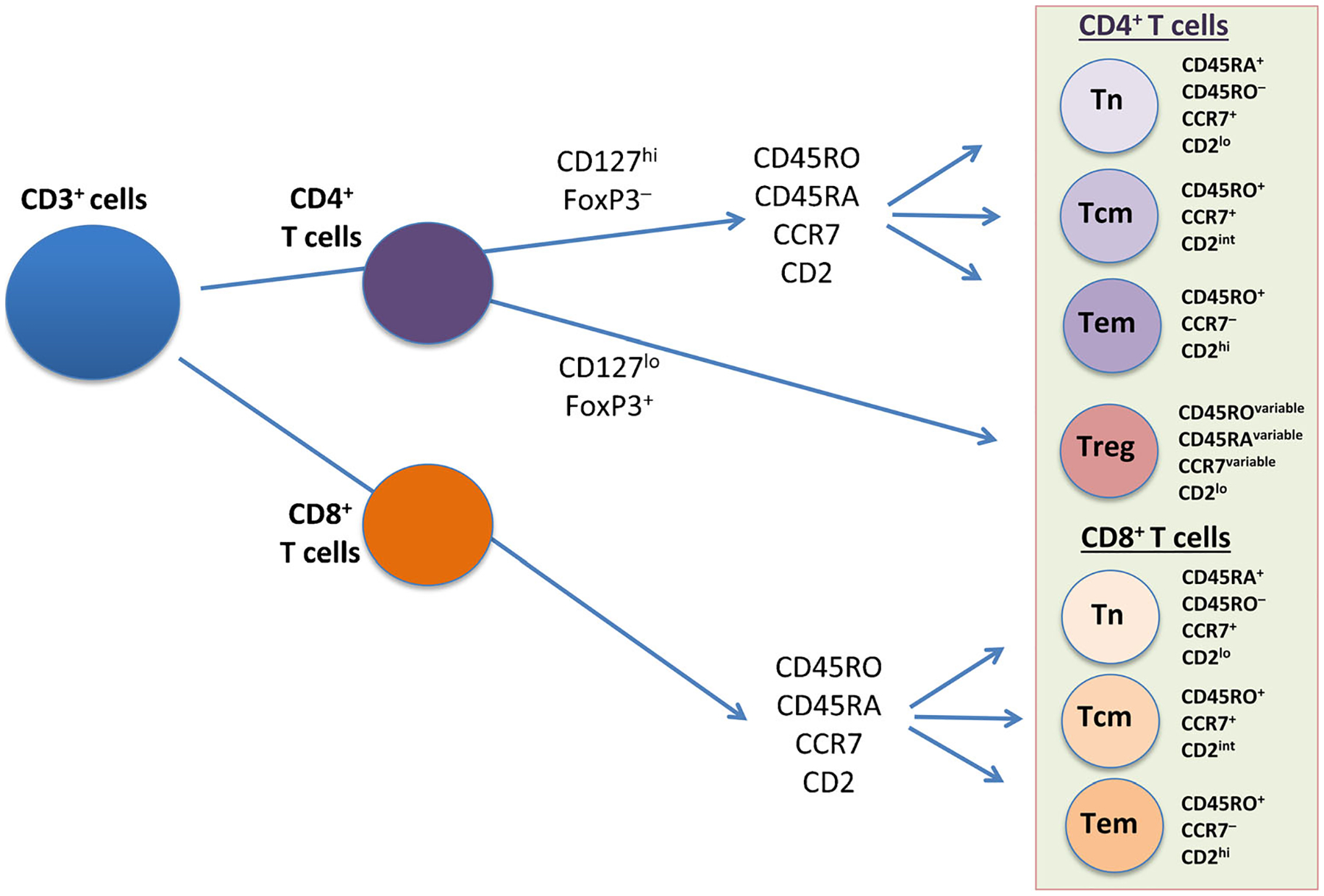
Relationships and identification of the major T cell lineages, including memory T cells. The schematic shows the major subsets of CD4+ and CD8+ T cells, identified by standard cell surface markers using multicolor flow cytometry and sequential gating [87]. Tn naïve T cell, Tcm central memory T cell, Tem effector memory T cell, Treg regulatory T cell
Tem cells are relatively long-lived, are able to undergo self-renewal using IL-7 and IL-15, possess immediate effector functionality like the Teff subset, and circulate between the blood, secondary lymphoid organs, and peripheral tissues, making the Tem population a highly effective, antigen-specific memory subset in autoimmunity [23•]. Recent work in the nonobese diabetic (NOD) mouse suggests that in the CD8+ compartment, beta-cell-specific T cells acquire the mature effector memory phenotype in infiltrated islets and then emigrate to peripheral lymphoid tissues, from whence they may recirculate to the pancreas and damage additional islets [24••]. In human studies, the precise phenotype of islet-reactive memory T cells is unclear, but based on their effector functionality, these cells can be assumed to be Tem cells [19, 20]. However, more work is required to define the subsets of memory T cells involved in autoimmunity. In T1D, there is evidence for the involvement of CD4+ Tcm cells [25••], as discussed further below. Further, analysis of autoreactive CD8+ T cells from T1D patients found greater proportions of stem memory (Tsm) T cells, which may serve as a reservoir for more differentiated memory T cells and could perpetuate the autoimmune destruction of beta cells [11••].
Strategies to Target Memory T Cells in T1D
In principle, several distinct strategies can be pursued to target memory T cells in T1D, including inhibition of homeostatic cytokines required to support and expand memory T cells, induction of hyporesponsiveness (e.g., anergy, exhaustion, or senescence), or T cell depletion. We will discuss strategies that have been tested, strategies that are in planning or in progress, and strategies that have a compelling rationale but are not yet ready for clinical translation.
Anti-Cytokine Agents
The homeostasis of memory CD4+ and CD8+ T cells is regulated by a combination of IL-7 and IL-15. In the CD8+ compartment, IL-7 primarily supports cell viability while IL-15 induces basal homeostatic proliferation, while memory CD4+ T cells require both IL-7 and IL-15 for basal homeostatic proliferation [26]. Importantly, lymphodepletion induced by immunosuppressive therapy during islet transplantation results in increases in IL-7 and IL-15 with vigorous homeostatic proliferation of autoreactive memory CD4+ and CD8+ T cells [19]. This has led to the concern that homeostatic proliferation after islet transplantation preferentially expands autoreactive memory T cells that can drive the recurrence of islet autoimmunity [19]. This concern is consistent with the notion that induction of lymphopenia (e.g., by viral infections) may lead to homeostatic proliferation and precipitation of autoimmunity [27]. For these reasons, inhibition of IL-7 and IL-15 signaling has attracted attention as a potential therapeutic strategy in autoimmunity and T1D.
Treatment with antibodies that target the alpha subunit of the IL-7 receptor (IL-7Rα) reverses new-onset autoimmune diabetes in the NOD mouse. Interestingly, anti-IL-7Rα therapy did not produce robust depletion of Tem cells but instead increased the proportions of PD-1-expressing Tems and Tregs [28••, 29••]. PD-1 is one of a growing list of immune checkpoint inhibitors that control immune responses and contribute to peripheral tolerance [30•]. PD-1 is of particular interest because it is upregulated following T cell activation and mediates downregulation of effector functions after binding to cognate ligands [31], thereby suppressing islet infiltration by CD4+ T cells in the NOD mouse model of T1D [31, 32•, 33, 34]. In a recent study, targeted expression of the PD-1 ligand PD-L1 in neo-islets in diabetic NOD mice led to decreased proliferation and increased apoptosis of infiltrating CD4+ T cells with robust reversal of hyperglycemia, suggesting that the PD-1/PD-L1 pathway is strongly tolerogenic in this model [35]. PD-1 upregulation on memory T cells by blockade of the IL-7 receptor indicates that therapeutic modulation of the autoreactive memory compartment can be achieved not only by depletion but also by induction of negative regulatory mechanisms. No clinical data with anti-IL-7Rα antibodies have been reported to date, but early stage clinical trials are in progress (e.g., see ClinicalTrials.gov identifier NCT02045732).
Antibodies targeting IL-15 or the IL-15 receptor have shown efficacy in a number of preclinical models of autoimmunity as well as in allograft survival, including islet allografts [reviewed in ref. 36]. These promising results led to clinical trials of an anti-IL-15 mAb (HuMax-IL15, later called AMG 714) in rheumatoid arthritis and psoriasis [37]. Efficacy in these indications was modest, and further progress has been slow. More recent work has shown that signaling through the heterotrimeric IL-15 receptor is complex. The soluble form of the IL-15 receptor alpha subunit can associate with IL-15 to produce a hyperagonist. Depending on their binding epitopes, some anti-IL-15 mAbs, such as DISC0280, similarly show agonist activity in vivo, even though in vitro, the antibodies inhibit IL-15-dependent T cell proliferation [38]. These results highlight the complexity of pursuing IL-15 as a therapeutic target. Nevertheless, the specific role of IL-15 in the homeostasis of CD8+ Tem cells continues to make this an attractive target in autoimmunity, but additional work will be required to optimize the therapeutic specificity of an IL-15 antagonist.
Targeting the CD2 Pathway
CD2 is expressed on T cells and facilitates T cell costimulation and interaction with dendritic cells, B cells, and endothelial cells [39, 40]. In addition, CD2 may facilitate cell migration to, and retention in, sites of acute inflammation. The primary ligand for CD2 in humans is CD58 (leukocyte function antigen-3 (LFA-3)), whereas in rodents, the primary ligand is CD48 [39, 41, 42]. CD48 is structurally and phylogenetically related to CD58, and both are found on a variety of cell types including antigen-presenting cells and endothelial cells. In humans, the highest levels of CD2 expression are on circulating effector memory T cells (Tem), with intermediate expression on central memory T cells (Tcm), and the lowest expression on naïve T cells and Tregs (Fig. 1) [41, 43, 44, 45••]. These characteristics make the CD2 pathway an especially attractive target in autoimmunity.
In animal studies, disruption of the CD2:CD48 pathway can prevent diabetic autoimmunity, prolong allograft acceptance, and promote immune tolerance. In the BioBreeding/Worcester rat model of spontaneous autoimmune diabetes (an animal model of human T1D), anti-CD2 MAb treatment results in long-term protection from spontaneous diabetes and lymphocytic insulitis and significant protection against adoptive transfer of disease [46]. This protection appears to be the result of a selective depletion of CD4+ T cells and reduced activation of effector T cells. In animal models of transplantation, blocking the CD2:CD48 pathway alone or in combination with other T cell molecules delays rejection and in some instances results in immune tolerance. In studies of murine cardiac allografts, anti-CD2 MAb therapy alone significantly prolongs graft survival, whereas coadministration with anti-CD48, anti-CD3, or CTLA4-Ig results in permanent acceptance of grafts in the absence of ongoing immune therapy [47–51]. In other studies, a brief course of anti-CD2 alone induced donor-specific tolerance in DBA/2 recipients of B6AF1 islet allografts [52, 53]. Tolerance was associated with specific reductions of CD2+ T cells and intragraft granzyme B and IL-10. These preclinical studies led to intense interest in modulating the CD2 pathway in autoimmunity and transplantation, with the hope that this approach may have the potential to engender immune tolerance. It should be noted, however, that disruption of the CD2:CD48 pathway has not been evaluated to date in the NOD mouse model with respect to prevention or reversal of diabetic autoimmunity.
Alefacept, a fusion protein consisting of two LFA-3 molecules bound to the Fc portion of IgG1 [39], binds CD2, which is expressed most prominently on CD4+ and CD8+ effector memory T (Tem) cells [40], the cells thought to be primarily responsible for beta-cell destruction in T1D [1]. Studies in psoriasis had demonstrated that alefacept selectively targets effector memory T cells and the mechanism of action was presumed to be a combination of partial depletion of these subsets and modulation of CD2-mediated costimulation [44]. In psoriasis, two or more courses of alefacept result in prolonged off-therapy remission, which may be a form of tolerance [54]. These results prompted the Immune Tolerance Network to conduct the T1DAL trial, which showed that two 12-week courses of alefacept in new-onset T1D result in preservation of endogenous insulin production, a significant reduction in insulin use, and a remarkable 50% decrease in rates of major hypoglycemia in drug- vs. placebo-treated patients at 12 months [45••].
Analysis of T cell subsets by flow cytometry revealed that alefacept treatment led to significant reductions in central and effector memory CD4+ and CD8+ T cells, preservation of Tregs, and favorable increases in Treg/Teff ratios, providing a plausible mechanism for the clinical effects of this drug [45••]. Preservation of Tregs—a novel finding in the T1DAL trial that had not been studied in the earlier psoriasis trials—was likely related to the lower expression of CD2 on Tregs compared to the higher levels on memory T cells [45••]. Analysis of the 24-month clinical and mechanistic results is ongoing. Of particular interest will be evidence of T cell modulation, including induction of T cell unresponsiveness and/or partial agonist effects, as has previously been suggested based on in vitro effects of alefacept on human PBMCs [41, 55]. Alefacept appears to be a very promising agent because it is well tolerated with no reports of drug-associated serious adverse events and no between-group (alefacept vs. placebo) differences in overall rates of adverse events [45••], suggesting that it may be an appropriate intervention in T1D, even in children.
Targeting CD3
The TCR is composed of two disulfide-bonded chains (α and β or γ and δ) and exists as a complex with three dimeric CD3 molecules: CD3γε, CD3δε, and CD3ζζ [55]. The intracellular domains of each of the CD3 chains contain immunoreceptor tyrosine-based activation motifs (ITAMs) that serve as the nucleating point for the intracellular signal transduction machinery upon TCR engagement [56]. Hence, CD3 plays a critical role in the antigen-driven activation and clonal expansion of antigen-specific T cells. Memory T cells have higher sensitivity to antigenic stimulation and less dependence on costimulation than naïve T cells [21], and therefore, targeting CD3 may be a strategy to modulate the memory T cell compartment in autoimmunity.
In the 1990s, Chatenoud and colleagues demonstrated remarkable efficacy of a short course of treatment with an anti-CD3 mAb in reversing diabetes and inducing immunologic tolerance in the NOD mouse [57]. Clinical use of anti-CD3 mAbs was initially hampered by significant toxicities, primarily T cell activation, and cytokine release syndrome (CRS), but this was substantially ameliorated by modifications in the Fc portion to eliminate Fc receptor binding [58]. This paved the way for clinical development, and in a landmark study, Herold and colleagues showed that the Fc receptor-nonbinding hOKT3γ1 (Ala-Ala) anti-CD3 antibody (later called teplizumab) preserved C-peptide and reduced HbA1c levels and insulin use in patients with new-onset T1D [59]. These results were confirmed independently using another anti-CD3 mAb (otelixizumab) [60], and both groups showed that these benefits were maintained for up to 3 years in a subset of patients [61, 62]. However, despite the improved characteristics of these biologics, the efficacy came at the cost of CRS during drug administration and Epstein-Barr virus (EBV) reactivation or EBV-related disease in a significant proportion of treated patients [62]. Subsequent larger trials failed to meet their primary endpoints, in retrospect perhaps because the endpoints selected were too stringent [63] or because the selected dose was too low [64, 65]. With this in mind, there may be a role for anti-CD3 therapies in T1D and a trial of teplizumab in those at risk of T1D is in progress (ClinicalTrials.gov identifier NCT01030861).
In a phase 2 trial of teplizumab, in which subjects received a second course of the drug at 12 months, C-peptide was significantly preserved at 24 months in the treated patients versus controls. An important finding in this trial (the AbATE trial) was that a subgroup of responders could be identified that had excellent C-peptide preservation while the nonresponders were almost indistinguishable from the controls [66•]. Response was predicted by better glycemic control and lower insulin use at baseline as well as subtle immunologic differences. However, the basis for drug response and the true mechanism of action of teplizumab remain unresolved. Earlier studies, particularly in the NOD model, had suggested that anti-CD3 treatment leads to selective depletion of pathogenic T cells with preservation of Tregs, but this mechanism has not been confirmed in the human studies [67]. A clue to the mechanism may come from the original observation that responders show a decrease in the CD4/CD8 ratio [59, 61], which is consistent with a more recent finding of an increase in CD8+ central memory T cells in responders in the Delay trial, in which T1D patients beyond the new-onset period were treated with teplizumab [68•]. While expansion of a memory CD8+ T cell population would seem to be counterintuitive, this may suggest that anti-CD3 has a partial agonist effect and is inducing some form of Teff modulation. This finding, which needs to be confirmed and extended by more detailed phenotypic analysis of the T cell subset involved, may provide validation for the notion that modulation of pathogenic memory T cells is critical for induction of tolerance in T1D.
Costimulation Blockade
Costimulation constitutes the key second signal required to activate T cells after a first encounter with antigen and is the bridge between innate and adaptive immunity. APCs process and present antigen-derived peptides via MHC-peptide complexes to TCRs. However, T cell activation requires a second signal via costimulatory receptors; in the absence of costimulation, the T cell becomes anergic (unresponsive) or may undergo apoptosis. The classic costimulatory ligand-receptor pair is comprised of the ligands CD80 and CD86 (also known as B7-1 and B7-2) on APCs and the receptor CD28 on naïve CD4+ and CD8+ T cells [69]. After T cell activation, a second CD80/CD86 receptor is expressed on T cells, CTLA4, which, unlike CD28, imparts an inhibitory signal [70]. Further work has uncovered an array of both costimulatory and coinhibitory pathways [30•, 71], many of which are amenable to pharmacologic intervention [72]. The first drug targeting costimulation was the fusion protein CTLA4-Ig [73], later known as abatacept and approved for rheumatoid arthritis, which blocks costimulation by acting as a soluble decoy receptor binding to CD80/CD86 and preventing ligation of CD28 [74].
Costimulation is primarily required for activation of naïve T cells, while memory T cells are less dependent on this process [21]. However, in a chronic autoimmune condition such as T1D, there is thought to be ongoing activation of naïve T cells, driven in part by the phenomenon of antigen spreading. It is known that at the time of diagnosis of T1D and even years afterwards, a significant proportion if islets retain insulin-secreting beta cells, and hence, there is ongoing exposure to autoantigens over many years [1]. It is unclear at what point in the natural history of T1D activation of fresh naïve T cells ceases and a stable pool of memory T cells is established; at this point, it can be assumed that costimulation becomes quantitatively less important and costimulation blockade would be predicted to become ineffective.
A trial of abatacept in new-onset T1D demonstrated significant preservation of C-peptide secretion at 24 months, although the degree of C-peptide preservation was modest and after 6 months appeared to parallel the decline seen in the placebo group despite continuous treatment for the duration of the 2-year trial [75]. Analysis of peripheral T cell subsets by flow cytometry revealed a modest but significant increase in naïve CD4+ T cells and a decrease in central memory CD4+ T cells, which appeared to correlate with C-peptide preservation [25••]. Of some concern was a parallel and significant decrease in Tregs [25••], which may have contributed to the observation that C-peptide responses began to decline soon after treatment began, albeit at an initially slower rate than in controls [75]. The effect on Tregs is not surprising, given that the development and peripheral survival of Tregs is CD28 dependent [74]. Also, since cell surface CTLA4 may be a core mechanism through which Tregs control APC function [76], there are unresolved questions about the effect of ongoing treatment with soluble CTLA4-Ig on Treg function. Nevertheless, the abatacept trial in T1D provided an important first insight into the potential for costimulatory blockade in T1D and warrants further study. A trial evaluating abatacept in the prevention of T1D in at-risk patients is currently ongoing (ClinicalTrials.gov identifier NCT01773707).
The specific effect of abatacept on CD4+ Tcm cells, with no effect on Tem cells, is intriguing. The authors speculate that this could be because of effects on migration or retention of Tcm cells in lymph nodes, possibly mediated by abatacept inhibition of transmigration of Tcm cells across CD86-expressing microvascular endothelial cells [25••]. However, in parallel with the decrease in circulating CD4+ Tcm was an increase in naïve CD4+ T cells, which leaves open the possibility that abatacept blocked activation of naïve T cells which was occurring in the new-onset setting, even though this is considered to be years after initiation of the autoimmune process in T1D. It is also notable that this is the second instance in which preservation of C-peptide was associated with a change in a Tcm population (the other is the association between anti-CD3 and CD8+ Tcm), which suggests that Tcm cells may in fact play an important role in the immunopathology of T1D.
T Cell Depletion
If memory T cells are important in driving and sustaining autoimmune responses, then T cell depletion should, at least in the short term, demonstrate some efficacy. In one of the earliest immune intervention trials, a pilot study of the combination of antithymocyte globulin (ATG) plus prednisone in recent-onset T1D gave a signal of efficacy in terms of improved HbA1c and reduced insulin dose in some of the treated patients [77]. These data together with other promising pilot clinical and preclinical results led to the randomized, double-blind START trial comparing ATG with placebo in patients with new-onset T1D. Surprisingly, there was no benefit in the ATG group compared to placebo at 12 months, and there was even a suggestion that C-peptide responses in the ATG-treated subjects showed accelerated decline in the first 6 months before stabilizing in the second 6 months [78•]. A clue to what may have happened with this intervention was the observation that virtually all treated patients experienced moderate to severe CRS during the drug administration period and serum sickness 7–10 days later, accompanied by brief but substantial increases in serum levels of IL-6 and acute-phase proteins, suggesting adverse immune activation early on. Unintended immune activation is also thought to be the mechanism for transient disease exacerbation seen in a pilot study of IL-2 plus rapamycin in T1D [79]. Also notable was the finding that despite profound CD4+ and CD8+ T cell depletion, including naïve and central memory subsets, effector memory T cells were resistant to depletion which, together with strong depletion of Tregs, led to an unfavorable Treg/Tem ratio in the first 6 months of the trial [78•].
The results of the START trial have led us to reevaluate the value of broad T cell-depleting therapies in T1D. It is useful to remember that a key feature of the BioBreeding rat, an accepted model for T1D susceptibility, is profound lymphopenia [80], and it has been suggested that lymphopenia or lymphodepletion predispose to autoimmunity, including T1D, because homeostatic expansion, driven by IL-7 and IL-15, enables autoreactive memory T cells to survive and proliferate [reviewed in ref. 81]. As noted elsewhere in this review, autoreactive memory T cells have been shown to expand following lymphopenia after islet transplantation [19], and lymphopenia-induced expansion of autoreactive clones results in rapid onset of autoimmune diabetes in a model of islet self-reactivity [27]. It is interesting to contrast the START trial with the recent report of a pilot study of the combination of ATG with granulocyte-colony stimulating factor (GCSF) in T1D, which suggested clinical efficacy in association with relative preservation of Tregs [82•]. It is unclear whether the effects seen in this trial reflect the lower dose of ATG that was used (one third the dose used in START) or the combination with GCSF. A larger, fully powered trial comparing the combination of ATG/GCSF with ATG alone and placebo, now underway (ClinicalTrials.gov identifier NCT02215200), should answer that question.
Memory T Cell-Specific Strategies
Anti-CD45RO Antibodies
A hallmark of memory T cells is expression of the RO isoform of CD45 (see Fig. 1), a transmembrane protein tyrosine phosphatase that plays a critical role in lymphocyte activation. A monoclonal antibody that recognizes both the RO and RB isoforms of CD45 (CD45RB is expressed on effector T cells) has been shown to selectively mediate apoptosis of CD4+ CD45RO/RB+ T cells, induce populations of CD4+ and CD8+ Tregs, and mediate long-term survival of human islet allografts in humanized NOD/SCID mice [83]. These are very attractive features of an intervention that specifically targets memory T cells, but to date, translation into the clinic has not been reported. One difficulty may be that there are eight isoforms of CD45 that are variously expressed on both T and B cells [83], and therefore, specificity will be important.
Kv1.3 Channel Inhibitors
Kv1.3 is a human K+ channel that regulates membrane potential and Ca2+ signaling in human T cells, and its expression is increased fourfold to fivefold in activated and memory CD4+ and CD8+ T cells, especially Tem cells [84, 85]. Kv1.3 is upregulated to 1500–2000 channels per cell in activated Tem cells (CD45RA−/CCR7−, see Fig. 1) and takes over as the dominant functional K+ channel in these cells, identifying Kv1.3 as a potential signature biomarker in activated Tem cells [reviewed in ref. 84]. Kv1.3 is highly expressed in postmortem multiple sclerosis (MS) brain inflammatory infiltrates, in both the perivenular infiltrates and in the parenchyma of demyelinated MS lesions. The Kv1.3+ cells are CCR7− CD4+ and CD8+ Tem cells, confirming earlier findings that activated memory T cells are a cardinal feature of MS lesions in the brain [84]. Similarly, in both RA and T1D, disease-associated autoreactive T cells are predominantly of the Tem phenotype with high Kv1.3 expression. Small-molecule Kv1.3 inhibitors were found to selectively inhibit cytokine production and proliferation of autoreactive Tem cells from RA and T1D patients in vitro and ameliorated autoimmunity in rat models of RA and T1D with no signs of systemic toxicity [85]. The results of a phase 1b trial in psoriasis were recently reported for the Kv1.3 inhibitor dalazatide (formerly ShK-186), indicating a signal of efficacy and good tolerability [86]. Dalazatide is the first of a novel class of Tem-specific agents with interesting potential in autoimmunity, but further phase 2 and 3 clinical developments will be required to assess the efficacy and safety of this drug in psoriasis and other autoimmune indications, including T1D.
Conclusions
Immunologic memory is a hallmark of autoimmunity and accounts for the persistence of autoimmune conditions and the difficulty in inducing long-term off-therapy remission, i.e., restoration of immunologic tolerance. T cell-driven, chronic autoimmune conditions, notably T1D, are characterized by the presence of autoreactive memory T cells, predominantly Tem cells but likely also other subsets of the memory compartment. These memory T cells appear to be comparatively resistant to suppression by Tregs, are difficult to eliminate by depletional therapies, have limited dependence on costimulation, and show preferential expansion by homeostatic proliferation following lymphodepletion. Nevertheless, in recent years, phase 2 trials of certain T cell-directed immunologic agents have indicated that it is possible to target the memory T cell compartment in T1D, by a combination of depletion and modulation, with concomitant preservation of islet function. Additional fundamental studies are needed to identify pathways uniquely involved in the survival and activation of memory T cells, followed by translational clinical trials evaluating approaches to target these pathways, perhaps in combination with other disease-specific therapies. Much work still needs to be done, but it is clear that efficient control of autoreactive memory T cells will be required if we are to realize the dream of reestablishing immunologic tolerance to prevent or reverse type 1 diabetes.
Acknowledgments
This publication was supported in part by the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH) under Award Number UM1AI109565. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of Interest Mario Ehlers declares that he has no conflict of interest. Mark Rigby is an employee of Janssen R&D, Pharmaceutical Companies of Johnson & Johnson.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. 2010;464:1293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.•.Lind M, Svensson AM, Kosiborod M, et al. Glycemic control and excess mortality in type 1 diabetes. N Engl J Med. 2014;371:1972–82. [DOI] [PubMed] [Google Scholar]; An important study showing that even with good glycemic control, T1D results in substantial excess mortality.
- 3.Steffes MW, Sibley S, Jackson M, Thomas W. Beta-cell function and the development of diabetes-related complications in the diabetes control and complications trial. Diabetes Care. 2003;26:832–6. [DOI] [PubMed] [Google Scholar]
- 4.Todd JA. Etiology of type 1 diabetes. Immunity. 2010;32:457–67. [DOI] [PubMed] [Google Scholar]
- 5.Wang P, Fiaschi-Taesch NM, Vasavada RC, et al. Diabetes mellitus—advances and challenges in human β-cell proliferation. Nat Rev Endocrinol. 2015;11:201–12. [DOI] [PubMed] [Google Scholar]
- 6.Coppieters KT, Wiberg A, Tracy AM, et al. Immunology in the clinic review series: focus on type 1 diabetes and viruses: the role of viruses in type 1 diabetes: a difficult dilemma. Clin Exp Immunol. 2012;168:5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schneider A, Rieck M, Sanda S, et al. The effector T cells of diabetic subjects aqre resistant to regulation via CD4+FOXP3+ regulatory T cells. J Immunol. 2008;181:7350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Danke NA, Yang J, Greenbaum C, et al. Comparative study of GAD65-specific CD4+ T cells in healthy and type 1 diabetic subjects. J Autoimmun. 2005;25:303–11. [DOI] [PubMed] [Google Scholar]
- 9.Monti P, Scirpoli M, Rigamonti A, et al. Evidence for in vivo primed and expanded autoreactive T cells as a specific feature of patients with type 1 diabetes. J Immunol. 2007;179:5785–92. [DOI] [PubMed] [Google Scholar]
- 10.•.Oling V, Reijonen H, Simell O, et al. Autoantigen-specific memory CD4+ T cells are prevalent early in progression to type 1 diabetes. Cell Immunol. 2012;273:133–9. [DOI] [PubMed] [Google Scholar]; One of several studies showing that islet-reactive CD4+ T cells are found in the peripheral blood of healthy subjects but that these cells tend to have a naïve phenotype versus the memory phenotype found in subjects with T1D autoimmunity.
- 11.••.Skowera A, Ladell K, McLaren JE, et al. β-cell-specific CD8 T cell phenotype in type 1 diabetes reflects chronic autoantigen exposure. Diabetes. 2015;64:916–25. [DOI] [PMC free article] [PubMed] [Google Scholar]; An important study showing that beta cell-specific CD8+ T cells in patients with T1D tend to have a stem memory phenotype and are characterized by a highly skewed oligoclonal T cell receptor repertoire.
- 12.Sutherland DE, Sibley R, Xu XZ, et al. Twin-to-twin pancreas transplantation: reversal and reenactment of the pathogenesis of type I diabetes. Trans Assoc Am Physicians. 1984;97:80–7. [PubMed] [Google Scholar]
- 13.Sibley RK, Sutherland DER, Goetz F, et al. Recurrent diabetes mellitus in the pancreas iso- and allograft. A light and electron microscopic and immunohistochemical analysis of four cases. Lab Invest. 1985;53:132–44. [PubMed] [Google Scholar]
- 14.Sutherland DER, Goetz FC, Sibley RK. Recurrence of diabetes in pancreas transplants. Diabetes. 1989;38 Suppl 1:85–7. [DOI] [PubMed] [Google Scholar]
- 15.Tyden G, Reinholt FP, Sundkvist G, et al. Recurrence of autoimmune diabetes mellitus in recipients of cadaveric pancreatic grafts. New Engl J Med. 1996;335:860–2. [DOI] [PubMed] [Google Scholar]
- 16.Roep BO, Stobbe I, Duinkerken G, et al. Auto- and alloimmune reactivity to human islet allografts trasnplanted into type 1 diabetic patients. Diabetes. 1999;48:484–90. [DOI] [PubMed] [Google Scholar]
- 17.Pinkse GGM, Tysma OHM, Bergen CAM, et al. Autoreactive CD8 T cells associated with β cell destruction in type 1 diabetes. Proc Natl Acad Sci U S A. 2005;102:18425–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laughlin E, Burke G, Pugliese A, et al. Recurrence of autoreactive antigen-specific CD4+ T cells in autoimmune diabetes after pancreas transplantation. Clin Immunol. 2008;128:23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Monti P, Scirpoli M, Maffi P, et al. Islet transplantation in patients with autoimmune diabetes induces homeostatic cytokines that expand autoreactive memory T cells. J Clin Invest. 2008;118:1806–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vendrame F, Pileggi A, Laughlin E, et al. Recurrence of type 1 diabetes after simultaneous pancreas-kidney transplantation, despite immunosuppression, is associated with autoantibodies and pathogenic autoreactive CD4 T-cells. Diabetes. 2010;59:947–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–63. [DOI] [PubMed] [Google Scholar]
- 22.•.Chang JT, Wherry EJ, Goldrath AW. Molecular regulation of effector and memory T cell differentiation. Nat Immunol. 2014;15:1104–15. [DOI] [PMC free article] [PubMed] [Google Scholar]; An excellent recent review of our understanding of memory T cell differentiation and function.
- 23.•.Devarajan P, Chen Z. Autoimmune effector memory T cells: the bad and the good. Immunol Res. 2013;57:12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]; A useful review of the memory T cell compartment in autoimmunity, with an emphasis on Tem cells.
- 24.••.Chee J, Ko HJ, Skowera A, et al. Effector-memory T cells develop in islets and report islet pathology in type 1 diabetes. J Immunol. 2014;192:572–80. [DOI] [PubMed] [Google Scholar]; An important study in the NOD model demonstrating that acquisition of the effector-memory phenotype by islet-specific CD8+ T cells occurs in infiltrated islets, followed by emigration to peripheral lymphoid tissue.
- 25.••.Orban T, Beam CA, Xu P, et al. Reduction in CD4 central memory T-cell subset in costimulation modulator abatacept-treated patients with recent-onset type 1 diabetes is associated with slower C-peptide decline. Diabetes. 2014;63:3449–57. [DOI] [PMC free article] [PubMed] [Google Scholar]; The first demonstration that CD4+ Tcm cells are down-modulated by costimulation blockade (abatacept) and that this correlates with C-peptide preservation.
- 26.Surh CD, Sprent J. Homeostasis of naïve and memory T cells. Immunity. 2008;29:848–62. [DOI] [PubMed] [Google Scholar]
- 27.Calzascia T, Pellegrini M, Lin A, et al. CD4 T cells, lymphopenia, and IL-7 in a multistep pathway to autoimmunity. Proc Natl Acad Sci U S A. 2008;105:2999–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.••.Penaranda C, Kuswanto W, Hofmann J, et al. IL-7 receptor blockade reverses autoimmune diabetes by promoting inhibition of effector/memory T cells. Proc Natl Acad Sci U S A. 2012;109:12668–73. [DOI] [PMC free article] [PubMed] [Google Scholar]; Together with the study by Lee et al. (ref. 29), the first demonstration that IL-7 receptor blockade robustly reverses diabetes in NOD mice by inhibiting Tem cells and upregulating PD-1 expression.
- 29.••.Lee LF, Logriono K, Tu GH, et al. Anti-IL-7 receptor-α reverses established type 1 diabetes in nonobese diabetic mice by modulating effector T-cell function. Proc Natl Acad Sci U S A. 2012;109:12674–9. [DOI] [PMC free article] [PubMed] [Google Scholar]; See comment relating to ref. 28.
- 30.•.Murakami N, Riella LV. Co-inhibitory pathways and their importance in immune regulation. Transplantation. 2014;98:3–14. [DOI] [PubMed] [Google Scholar]; An excellent recent review of co-inhibitory and co-stimulatory pathways in immune regulation.
- 31.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.•.Pauken KE, Jenkins MK, Azuma M, Fife BT. PD-1, but not PD-L1, expressed by islet-reactive CD4+ T cells suppresses infiltration of the pancreas during type 1 diabetes. Diabetes. 2013;62:2859–69. [DOI] [PMC free article] [PubMed] [Google Scholar]; One of several studies indicating that the PD-1 pathway may be important in maintaining peripheral tolerance in the NOD model of diabetes autoimmunity.
- 33.Ansari MJ, Salama AD, Chitnis T, et al. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med. 2003;198:63–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fife BT, Guleria I, Gubbels Bupp M, et al. Insulin-induced remission in new-onset NOD mice is maintained by the PD-1-PD-L1 pathway. J Exp Med. 2006;203:2737–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li R, Lee J, Kim MS, et al. PD-L1-driven tolerance protects neurogenin3-induced islet neogenesis to reverse established type 1 diabetes in NOD mice. Diabetes. 2015;64:529–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waldmann TA. Targeting the interleukin-15 system in rheumatoid arthritis. Arthritis Rheum. 2005;52:2585–8. [DOI] [PubMed] [Google Scholar]
- 37.Baslund B, Tvede N, Danneskiold-Samsoe B, et al. Targeting interleukin-15 in patients with rheumatoid arthritis. A proof-of-concept study. Arthritis Rheum. 2005;52:2686–92. [DOI] [PubMed] [Google Scholar]
- 38.Finch DK, Midha A, Buchanan CL, et al. Identification of a potent anti-IL-15 antibody with opposing mechanisms of action in vitro and in vivo. Br J Pharmacol. 2011;162:480–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krueger GG. Selective targeting of T cell subsets: focus on alefacept—a remittive therapy for psoriasis. Expert Opin Biol Ther. 2002;2:431–41. [DOI] [PubMed] [Google Scholar]
- 40.Chamian F, Lin SL, Lee E, et al. Alefacept (anti-CD2) causes a selective reduction in circulating effector memory T cells (Tem) and relative preservation of central memory T cells (Tcm) in psoriasis. J Transl Med. 2007;5:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haider AS, Lowes MA, Gardner H, et al. Novel insight into the agonistic mechanism of alefacept in vivo: differentially expressed genes may serve as biomarkers of response in psoriasis patients. J Immunol. 2007;178:7442–9. [DOI] [PubMed] [Google Scholar]
- 42.Punch JD, Lin J, Bluestone J, et al. CD2 and CD3 receptor-mediated tolerance: constraints on T cell activation. Transplantation. 1999;67:741–8. [DOI] [PubMed] [Google Scholar]
- 43.Chamian F, Lowes MA, Lin SL, et al. Alefacept reduces infiltrating T cells, activated dendritic cells, and inflammatory genes in psoriasis vulgaris. Proc Natl Acad Sci U S A. 2005;102:2075–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ellis CN, Krueger GG. Treatment of chronic plaque psoriasis by selective targeting of memory effector T lymphocytes. N Engl J Med. 2001;345:248–55. [DOI] [PubMed] [Google Scholar]
- 45.••.Rigby MR, DiMeglio LA, Rendell MS, et al. Targeting of memory T cells with alefacept in new-onset type 1 diabetes (T1DAL study): 12 month results of a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Diabetes Endocrinol. 2013;1:284–94. [DOI] [PMC free article] [PubMed] [Google Scholar]; The first demonstration in the clinic that targeting the CD2 pathway leads to robust depletion of Tem cells, an increase in the Treg/Tem ratio, and preservation of C-peptide in new-onset T1D.
- 46.Barlow AK, Like AA. Anti-CD2 monoclonal antibodies prevent spontaneous and adoptive transfer of diabetes in the BB/Wor rat. Am J Pathol. 1992;141:1043–51. [PMC free article] [PubMed] [Google Scholar]
- 47.Bai Y, Fu S, Honig S, et al. CD2 is a dominant target for allogeneic responses. Am J Transplant. 2002;2:618–26. [DOI] [PubMed] [Google Scholar]
- 48.Chavin KD, Qin L, Lin J, et al. Combined anti-CD2 and anti-CD3 receptor monoclonal antibodies induce donor-specific tolerance in a cardiac transplant model. J Immunol. 1993;151:7249–59. [PubMed] [Google Scholar]
- 49.Chavin KD, Qin L, Lin J, et al. Combination anti-CD2 and anti-CD3 monoclonal antibodies induce tolerance while altering interleukin-2, interleukin-4, tumor necrosis factor, and transforming growth factor-beta production. Ann Surg. 1993;218:492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chavin KD, Qin L, Lin J, et al. Anti-CD2 and anti-CD3 monoclonal antibodies synergize to prolong allograft survival with decreased side effects. Transplantation. 1993;55:901–8. [DOI] [PubMed] [Google Scholar]
- 51.Chavin KD, Qin L, Lin J, et al. Anti-CD2 monoclonal antibodies synergize with anti-CD3 to prolong allograft survival and decrease cytokine production. Transplant Proc. 1993;25:823–4. [PubMed] [Google Scholar]
- 52.Kapur S, Khanna A, Sharma VK, et al. CD2 antigen targeting reduces intragraft expression of mRNA-encoding granzyme B and IL-10 and induces tolerance. Transplantation. 1996;62:249–55. [DOI] [PubMed] [Google Scholar]
- 53.Kapur S, Sharma V, Khanna A, et al. Regulation of the anti-allograft response by targeting the CD2 antigen: a potential strategy for the creation of transplant tolerance. Surg Technol Int. 1996;5:233–40. [PubMed] [Google Scholar]
- 54.Krueger GG, Callis KP. Development and use of alefacept to treat psoriasis. J Am Acad Dermatol. 2003;49:S87–97. [DOI] [PubMed] [Google Scholar]
- 55.Miller GT, Hochman PS, Meier W, et al. Specific interaction of lymphocyte function-associated antigen 3 with CD2 can inhibit T cell responses. J Exp Med. 1993;178:211–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuhns MS, Davis MM, Garcia KC. Deconstructing the form and function of the TCR/CD3 complex. Immunity. 2006;24:133–9. [DOI] [PubMed] [Google Scholar]
- 57.Chatenoud L, Thervet E, Primo J, et al. Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc Natl Acad Sci U S A. 1994;91:123–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Daifotis AG, Koenig S, Chatenoud L, et al. Anti-CD3 clinical trials in type 1 diabetes mellitus. Clin Immunol. 2013;149:268–78. [DOI] [PubMed] [Google Scholar]
- 59.Herold KC, Hagopian W, Auger JA, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346:1692–8. [DOI] [PubMed] [Google Scholar]
- 60.Keymeulen B, Vandemeulebroucke E, Ziegler AG, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med. 2005;352:2598–608. [DOI] [PubMed] [Google Scholar]
- 61.Herold KC, Gitelman SE, Masharani U, et al. A single course of anti-CD3 monoclonal antibody hOKT3γ1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes. 2005;54:1763–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Keymeulen B, Walter M, Mathieu C, et al. Four-year metabolic outcome of a randomised controlled CD3-antibody trial in recent-onset type 1 diabetic patients depends on their age and baseline residual beta cell mass. Diabetologia. 2010;53:614–23. [DOI] [PubMed] [Google Scholar]
- 63.Sherry N, Hagopian W, Ludvigsson J, et al. Teplizumab for treatment of type 1 diabetes (Protégé study): 1-year results from a randomised, placebo-controlled trial. Lancet. 2011;378:487–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ambery P, Donner TW, Biswas N, et al. Efficacy and safety of low-dose otelixizumab anti-CD3 monoclonal antibody in preserving C-peptide secretion in adolescent type 1 diabetes: DEFEND-2, a randomized, placebo-controlled, double-blind, multi-centre study. Diabet Med. 2014;31:399–402. [DOI] [PubMed] [Google Scholar]
- 65.Aronson R, Gottlieb PA, Christiansen JS, et al. Low-dose otelixizumab anti-CD3 monoclonal antibody DEFEND-1 study: results of the randomized phase III study in recent-onset human type 1 diabetes. Diabetes Care. 2014;37:2746–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.•.Herold KC, Gitelman SE, Ehlers MR, et al. Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial: metabolic and immunologic features at baseline identify a subgroup of responders. Diabetes. 2013;62:3766–74. [DOI] [PMC free article] [PubMed] [Google Scholar]; The first trial in new-onset T1D to clearly show that response to anti-CD3 therapy is heterogenous and that a responder group can be identified.
- 67.Penaranda C, Tang Q, Bluestone JA. Anti-CD3 therapy promotes tolerance by selectively depleting pathogenic cells while preserving regulatory T cells. J Immunol. 2011;187:2015–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.•.Herold KC, Gitelman SE, Willi SM, et al. Teplizumab treatment may improve C-peptide responses in participants with type 1 diabetes after the new-onset period: a randomised controlled trial. Diabetologia. 2013;56:391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]; The first demonstration that CD8+ Tcm cells are modulated in patients with recent-onset T1D who are responders to anti-CD3 therapy.
- 69.Salomon B, Bluestone JA. Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu Rev Immunol. 2001;19:225–52. [DOI] [PubMed] [Google Scholar]
- 70.Linsley PS, Brady W, Urnes M, et al. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. 1991;174:561–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ford ML, Adams AB, Pearson TC. Targeting co-stimulatory pathways: transplantation and autoimmunity. Nat Rev Nephrol. 2014;10:14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Webster RM. The immune checkpoint inhibitors: where are we now? Nat Rev Drug Discov. 2014;13:883–4. [DOI] [PubMed] [Google Scholar]
- 73.Linsley PS, Wallace PM, Johnson J, et al. Immunosuppression in vivo by a soluble form of the CTLA-4 T cell activation molecule. Science. 1992;257:792–5. [DOI] [PubMed] [Google Scholar]
- 74.Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev. 2008;224:166–82. [DOI] [PubMed] [Google Scholar]
- 75.Orban T, Bundy B, Becker DJ, et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet. 2011;378:412–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol. 2010;11:7–13. [DOI] [PubMed] [Google Scholar]
- 77.Eisenbarth GS, Srikanta S, Jackson R, et al. Anti-thymocyte globulin and prednisone immunotherapy of recent onset type 1 diabetes mellitus. Diabetes Res. 1985;2:271–6. [PubMed] [Google Scholar]
- 78.•.Gitelman SE, Gottlieb PA, Rigby MR, et al. Antithymocyte globulin treatment for patients with recent-onset type 1 diabetes: 12-month results of a randomised, placebo-controlled, phase 2 trial. Lancet Diabetes Endocrinol. 2013;1:306–16. [DOI] [PMC free article] [PubMed] [Google Scholar]; The first adequately powered trial in new-onset T1D showing that treatment with antithymocyte globulin monotherapy does not deplete Tem cells and fails to preserve C-peptide.
- 79.Long SA, Rieck M, Sanda S, et al. Rapamycin/IL-2 combination therapy in patients with type 1 diabetes augments Tregs yet transiently impairs beta-cell function. Diabetes. 2012;61:2340–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mordes JP, Bortell R, Doukas J, et al. The BB/Wor rat and the balance hypothesis of autoimmunity. Diabetes Metab Rev. 1996;12:103–9. [DOI] [PubMed] [Google Scholar]
- 81.Smilek DE, Ehlers MR, Nepom GT. Restoring the balance: immunotherapeutic combinations for autoimmune disease. Dis Model Mech. 2014;7:503–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.•.Haller MJ, Gitelman SE, Gottlieb PA, et al. Anti-thymocyte globulin/G-CSF treatment preserves β cell function in patients with established type 1 diabetes. J Clin Invest. 2015;125:448–55. [DOI] [PMC free article] [PubMed] [Google Scholar]; An important pilot study demonstrating that combination therapy with low-dose antithymocyte globulin plus G-CSF in new-onset T1D appears to preserve C-peptide, possibly by inducing a favorable Treg/Teff ratio.
- 83.Gregori S, Mangia P, Bacchetta R, et al. An anti-CD45RO/RB monoclonal antibody modulates T cell responses via induction of apoptosis and generation of regulatory T cells. J Exp Med. 2005;201:1293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rus H, Pardo CA, Hu L, et al. The voltage-gated potassium channel Kv1.3 is highly expressed on inflammatory infiltrates in multiple sclerosis brain. Proc Natl Acad Sci U S A. 2005;102:11094–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Beeton C, Wulff H, Standifer NE, et al. Kv1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc Natl Acad Sci U S A. 2006;103:17414–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kineta Announces Promising Top-line Clinical Results for Dalazatide; Target Implicated in Broad Array of Autoimmune Diseases. http://www.kinetabio.com/press_releases/PressRelease20150505_dalazatide.pdf: Kineta, Inc.; 2015. [Google Scholar]
- 87.Maecker HT, McCoy JP, Nussenblatt R. Standardizing immunophenotyping for the Human Immunology Project. Nat Rev Immunol. 2012;12:191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]