

Abstract
Toxoplasma gondii (T. gondii) is a globally prevalent zoonotic parasite causing severe health and economic impacts. Despite decades of research, no commercial vaccine provides comprehensive protection against both acute and chronic toxoplasmosis. DNA vaccines represent a promising strategy, but their application is hindered by low delivery efficiency and limited immunogenicity. Here, we developed and evaluated pVAX1-TgIMC1-loaded PLGA and chitosan (CS) nanospheres as potential vaccine candidates. Immunization studies in mice showed that pVAX1-TgIMC1/PLGA and pVAX1-TgIMC1/CS nanospheres induced robust humoral and cellular immune responses, significantly enhancing specific IgG levels and cytokine production IFN-γ and IL-17 compared to the naked DNA vaccine. Both nanospheres also promoted dendritic cell maturation and T-cell activation, resulting in reduced parasite burdens in cardiac tissues post-challenge. Notably, the PLGA nanospheres exhibited superior protection against acute toxoplasmosis, while CS nanospheres provided additional advantages in antigen stability and delivery. The nanospheres were non-toxic, as confirmed by biochemical markers and histopathological analysis. These findings highlight pVAX1-TgIMC1/PLGA and pVAX1-TgIMC1/CS nanospheres as promising candidates for T. gondii vaccine development, warranting further optimization and validation in broader animal models.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12917-025-04961-z.
Keywords: Toxoplasma gondii, Inner membrane complex 1, Nanomaterial nanospheres, Immunoprotection
Introduction
Widely distributed globally, Toxoplasma gondii (T. gondii) is an obligate intracellular protozoan parasite that could infect nearly all warm-blooded mammals, including humans [1, 2]. It is estimated that approximately 30% of the global population have been infected with this parasite [3]. Transmission often occurs through multiple routes, including consumption of undercooked meat containing T. gondii tissue cysts or ingestion of food and water contaminated with cat feces, posing serious threats to public health and food safety [4, 5]. In immunocompetent individuals, infections are often asymptomatic, but immunocompromised individuals and fetuses are at risk of severe health complications, including encephalitis and congenital defects [6–8]. The impact of T. gondii infection on livestock is similarly concerning. Infected animals often experience reproductive disorders, such as abortion, stillbirth, and the birth of weak offspring, leading to significant losses in production efficiency [9, 10]. Moreover, infected animals may exhibit symptoms such as weakness, anorexia, and behavioral changes. Severe cases can lead to systemic inflammation, multi-organ failure, or death, particularly in young or immunocompromised animals. These issues are especially pronounced in economically significant species like sheep and pigs, further exacerbating economic burdens and posing challenges to livestock industries [11].
To date, no effective strategy for the complete eradication of T. gondii infection has been achieved. Current treatment options, including sulfonamides and folate antagonists, are associated with significant side effects and cannot eliminate tissue cyst-residing bradyzoites [12, 13]. Vaccines remain the most effective approach for preventing toxoplasmosis. However, the only commercially available vaccine, Toxovax® (Ovilis Toxovax®), is approved solely for preventing abortion in sheep [14]. Moreover, its efficacy against tissue cyst formation is debated, and it lacks applicability to other host species and livestock. Developing a safe, effective, and broadly protective vaccine against T. gondii has thus become a pressing scientific and public health challenge to reduce infection rates and economic losses. For this study, we constructed a DNA vaccine using the pVAX1 plasmid vector to express the Toxoplasma gondii IMC1 protein (TgIMC1). The resulting plasmid construct is designated as pVAX1-TgIMC1, which encodes and expresses the recombinant TgIMC1 protein (rTgIMC1) in host cells.
In recent years, various candidate vaccines have been explored, including live-attenuated vaccines, subunit vaccines, DNA vaccines, and recombinant protein vaccines [15, 16]. Among these, live attenuated vaccines offer strong immunogenicity but carry safety risks such as virulence reversion [17]. Subunit vaccines, while safer, often lack sufficient immunogenicity and rely heavily on adjuvants [18]. In contrast, DNA vaccines can simultaneously induce both humoral and cellular immune responses, significantly enhancing overall immunological efficacy [19]. They also allow rapid adaptation to new pathogens or mutations and avoid immune interference from viral vectors [20]. With advantages such as high safety, stability, short production cycles, and low cost, DNA vaccines have garnered significant attention in modern vaccine development. At the same time, selecting the right vaccine antigen is crucial. The Toxoplasma gondii IMC1 protein is a key component of the substructure within the inner membrane complex (IMC), known as alveolin, which primarily provides mechanical support and maintains parasite morphological stability during the formation of daughter parasites [21, 22]. In its immature stage, IMC1 exhibits high solubility in detergents such as deoxycholate. However, as the daughter cells mature, approximately 5 kDa of the C-terminus is cleaved by specific proteases, transitioning the protein from a soluble state into a detergent-insoluble, robust network structure [23]. This transformation enables IMC1 to provide enhanced stability and mechanical support during sporozoite maturation and is closely associated with anchoring the parasite’s motility apparatus, such as the actin-myosin motor complex, ensuring T. gondii’s ability to grow and invade within and outside host cells [24]. Given its critical function, TgIMC1 is expected to be a candidate antigen for T. gondii vaccine development. Therefore, we constructed a DNA vaccine plasmid (pVAX1-TgIMC1) encoding this protein.
Polylactic-co-glycolic acid (PLGA) and chitosan (CS) are two widely utilized biodegradable materials with distinct yet complementary advantages in immunological applications. PLGA effectively protects antigens from environmental degradation, maintaining their stability and bioactivity. Its controlled degradation rate enables sustained release and precise delivery, prolonging immune stimulation and enhancing immune responses [25, 26]. Similarly, CS, derived from the natural polysaccharide chitin, offers excellent biocompatibility and biodegradability. Its cationic properties facilitate interactions with cell membranes and mucosal surfaces, increasing antigen retention and boosting immune response intensity. Additionally, CS enhances antigen uptake and activation by dendritic cells and macrophages, promotes cytokine secretion, and supports immune cell maturation. Together, these properties position PLGA and CS as promising materials for advanced vaccine delivery systems, capable of improving antigen stability, delivery efficiency, and immunogenicity [27, 28].
In this study, a novel nanoparticle-based DNA vaccine was constructed using the pVAX1-TgIMC1 plasmid construct encoding the TgIMC1 protein to prevent and control toxoplasmosis. Recombinant TgIMC1 protein (rTgIMC1) was expressed in a prokaryotic system and the pVAX1-TgIMC1 plasmid construct was encapsulated in PLGA and CS nanoparticles to develop the vaccine formulations. Following a specific immunization protocol, pVAX1-TgIMC1/PLGA and pVAX1-TgIMC1/CS nanoparticles were administered to mice. Subsequently, spleens of immunized mice were collected for histological analysis, and antibody levels, cytokine profiles, lymphocyte proliferation, and cardiac parasite burden were measured. The immunoprotective efficacy of the nanoparticle vaccine was evaluated based on these experimental findings.
Materials and methods
Our approach involves: (1) construction of the pVAX1-TgIMC1 DNA vaccine plasmid, (2) expression and purification of recombinant TgIMC1 protein for characterization, and (3) encapsulation of the DNA vaccine in biodegradable nanoparticles for enhanced delivery and immunogenicity.
Parasites, cells, and animals
The Toxoplasma gondii RH (Type I) strain was used to infect mice. The Toxoplasma gondii RH (Type I) strain was obtained from Dr. Xiangrui Li at Nanjing Agricultural University, China, where it was originally isolated from a human case and maintained by serial passage in mice. This strain is preserved in liquid nitrogen at the Laboratory of Veterinary Molecular and Immunological Parasitology at Ningxia University, China. Female BALB/c mice (6–8 weeks old, 18–22 g), were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. and housed in a specific pathogen-free (SPF) barrier system with ad libitum access to food and water. To maintain the T. gondii RH (Type I) strain, mice were inoculated intraperitoneally twice a week. Tachyzoites were harvested from the peritoneal lavage fluid of the inoculated mice, washed by centrifugation in phosphate-buffered saline (PBS), and resuspended to a final concentration of 1 × 108 tachyzoites per ml in PBS for subsequent inoculations [29].
Human embryonic kidney 293-T cells (HEK 293-T) purchased from Shanghai Institute of Cell Biology, Chinese Academy of Sciences, containing 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA, USA) and 1% double antibiotics (penicillin-streptomycin solution, Gibco, Carlsbad, CA, USA) were cultured at 37 °C in 5% CO2 atmosphere.
Specific pathogen-free (SPF) mice (18–22 g, including female BALB/c mice aged 6–8 weeks) and Sprague-Dawley rats (200–220 g, 8–10 weeks old) were obtained from the Animal Research Center of Charles River company and used in the study. All mice were housed under identical SPF conditions. Mice were euthanized by gradual-fill carbon dioxide asphyxiation. All the experimental design, animal management, injection, and operations were strictly followed the Ethics Procedures and Guidelines of the People’s Republic of China and were supervised by the Animal Ethics Committee, Ningxia University, Yinchuan, P.R. China. The mouse license number was NXU-2023-083, and the rat license number was NXU-2023-086.
Gene cloning and plasmid construction
To enhance the expression of the rTgIMC1 protein in animals, the TgIMC1 gene sequence (TGRH88_075820) was submitted for codon optimization using the online tool at www.novopro.cn/tools/codon-optimization.html. and the optimized species was mammal. The optimized gene sequence was then synthesized by Sangon Biotech (Shanghai, China). The obtained gene fragment was PCR-amplified using the primers IMC1-opt-MF: 5ʹ-GGTACCATGTTTAAAGATTGTGCAGACCCATGT-3ʹand IMC1-opt-MR: 5ʹ-GGATCCTTAGCACTGGCAGCGAAAAC-3ʹ. The PCR reaction setup, as per the manufacturer’s instructions (Vazyme Biotech, Nanjing, China), included 12.5 µl of 2 × Phanta Max Master Mix, 1 µl each of IMC1-opt-MF and IMC1-opt-MR primers, 500 ng of cDNA template, and ddH₂O added to a final volume of 25 µl. The cDNA template was synthesized from total RNA extracted from 10^7 T. gondii RH tachyzoites using reverse transcription (Takara Biotechnology, Dalian, China). The thermal cycling conditions were as follows: an initial denaturation at 95 °C for 3 min, followed by 35 cycles of denaturation at 95 °C for 15 s, annealing at 58 °C for 15 s, and extension at 72 °C for 1 min, with a final extension at 72 °C for 5 min. The PCR products were verified using 1% agarose gel electrophoresis, and the correct band was purified with a gel extraction kit (OMEGA Bio-Tek, Norcross, GA, USA). After restriction digestion with KpnI and BamHI (Takara Biotechnology, Dalian, China), the purified gene fragment was ligated into a linearized pVAX1 vector to create the pVAX1-TgIMC1 recombinant plasmid. The resulting plasmid was verified by double enzyme digestion and subsequently transformed into E. coli DH5α competent cells. Single colonies were selected and cultured to obtain genetically engineered bacteria. Following large-scale cultivation, Endotoxin-free DNA plasmids were extracted using an Endo-free DNA plasmid extraction kit (Vazyme Biotech, Nanjing, China) and verified with the ToxinSensor™ Chromogenic LAL Endotoxin Assay Kit (GeneScript, Piscataway, NJ, USA), Ensure that the obtained plasmid endotoxin is at a low level. The concentration of the obtained plasmid was determined using a Nanodrop spectrophotometer (Thermo Scientific, Waltham, MA, USA) before storage at -20 °C.
Preparation of antibodies targeting T. gondii soluble antigens
To obtain the soluble tachyzoite antigen (STAg) of Toxoplasma gondii, the collected peritoneal lavage fluid was filtered with a 5 μm filter membrane (Millipore, Billerica, MA, USA) [30]. After centrifugation at 2500 rpm for 5 min, 5 × 106 T. gondii RH tachyzoites were re-dissolved in 1000 µl PBS. Then protease and phosphatase inhibitors were added according to the instructions (Beyotime, Shanghai, China), tip sonication was performed in pulse mode (2 s on, 2 s off) at 30 W output power for 10 min total. The total protein concentration was determined by BCA assay (Thermo Scientific, Waltham, MA, USA) prior to storage at -20 °C.
Two Sprague-Dawley rats were immunized subcutaneously with 200 µg STAg emulsified in complete Freund’s adjuvant. Two weeks later, the rats received four booster injections of 200 µg STAg emulsified in incomplete Freund’s adjuvant at weekly intervals. Each injection was administered subcutaneously at multiple dorsal sites. One week after the final immunization, blood was collected from the orbital plexus, and sera containing polyclonal antibodies against T. gondii STAg were obtained.
Detection of pVAX1-TgIMC1 in vitro
HEK 293-T cells were cultured in six-well plates and transfected with Endo-free pVAX1-TgIMC1 plasmid using Lipofectamine 2000 reagent (Invitrogen Biotechnology, Shanghai, China). After 48 h of transfection, the culture supernatant was discarded, and the cells were washed three times with PBS. The cells were then fixed with 1 ml of 4% paraformaldehyde at 4 °C for 12 h to maintain structural integrity, followed by permeabilization with Tris-buffered saline containing 0.1% Triton X-100 (TBSx) to increase membrane permeability and facilitate antibody entry. To minimize nonspecific binding, the cells were blocked with TBSx containing 5% bovine serum albumin (BSA). After blocking, the cells were incubated with anti-STAg serum (primary antibody, 1:100) diluted in TBSx with 5% BSA to detect the target antigen. Unbound primary antibodies were removed by washing the cells with TBSx for 5 min, and the cells were subsequently incubated with CY3-conjugated anti-rat IgG (secondary antibody, 1:1000, Sigma, Saint Louis, MO, USA) for specific labeling. To visualize nuclei, 500 µl of DAPI staining solution (Beyotime, Shanghai, China) was added to each well. Finally, images were captured and analyzed using a laser scanning confocal microscope (Nikon Corporation, Tokyo, Japan).
To confirm the in vitro expression of the constructed plasmid, transfected HEK 293-T cells were incubated at 37 °C for 48 h. After incubation, the cells were lysed in RIPA lysis buffer containing protease and phosphatase inhibitors (Beyotime, Shanghai, China) and disrupted using probe sonication (Scientz Biotechnology, Ningbo, China) under the parameters described in the antibody preparation section. Following sonication, 10× SDS-PAGE loading buffer was added to the lysates, thoroughly mixed, and heated at 95 °C for 5 min to fully denature the proteins. The protein samples were then separated by electrophoresis on a 12% SDS-PAGE gel and transferred onto a PVDF membrane for Western blot analysis. Detection was performed using rat anti-TgIMC1 protein serum (1:500) as the primary antibody and goat anti-rat IgG (1:1000) as the secondary antibody. Untransfected cells were used as a negative control, and protein bands were visualized using an enhanced chemiluminescence (ECL) substrate.
Preparation of nanospheres
To synthesize PLGA nanospheres, 100 mg of PLGA (molecular weight 40,000–75,000 Da, LA/GA: 65/35, Sigma, Saint Louis, MO, USA) was dissolved in 1 ml of dichloromethane (DCM; Sigma, Saint Louis, MO, USA) in a 50 ml centrifuge tube with a magnetic stir bar. Separately, a 2% polyvinyl alcohol (PVA; MW 31,000–75,000 Da, Sigma, Saint Louis, MO, USA) solution was prepared by dissolving PVA in double-distilled water, followed by filtration through a 0.22 μm membrane (Millipore, Billerica, MA, USA). At room temperature, 2 ml of the PVA solution was stirred at 400 rpm, and 4 mg of endotoxin-free DNA plasmid (1 mg/ml) was added dropwise using a pipette. After vortexing for 5 min, the organic solution was added dropwise to the aqueous solution while stirring at 400 rpm, followed by tip sonication at 4 °C (140 W power, 2-second pulses for a total of 5 min) to form a water-in-oil (w/o) emulsion. The w/o emulsion was then added dropwise to 3 ml of 6% PVA solution under the same stirring and sonication conditions to form a water-in-oil-in-water (w/o/w) emulsion, which was stirred overnight at 400 rpm to evaporate the DCM. The w/o/w emulsion was centrifuged at 40,000 rpm for 50 min at 4 °C, and the resulting PLGA nanospheres (pellet) were collected, filtered through a 0.22 μm membrane, and frozen. After lyophilization in a vacuum freeze dryer (Labconco, Kansas City, MO, USA), the PLGA nanospheres were stored in a sealed container at 4 °C until further use.
For the preparation of the chitosan solution, 20 mg of chitosan was dissolved in 10 ml of 1.0% acetic acid solution, and the pH was adjusted to 5.0 using 1 M NaOH solution. The chitosan solution was placed on a magnetic stirrer and maintained at 30 °C. Endo-free pVAX1-TgIMC1 plasmid (4 mg) was added dropwise, followed by 2 ml of 2 mg TPP solution, with continuous stirring for an additional 30 min after the addition. The mixture was then sonicated at 4 °C with an ultrasonic power of 140–160 W, alternating 1–2 s on and 1–2 s off, for a total duration of 4–5 min. After sonication, the mixture was centrifuged at 40,000 rpm for 15 min. The chitosan nanospheres (precipitation) were collected in double-distilled water, while the supernatant was retained for further analysis. After filtration through a 0.22 μm membrane, the collected PLGA nanoparticles were frozen. Once completely solidified, the nanoparticles were transferred to a vacuum freeze dryer and lyophilized until fully dried. The lyophilized CS nanospheres were stored under sealed conditions at -20 °C until further use.
Characterization of synthesized nanospheres
The prepared pVAX1-TgIMC1 nanoparticles were sent to the College of Life Sciences, Nanjing Agricultural University for imaging using a scanning electron microscope (SEM). Based on the SEM images, the average diameter of the pVAX1-TgIMC1 nanovaccine was analyzed using ImageJ software. To determine the encapsulation efficiency, the supernatant was collected after ultracentrifugation, and the total volume was recorded. The concentration of free protein in the supernatant was measured using the BCA assay. The loading capacity (LC) and encapsulation efficiency (EE) of PLGA and nanospheres were then calculated based on Eqs. (1) and (2), respectively.
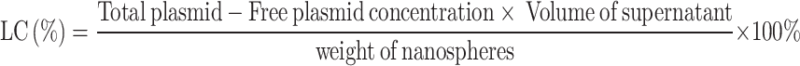 |
1 |
 |
2 |
Assessment of nanosphere toxicity
Thirty-five mice were randomly divided into seven groups (n = 5 per group) as follows: Blank group (immunized with PBS), Control group (immunized with 300 µg of empty pVAX1 plasmid), pVAX1/CS group (immunized with pVAX1/CS nanoparticles), pVAX1/PLGA group (immunized with pVAX1/PLGA nanoparticles), pVAX1-TgIMC1 group (immunized with pVAX1-TgIMC1 plasmid), pVAX1-TgIMC1/CS group (immunized with pVAX1-TgIMC1/CS nanoparticles), and pVAX1-TgIMC1/PLGA group (immunized with pVAX1-TgIMC1/PLGA nanoparticles). Each mouse received an intramuscular injection at a dose three times the standard immunization amount. Three days after the first immunization, a booster immunization was administered. Three days after the second immunization, blood was collected from the retro-orbital sinus, and serum was separated. Serum creatinine (Cr) and blood urea nitrogen (BUN) levels were measured using creatinine oxidase and urease-indophenol methods, respectively. The mental status and activity levels of the mice were monitored throughout the experiment.
Immunization and challenging schedules in mice
Female mice, weighing 18–22 g, were purchased from Beijing Vital River Laboratory Animal Technology Co. and housed in an SPF barrier system with ad libitum access to food and water. The mice were randomly allocated into seven experimental groups as detailed in Table 1: Blank group, Control group, pVAX1/CS group, pVAX1/PLGA group, pVAX1-TgIMC1 group, pVAX1-TgIMC1/CS group, and pVAX1-TgIMC1/PLGA group. All groups received two intramuscular injections administered at ten-day intervals through multiple injection points in the leg muscles. Pre-immunization blood samples were collected via retro-orbital bleeding to establish baseline serum parameters, which were subsequently stored at -20 °C until analysis.
Table 1.
Immunization strategy in mice
| Group | Day10 and Day20 | Day20 |
|---|---|---|
| Blank | 100 µl PBS | 100 T. gondii RH tachyzoites intraperitoneally per mouse |
| Control | 100 µl pVAX1empty DNA vaccine | |
| CS | 100 µl pVAX1-loaded CS nanoparticles | |
| PLGA | 100 µl pVAX1-loaded PLGA nanoparticles | |
| pVAX1-TgIMC1 | 100 µl pVAX1-TgIMC1 DNA vaccine | |
| pVAX1-TgIMC1/CS | 100 µl pVAX1-TgIMC1-loaded CS nanoparticles | |
| pVAX1-TgIMC1/PLGA | 100 µl pVAX1-TgIMC1-loaded PLGA nanoparticles |
Antibody and cytokine investigation
The levels of IgG, IgG1 and IgG2a in serum samples of mice were determined by indirect enzyme-linked immunosorbent assay (ELISA). Briefly, 96-well plates were coated overnight at 4 °C with 1 µg of soluble T. gondii antigen (STAg) diluted in carbonate buffer. After incubation, the plates were washed with TBS containing 0.05% Tween-20 (TBS-T) and blocked for 1 h at room temperature with 5% bovine serum albumin (BSA) in TBS-T. Following three washes with TBS-T, mouse serum samples, diluted 1:100 in TBS-T with 5% BSA, were added to the wells and incubated for 1 h. The plates were then washed three additional times with TBS-T before adding HRP-conjugated goat anti-mouse IgG, IgG1, or IgG2a antibodies (diluted 1:5000), which were incubated at 37 °C for 1 h. After a final wash, 100 µl of TMB substrate solution was added to each well for color development, and the reaction was terminated using 2 mol/L H₂SO₄. The absorbance was measured at 450 nm using a microplate reader.
To evaluate cytokine levels post-boost, retro-orbital blood was collected, and serum levels of IFN-γ, TGF-β, IL-4, IL-6, IL-10, IL-17, and IL-12 were quantified using a sandwich ELISA kit (Elabscience, China).
Flow cytometry analysis
On day 10 post-primary and secondary immunizations, five mice per group were euthanized by CO₂ asphyxiation, and splenocytes were isolated using commercial spleen lymphocyte separation kit (Solarbio, Beijing, China). The isolated splenocytes were cultured overnight in DMEM with 10% FBS and 1% penicillin-streptomycin. Non-adherent cells were removed the next day, and adherent cells were washed with PBS three times and collected. The cells were then stained at 4 °C in the dark for 40 min using anti-mouse CD11c-APC, CD86-FITC, and CD83-PE antibodies. Following staining, cells were washed three times with PBS and analyzed using flow cytometry.
To investigate changes in CD83, CD86, and MHC molecules in splenic dendritic cells (DCs), isolated lymphocytes were subjected to dual staining. The cells were incubated with anti-mouse CD11c-APC, CD86-FITC, CD83-PE, or CD11c-PE, MHC-I-FITC, and MHC-II-APC antibodies (eBioscience, San Diego, CA, USA) at 4 °C for 40 min in the dark. After incubation, the cells were washed three times with PBS, centrifuged, and collected. The stained cells were then analyzed using flow cytometry.
In the flow cytometry analysis, we employed a tiered gating strategy to address different biological questions. For total population analysis, live single cells were first selected by FSC-A/SSC-A and FSC-H/FSC-W gating, followed by identification of CD11c+ cells, with percentages of CD86+ or MHC II+ cells calculated relative to total splenocytes to assess overall immune activation. For subset-specific analysis, we further gated on CD11c+ cells with high FSC/SSC characteristics (representing mature, activated DCs) and reported percentages relative to the parent CD11c+ population to evaluate DC-specific functionality. This approach allowed us to distinguish between systemic immune response magnitude and specialized DC subpopulation characteristics. All gating boundaries were established using fluorescence-minus-one (FMO) and isotype controls, with consistent gates applied across samples using the CytoExpert software (version 2.3, Beckman Coulter, Brea, CA, USA).
Lymphocyte proliferation of spleen
On day 28, splenic cells were isolated from mice using the previously described method. A total of 5 × 10⁵ cells per well were seeded into 96-well plates, with each well stimulated with rTgIMC1 (10 µg/ml). The plates were incubated at 37 °C with 5% CO₂ for 48 h. Following incubation, 10 µl of CCK-8 reagent (BioTek, Burlington, VT, USA) was added to each well, and the plates were further incubated for 4 h. Optical density (OD) values were then measured at 450 nm using a microplate reader.
T. gondii burden in animals
Fourteen days after booster immunization, all mice were intraperitoneally challenged with 100 tachyzoites of the virulent T. gondii RH strain. Four days post-challenge, spleens were collected from three euthanized mice per group. Spleen tissues were fixed in formalin for 24–48 h, dehydrated with graded ethanol, cleared in xylene for 40 min, and embedded in paraffin. Using a Minux® rotary microtome S710 (RWD Life Science, Shenzhen, China), 4–6 μm sections were prepared, mounted on slides, and dried at 37 °C for 24 h. After deparaffinization and antigen retrieval with EDTA buffer, sections were treated with 3% H₂O₂ for 25 min to block endogenous peroxidase activity. The sections were then blocked with 5% bovine serum albumin at 37 °C for 30 min and incubated overnight at 4 °C with anti-T. gondii antibody (1:500 dilution, Abcam, Cambridge, UK). Following three washes with PBS, sections were incubated at room temperature for 1 h with CY3-conjugated goat anti-rabbit IgG (1: 1000 dilution, ABclonal, Wuhan, China). After washing with TBS, the sections were counterstained with DAPI, mounted, and examined under a fluorescence microscope to assess the distribution of T. gondii in the spleen tissues.
To assess parasite burden, five mice per group were euthanized by CO₂ asphyxiation, and spleen tissues were collected. Genomic DNA was extracted from the tissues using a commercial kit, and parasite load in cardiac tissue was quantified by absolute quantitative PCR targeting the Toxoplasma gondii 529 bp repeat sequence. Plasmids containing the 529 bp sequence served as reference standards for evaluating infection intensity in cardiac tissue. Each DNA sample was tested in triplicate.
Results
Expression of rTgIMC1 and Western blot analysis
The pVAX1-TgIMC1 plasmid was successfully constructed in this study. To verify the plasmid, Double digestion with KpnI and BamHI yielded two fragments: 2,987 bp (pVAX1 vector) and 1,836 bp (optimized pVAX1-TgIMC1 insert) (Fig. 1). Subsequently, the plasmid was sent to Sangon Biotech for sequencing, and the results confirmed that the inserted fragment within the plasmid is the open reading frame (ORF) of pVAX1-TgIMC1. These findings demonstrate that the pVAX1-TgIMC1 plasmid was correctly constructed.
Fig. 1.

A Double digestion analysis of the recombinant plasmid pVAX1-TgIMC1. M: DL5000 marker; Lane 1: Double digestion with KpnI and BamHI. B Immunofluorescence analysis of pVAX1-TgIMC1 expression in HEK 293-T cells. The scale bar represents 25 μm. C Western blotting of cell lysates probed with rat polyclonal antibodies against Toxoplasma gondii STAg. Cell lysates of HEK 293-T cell transfected with pVAX1-TgIMC1 (Line 1) and pVAX1 (Line 2) plasmids. Line M, MW marker proteins
The endotoxin levels in the extracted plasmid were measured using an endotoxin assay kit, with values found to be below 0.01 EU/ml. After transfecting HEK 293-T eukaryotic cells with endotoxin-free pVAX1-TgIMC1 plasmid, results are shown in Fig. 1B. Cells transfected with pVAX1-TgIMC1 exhibited specific red fluorescence, whereas cells transfected with the control pVAX1 plasmid showed no red fluorescence, indicating the successful expression of the recombinant TgIMC1 protein. Western blot analysis using home-made rat anti-TgIMC1 serum revealed a single band with a molecular weight of approximately 100 kDa (Fig. 1C), while no corresponding band was detected in cells transfected with the control pVAX1 plasmid. These results confirm that the recombinant TgIMC1 protein was successfully expressed via the pVAX1-TgIMC1 plasmid.
Characterization of nanospheres
After encapsulating the pVAX1-TgIMC1 plasmid in PLGA and chitosan (CS) nanoparticles, the morphology of the nanoparticles was examined using SEM. SEM analysis showed that both PLGA and CS nanoparticles were spherical, with small round protrusions on their surfaces (Fig. 2). Based on randomly selected SEM images of five nanoparticles, the synthesized PLGA nanoparticles had an average diameter of 68.04 ± 5.21 nm, while the synthesized CS nanoparticles had an average diameter of 84.12 ± 7.23 nm. Results from three independent experiments indicated that the loading capacity (LC) of PLGA and CS nanospheres was 0.99 ± 0.12% and 6.42 ± 0.65%, respectively, while their encapsulation efficiency (EE) was 70.18 ± 1.22% and 62.40 ± 2.02%, respectively.
Fig. 2.

The pVAX1-TgIMC1 plasmid was encapsulated into poly (lactic-co-glycolic acid) (PLGA) (A) and chitosan (CS) (B) nanoparticles, and their morphology was observed using SEM. PLGA and CS nanoparticles were synthesized using solvent evaporation (w/o/w) and ionic gelation techniques, respectively. After complete lyophilization, the magnification was set to ×30,000, with scale bars representing 500 nm
To study the release profiles of pVAX1-TgIMC1/PLGA and pVAX1-TgIMC1/CS nanoparticles, comparisons were made with blank PLGA and CS nanoparticles. As shown in Fig. 3, a sharp increase in release was observed for both nanoparticles within the first two days. pVAX1-TgIMC1/CS nanoparticles exhibited a more gradual release curve compared to pVAX1-TgIMC1/PLGA nanoparticles. After day 4, the release profile of pVAX1-TgIMC1/PLGA nanoparticles stabilized, but a slow-release trend was observed for both nanoparticle types until day 5. In vitro results showed that both obtained nanospheres could provide a stable release for one week, thereby enhancing the effectiveness of immunization.
Fig. 3.
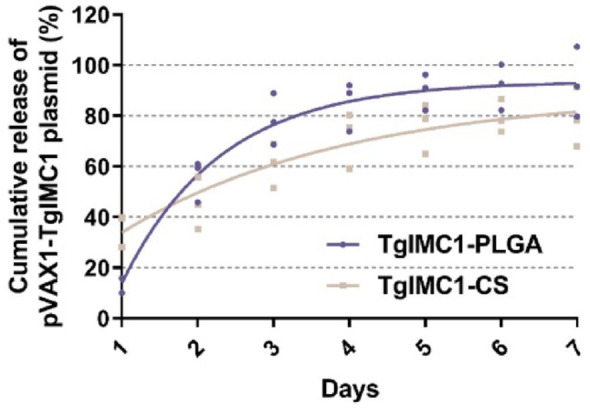
The release curves of pVAX1-TgIMC1 plasmid encapsulated in PLGA and CS nanoparticles were measured in vitro over seven days. Comparisons were made with blank PLGA and CS nanoparticles. The total plasmid content in the supernatant was determined using a Nanodrop spectrophotometer. Three independent experiments were conducted, with each sample measured once. The values are presented as mean ± standard deviation (SD) (n = 3)
The toxicity of PLGA and CS nanoparticles containing the pVAX1-TgIMC1 plasmid was evaluated by measuring Cr (Fig. 4A) and BUN levels (Fig. 4B) using the creatinine oxidase and urease-indole method. Results showed that Cr and BUN levels were within acceptable ranges, with no statistically significant differences (p > 0.05) between experimental groups and the blank or control groups. Additionally, no abnormal changes were observed in the physical health or mental status of experimental animals during the study. These findings confirm that the synthesized nanoparticles are non-toxic.
Fig. 4.

The toxicity of pVAX1-TgIMC1 plasmid encapsulated in PLGA and chitosan nanoparticles was analyzed. Serum samples were collected from each group, and Cr (A) and blood urea nitrogen (BUN) (B) levels were measured according to the instructions provided with commercial kits. Each serum sample was tested once. Statistical significance was evaluated using one-way ANOVA, followed by Dunnett’s test. After Bonferroni correction, pairwise comparisons of pVAX1-TgIMC1, pVAX1-TgIMC1/PLGA, and pVAX1-TgIMC1/CS groups were performed. Results are presented as mean ± SD (n = 5)
Antibodies determination
The immunogenicity of pVAX1-TgIMC1/PLGA and pVAX1-TgIMC1/CS nanoparticles was evaluated by comparing single and booster immunizations with either the pVAX1-TgIMC1 plasmid alone or the nanoparticle-encapsulated plasmid. The results demonstrated that both pVAX1-TgIMC1/PLGA and pVAX1-TgIMC1/CS groups induced significantly higher IgG levels after both primary and booster immunizations compared to the control group (p < 0.001, Fig. 5A). Notably, animals immunized with the naked pVAX1-TgIMC1 plasmid alone also exhibited a significant increase in total IgG levels compared to the control group (p < 0.01), albeit lower than the nanoparticle groups. Following the primary immunization, animals immunized with pVAX1-TgIMC1/CS nanoparticles exhibited significantly higher IgG levels compared to those immunized with pVAX1-TgIMC1/PLGA nanoparticles (p < 0.01).
Fig. 5.

The total IgG (A), IgG1 (B), and IgG2a (C) levels in the serum of immunized animals were measured at weeks 0, 2, and 4. Each serum sample was tested once. Statistical significance was evaluated using one-way ANOVA, followed by Dunnett’s test and Bonferroni correction. Pairwise comparisons of pVAX1-TgIMC1, pVAX1-TgIMC1/PLGA, and pVAX1-TgIMC1/CS groups were performed. The results are presented as OD450 ± standard deviation (SD) (n = 5). Significant differences compared to the blank or control groups were indicated (*p < 0.05, **p < 0.01, ***p < 0.001)
Furthermore, as shown in Fig. 5B and C, both pVAX1-TgIMC1/PLGA and pVAX1-TgIMC1/CS groups generated higher levels of T. gondii-specific IgG1 and IgG2a compared to the control and naked pVAX1-TgIMC1 plasmid groups. The naked pVAX1-TgIMC1 plasmid alone also induced detectable IgG1 and IgG2a responses (p < 0.05 vs. control), but these were significantly surpassed by the nanoparticle formulations (p < 0.001). Notably, regardless of the number of immunizations, pVAX1-TgIMC1/CS and pVAX1-TgIMC1/PLGA nanoparticle immunizations resulted in a statistically significant increase in IgG1 levels compared to naked rTgIMC1 protein (p < 0.001, Fig. 5B).
Cytokines determination
Cytokine production was measured from serum collected after primary and booster immunizations. Compared to blank or control groups, the use of naked pVAX1-TgIMC1 plasmid or its nanoparticle-encapsulated forms effectively stimulated the production of cytokines IFN-γ (Fig. 6A), IL-4 (Fig. 6B), IL-10 (Fig. 6C), and IL-17 (Fig. 6D). As shown in Fig. 6A and B, and 6E, mice immunized with pVAX1-TgIMC1/PLGA and pVAX1-TgIMC1/CS nanospheres exhibited significantly increased secretion of IFN-γ, IL-4, and IL-17 compared to the control group (p < 0.001).
Fig. 6.

Cytokine secretion levels for IFN-γ (A), IL-4 (B), IL-10 (C), IL-17 (D), IL-6 (E), and TGF-β (F) were measured in serum collected at weeks 0, 2, and 4 using ELISA kits. Each serum sample was tested once. Statistical significance was evaluated using one-way ANOVA, followed by Dunnett’s test and Bonferroni correction. Pairwise comparisons of pVAX1-TgIMC1, pVAX1-TgIMC1/PLGA, and pVAX1-TgIMC1/CS groups were conducted. Results are expressed as mean ± SD (n = 5). Significant differences compared to blank or control groups are indicated as *p < 0.05, **p < 0.01, ***p < 0.001
The results demonstrated that the level of IL-10 in the pVAX1-TgIMC1/CS group was significantly higher than in the pVAX1-TgIMC1/PLGA group (p < 0.001, Fig. 6D). In contrast, IL-6 levels were significantly downregulated in both nanoparticle groups compared to the control, with the pVAX1-TgIMC1/PLGA group showing the most pronounced reduction (p < 0.001, Fig. 6C). No statistically significant differences in TGF-β levels were observed between the control and immunized groups (p > 0.05, Fig. 6F).
Vaccine boosted spleen dendritic cell activity
CD83 and CD86 are key molecules for assessing the maturation and antigen-presenting functions of DCs, with their high expression often closely associated with more effective T-cell activation and enhanced immune responses. To evaluate the impact of nanoparticles on DC maturation, splenic cells were collected after both the primary and booster immunizations from immunized animals. DCs were isolated from the splenic cell suspension using density gradient centrifugation, followed by flow cytometry analysis of CD83 and CD86 expression. As shown in Figs. 7A-C, both pVAX1-TgIMC1/PLGA and pVAX1-TgIMC1/CS nanospheres significantly increased the expression frequency of CD83 and CD86 following both primary and booster immunizations compared to the control group (p < 0.01; Figs. 7A, B). Furthermore, data analysis indicated that after the booster immunization, pVAX1-TgIMC1/PLGA nanoparticles were more effective in inducing CD83 and CD86 expression compared to the naked pVAX1-TgIMC1 plasmid. These findings demonstrate that both the pVAX1-TgIMC1 plasmid and its nanoparticle forms play important roles in inducing the expression of DC surface molecules, with effects varying by nanoparticle type and immunization frequency.
Fig. 7.

Flow cytometry was used to analyze CD83+ (A) and CD86+ (B) molecules on DC surfaces in the spleens of immunized mice. Five mice per group were sacrificed, and their splenic cells were analyzed. C Shows the relative numbers of CD11c+CD83+ and CD11c+CD86+ cells. Results are presented as mean ± standard deviation (SD) (n = 5). Statistical significance was evaluated using one-way ANOVA, followed by Dunnett’s test. After Bonferroni correction, pairwise comparisons of rTgIMC1 protein, pVAX1-TgIMC1/PLGA, and pVAX1-TgIMC1/CS groups were performed using ANOVA. Significant differences compared to the control group are indicated as *p < 0.05, **p < 0.01, and ***p < 0.001
Since MHC-I primarily presents endogenous antigens to CD8+ T cells, thereby triggering cytotoxic immune responses, while MHC-II presents exogenous antigens to CD4+ T cells, facilitating the regulation of both humoral and cellular immune responses. To explore their impact on these pathways, the effects of pVAX1-TgIMC1/CS and pVAX1-TgIMC1/PLGA nanoparticles on antigen presentation were investigated. Splenocytes were harvested and DCs were enriched by discontinuous density gradient centrifugation using OptiPrep (Sigma-Aldrich). Briefly, single-cell suspensions were layered over 14.5% OptiPrep solution and centrifuged at 600×g for 15 min at 4 °C. The low-density interface containing DCs was collected and washed twice with PBS. The expression of MHC-I and MHC-II on CD11c+ DCs was then analyzed by flow cytometry. As shown in Figs. 8A-C, booster immunization with the naked pVAX1-TgIMC1 plasmid and pVAX1-TgIMC1/CS nanoparticles induced significantly higher levels of MHC-I expression on DCs compared to the control group (p < 0.01). Furthermore, both the pVAX1-TgIMC1/PLGA and pVAX1-TgIMC1/CS groups exhibited significantly enhanced expression levels of MHC-II molecules on DCs following both primary and booster immunizations compared to the control group (p < 0.01). The results also demonstrated that the expression levels of MHC-II in the pVAX1-TgIMC1/PLGA and pVAX1-TgIMC1/CS groups were higher than those in the pVAX1-TgIMC1 group after both immunizations. These findings suggest that the rTgIMC1 protein and its nanoparticle formulations effectively activate the antigen-presenting function of dendritic cells.
Fig. 8.

Flow cytometry was used to analyze MHC molecule expression on DCs. Five animals per group were sacrificed, and splenic cells were studied. Bar graphs display the ratios of MHC-I (A) and MHC-II molecules (B) on splenic DCs, while dot plots (C) show the percentages of CD11c+ MHC-I+ and CD11c+ MHC-II+ cells. Results are presented as mean ± SD (n = 5). Statistical significance was evaluated using one-way ANOVA, followed by Dunnett’s test. After Bonferroni correction, pairwise comparisons of pVAX1-TgIMC1, pVAX1-TgIMC1/PLGA, and pVAX1-TgIMC1/CS groups were conducted. Significant differences compared to the control group are indicated as *p < 0.05, **p < 0.01, and ***p < 0.001
Vaccine enhances T cell growth
To determine whether nanoparticles enhance T lymphocyte responses, splenic cells were isolated three days after booster immunization, and their proliferation response to recombinant rTgIMC1 protein stimulation was measured. As shown in Fig. 9, animals immunized with pVAX1-TgIMC1/CS and TgIMC1- PLGA nanoparticles exhibited significant proliferation compared to control groups. Compared to the naked pVAX1-TgIMC1 plasmid, pVAX1-TgIMC1/CS nanoparticles significantly promoted splenic lymphocyte proliferation (p < 0.01), indicating that chitosan nanoparticles have an advantage in inducing T lymphocyte proliferation.
Fig. 9.
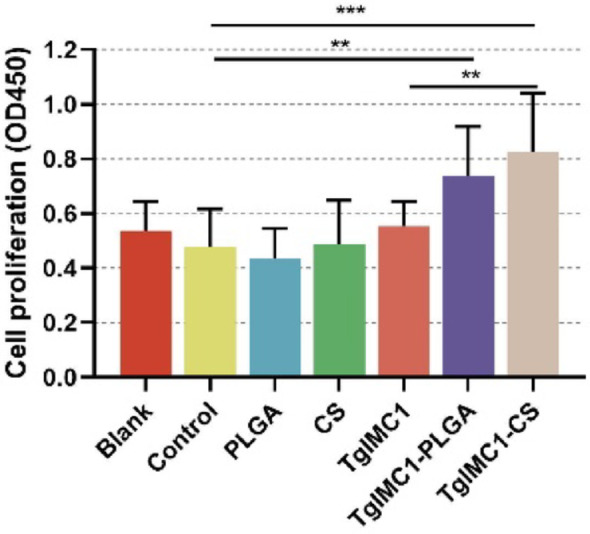
Splenic cells from three mice per group were collected and divided into three portions. Splenic cells were stimulated with rTgIMC1 (10 µg/ml) for 72 h prior to proliferation assay. Each lymphocyte portion was tested once. Statistical significance was evaluated using one-way ANOVA, followed by Dunnett’s test. Results are presented as mean ± SD (n = 9). After Bonferroni correction, pairwise comparisons of pVAX1-TgIMC1, pVAX1-TgIMC1/PLGA, and pVAX1-TgIMC1/CS groups were conducted. Significant differences compared to the control group are indicated as **p < 0.01 and ***p < 0.001
To assess the influence of PLGA and chitosan nanoparticles on T lymphocyte responses, CD4+ and CD8+ T lymphocyte expression levels in splenic cells were analyzed using flow cytometry. As shown in Figs. 10A, B, compared to the control group, both pVAX1-TgIMC1/CS and pVAX1-TgIMC1/PLGA nanoparticles significantly increased the levels of splenic CD4⁺ T and CD8⁺ T lymphocytes following the primary immunization (p < 0.05, Figs. 10A, B). The results further indicated that after both the primary and booster immunizations, the expression of CD4⁺ T cells in the pVAX1-TgIMC1/PLGA and pVAX1-TgIMC1/CS groups was significantly higher than that in the pVAX1-TgIMC1 group. Moreover, following the booster immunization, the expression of CD8⁺ T cells in the pVAX1-TgIMC1/PLGA and pVAX1-TgIMC1/CS groups was also significantly elevated compared to the pVAX1-TgIMC1 group (p < 0.001).
Fig. 10.

CD4+ (A) and CD8+ (B) T lymphocytes in the splenic cells of five mice per group were analyzed using flow cytometry. Dot plots (C) show the relative percentages of CD3e+CD4+ and CD3e+CD8+ cells. Results are expressed as mean ± SD (n = 5). Statistical significance was assessed using one-way ANOVA, followed by Dunnett’s test. After Bonferroni correction, pairwise comparisons of pVAX1-TgIMC1, pVAX1-TgIMC1/PLGA, and pVAX1-TgIMC1/CS groups were conducted. Significant differences compared to the control group are indicated as *p < 0.05, **p < 0.01, ***p < 0.001
Immunohistochemistry of spleen in immune mice and cardiac Toxoplasma load
Fourteen days after the booster immunization, mice were challenged with an intraperitoneal injection of 100 tachyzoites of the highly virulent T. gondii RH strain. Four days post-challenge, the mice were euthanized under the supervision of the Ethics Committee of Ningxia University, and spleen tissues were collected and processed into paraffin sections. As shown in Fig. 11, mice immunized with rTgIMC1 protein, pVAX1-TgIMC1/PLGA, and pVAX1-TgIMC1/CS nanospheres exhibited reduced T. gondii infection in the spleen. These results suggest that nanosphere-based immunization provides protective immunity against toxoplasmosis in mice, effectively inhibiting the growth of T. gondii tachyzoites to a certain extent.
Fig. 11.

Four days post-challenge, three mice were euthanized for spleen collection (n = 3). Paraffin-embedded sections were incubated with rabbit anti-T. gondii primary antibody (1:500, Abcam, UK) followed by CY3-conjugated goat anti-rabbit IgG secondary antibody (1:1000, ABclonal, China). One representative section per spleen was analyzed, with random high-magnification fields selected for observation (scale bar = 100 μm). The specific antibody staining confirms successful T. gondii detection in infected tissues, with appropriate controls validating the signal specificity
Fourteen days after booster immunization (day 29), the mice were challenged with 100 tachyzoites of the virulent T. gondii RH strain, and parasite burdens in the heart tissue were evaluated on day 6 post-infection using qPCR. As shown in Fig. 12, parasite loads were significantly reduced in the pVAX1-TgIMC1, pVAX1-TgIMC1/PLGA, and pVAX1-TgIMC1/CS nanoparticle groups compared to the control group (p < 0.001). Moreover, pVAX1-TgIMC1/CS nanoparticles showed stronger anti-toxoplasmosis effects than the naked pVAX1-TgIMC1 plasmid (p < 0.001), with nearly a twofold reduction in parasite burden. These findings highlight the crucial role of nanomaterials in enhancing immune protection.
Fig. 12.

The parasite burden in heart tissue was measured as the copy number of a 529 bp T. gondii fragment using qPCR. Two weeks after booster immunization, animals were challenged with 100 tachyzoites, and the heart tissue was collected six days post-infection. Each sample was tested three times (n = 15). Statistical significance was analyzed using one-way ANOVA, followed by Dunnett’s test. Pairwise comparisons of pVAX1-TgIMC1, pVAX1-TgIMC1/PLGA, and pVAX1-TgIMC1/CS groups were conducted using independent t-tests. Values are presented as mean ± SD (n = 15). Significant differences compared to the control group are indicated (**p < 0.01, ***p < 0.001)
Discussion
This study aimed to develop a novel DNA vaccine delivery system by encapsulating the pVAX1-TgIMC1 plasmid with nanomaterials, offering a new strategy for the prevention and control of Toxoplasma gondii infections. T. gondii is a globally distributed zoonotic protozoan pathogen capable of establishing chronic infections in various hosts, posing significant health and economic threats [4, 31, 32]. Current efforts to prevent and control T. gondii infections face significant challenges in both clinical and agricultural contexts, and no commercially available vaccine provides comprehensive and long-lasting protection [33–35]. DNA vaccines have garnered extensive attention over the past two decades due to their ability to express foreign antigens directly in host cells, eliciting specific humoral and cellular immune responses, thus offering a promising approach for the development of T. gondii vaccines [36, 37]. However, the practical application of DNA vaccines remains hindered by limitations such as low delivery efficiency, susceptibility of plasmids to degradation, and suboptimal immunogenicity [38–40].
In recent years, nanomaterials have made remarkable progress in drug and vaccine delivery. These materials not only improve antigen stability and delivery efficiency but also allow for the modulation of immunological properties by controlling nanoparticle size, surface charge, and surface ligands [41, 42]. Among these, poly (lactic-co-glycolic acid) (PLGA), a biodegradable polymer, has been widely utilized in drug and vaccine delivery due to its ability to protect DNA from enzymatic degradation and provide sustained antigen release [43, 44]. However, the inherently negative surface charge of PLGA nanoparticles limits their mucosal adhesion and cellular uptake, posing challenges to antigen delivery efficiency and immune response activation [45, 46]. In contrast, chitosan, a cationic polysaccharide, provides several advantages. It binds tightly to DNA, facilitates cellular uptake, and protects DNA from nuclease degradation. Moreover, its biocompatibility and low toxicity make it an ideal non-viral delivery vector capable of stimulating immune responses via innate immune pathways such as cGAS-STING [47–49]. Additionally, chitosan exhibits antimicrobial properties, which may further enhance the protective efficacy of vaccines by inhibiting the growth of pathogenic microorganisms [50].
In this study, pVAX1-TgIMC1/PLGA and pVAX1-TgIMC1/CS nanoparticles were successfully constructed and systematically evaluated for their immunological properties and biosafety. Using optimized solvent evaporation and ionic gelation techniques, the resulting PLGA and chitosan nanoparticles exhibited regular spherical morphologies with particle sizes typically under 200 nm. This size range is conducive to uptake by antigen-presenting cells such as DCs and MACROPHAGES, enhancing antigen presentation and the induction of specific immune responses [51, 52]. Previous studies have indicated that nanoparticles smaller than 200 nm are more effectively internalized by DCs, thereby enhancing immunogenicity [53]. Additionally, lyophilization effectively removed residual organic solvents, improved storage stability, and reduced potential toxicity associated with solvent residues [54]. Experimental results demonstrated no significant toxicity in animal models immunized with the nanoparticles, as evidenced by unaltered serum biochemical parameters (e.g., BUN, Cr) and normal behavior compared to controls. These findings confirm the biosafety of the nanoparticle-based delivery strategy and are consistent with prior reports of the low toxicity and high biocompatibility of PLGA and chitosan nanoparticles.
The immune protection against T. gondii involves both humoral and cellular immunity. Humoral immunity, mediated by B cells, protects hosts through the secretion of specific antibodies such as IgG, while cellular immunity relies on T cell responses and the secretion of cytokines such as IFN-γ, IL-4, and IL-17 to eliminate parasites or inhibit their proliferation [55–57]. In this study, mice immunized with pVAX1-TgIMC1/PLGA and pVAX1-TgIMC1/CS nanoparticles exhibited significantly higher levels of IgG and its subclasses, IgG1 and IgG2a, compared to controls immunized with the rTgIMC1 protein alone. IgG1 is associated with Th2-type immune responses, while IgG2a is linked to Th1-type responses [58]. The simultaneous increase in IgG1 and IgG2a levels indicates that these nanoparticles can activate both Th1- and Th2-associated immune pathways, resulting in a balanced mixed immune response. This result aligns with studies suggesting that nanoparticle-based vaccines can induce multifaceted immune responses [59–61], highlighting the potential of nanomaterial-mediated DNA vaccine delivery to achieve comprehensive, balanced, and durable humoral immune protection. Enhanced humoral responses are crucial for neutralizing T. gondii invasion-related molecules, mitigating acute infections, and reducing cyst formation, thereby alleviating clinical symptoms and tissue damage [20, 62].
The immune responses elicited by T. gondii infection are regulated by a complex network of cytokines involving both innate and adaptive immunity. IFN-γ is recognized as a central cytokine in controlling T. gondii proliferation, as it activates MACROPHAGES and promotes lysosome-phagosome fusion to inhibit parasite replication [63, 64]. In this study, IFN-γ levels were significantly elevated in mice immunized with nanoparticles, indicating a strong Th1-dominant cellular immune response. Concurrently, IL-4 levels were also upregulated. IL-4 plays a crucial role in maintaining immune homeostasis, assisting in Th2 response development, and interacting with IFN-γ to provide comprehensive host protection [65, 66]. Notably, IL-10, an immunosuppressive cytokine that regulates excessive inflammation and prevents immunopathological damage, was upregulated in the nanovaccine groups, while TGF-β levels remained unchanged [67]. This upregulation of IL-10 suggests that the immune system actively promotes immune tolerance while preventing excessive inflammatory activation, thereby minimizing tissue damage and optimizing the activation of B cells and T cells. Furthermore, IL-6, a pro-inflammatory cytokine linked to tissue damage during acute T. gondii infections, was elevated in the rTgIMC1 protein-only group but remained at baseline levels in the nanoparticle groups, indicating that the nanoparticles mitigated excessive inflammatory responses [68–70]. The significant elevation of IL-17 levels in nanoparticle-immunized mice suggests that these formulations effectively activated Th17-associated immune responses, which play a critical role in enhancing neutrophil recruitment and pathogen clearance during acute infection [71, 72].
The maturation and activation of DCs are essential for initiating and sustaining adaptive immune responses [73]. Enhanced expression of costimulatory molecules such as CD83 and CD86, as well as major histocompatibility complex molecules (MHC-I and MHC-II), promotes efficient antigen presentation and T cell activation [74, 75]. In this study, all nanoparticle-immunized groups demonstrated significant increases in CD83 and CD86 expression, indicating effective promotion of DC maturation and activation. This enhancement may be attributed to the physicochemical properties of nanoparticles, such as size and surface charge, which facilitate DC uptake and antigen processing [76]. Increased MHC-II expression further highlights the role of nanoparticles in activating CD4+ T cell-mediated immune responses.
While our nanoparticle vaccines achieved significant reduction in acute-phase parasite burden (Fig. 11), the presence of residual Toxoplasma signals in vaccinated spleens reflects a well-characterized phenomenon in subunit vaccine development. Similar to other protein-based vaccines, our formulation demonstrates substantial but incomplete parasite clearance during early infection stages - an expected outcome given the biological complexity of establishing sterilizing immunity against intracellular pathogens. The necessary ethical protocol requiring euthanasia at onset of clinical symptoms (4 days post-challenge) prevented evaluation of later-stage immunity, though this early endpoint is standard for acute toxoplasmosis models to minimize animal suffering. Three complementary approaches could further elucidate this question: (1) utilizing engineered mouse models permitting extended observation, (2) establishing in vitro systems to track parasite survival in vaccinated host cells over time, and (3) implementing advanced imaging modalities to monitor infection progression non-invasively. Such studies would maintain ethical standards while revealing the vaccine’s full potential for sustained parasite control.
The evaluation of immune protection efficacy is a critical aspect of vaccine development. In this study, challenge experiments using the highly virulent RH strain demonstrated significantly reduced T. gondii burdens in the hearts of mice immunized with pVAX1-TgIMC1/PLGA and pVAX1-TgIMC1/CS nanoparticles compared to controls. Histopathological analysis of spleen tissues further confirmed these results, revealing reduced parasite invasion and tissue damage in the nanoparticle groups.
In conclusion, the pVAX1-TgIMC1/PLGA and pVAX1-TgIMC1/CS nanoparticles developed in this study demonstrated excellent structural and stability characteristics while exhibiting significant immunological advantages. These nanoparticle formulations effectively induced robust humoral and cellular immune responses, promoted DC maturation, and activated multiple T cell subsets, thereby significantly reducing acute-phase T. gondii burdens. Three key limitations should be noted: (1) The acute challenge model precluded assessment of chronic infection control; (2) Ethical endpoints at 4 days post-infection limited observation of memory immune clearance; (3) Residual parasite detection (Fig. 11) suggests the need for adjuvant optimization. Nevertheless, our PLGA/CS system represents a major advancement as it is the first nanovaccine demonstrating against T. gondii, the nanoparticle delivery strategy established here provides a solid foundation for the future optimization and practical application of T. gondii DNA vaccines.
Conclusion
In summary, this study validated the potential application of PLGA and CS nanoparticles in the development of Toxoplasma gondii vaccines. The experimental results demonstrated that nanoparticle-based delivery systems exhibit excellent performance in improving vaccine stability, delivery efficiency, and immunogenicity. In vivo experiments revealed that the constructed pVAX1-TgIMC1/PLGA and pVAX1-TgIMC1/CS nanoparticles significantly enhanced the production of specific antibodies, induced a balanced Th1 and Th2 mixed immune response, and promoted dendritic cell (DC) maturation and antigen presentation. These findings highlight the promise of nanomaterial-based DNA vaccine strategies as an effective approach for controlling T. gondii infections. Additionally, the pVAX1-TgIMC1/PLGA and pVAX1-TgIMC1/CS nanoparticles, referred to as nanovaccines, were shown to significantly inhibit parasite invasion in the spleen and reduce parasite burdens in the host. These results suggest that pVAX1-TgIMC1/PLGA and pVAX1-TgIMC1/CS nanoparticles are promising candidates for preventing acute toxoplasmosis. However, given the complexity of chronic T. gondii infections and their long-term impact on host immunity, further optimization of vaccine formulations and validation in larger-scale animal models will be necessary to establish a stronger basis for clinical application.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We sincerely thank Professor Li Xiangrui from Nanjing Agricultural University, Nanjing, China, for providing the Toxoplasma gondii RH strain (Type I).
Abbreviations
- PLGA
poly (lactic-co-glycolic acid)
- BUN
blood urea nitrogen
- Cr
creatinine
- DCM
dichloromethane
- DCs
dendritic cells
- DMEM
Dulbecco’s Modified Eagle’s Medium
- EE
encapsulation efficiency
- ELISA
enzyme linked immunosorbent
- FBS
fetal bovine serum
- IFN
interferon-gamma
- IL
interleukin
- PVDF
polyvinylidene fluoride
- PYR
pyrimethamine
- qPCR
quantitative real-time PCR
- SEM
scanning electron microscope
- SPF
specific pathogen free
- STAg
soluble tachyzoite antigens
- TGF
transforming growth factor
- TgIMC1
T. gondii Inner Membrane Complex Protein 1
- TMB
3,3’,5,5’-tetramethylbenzidine
Author contributions
ZY designed the research. YY provided financial support. PZ and YF conducted the research with assistance from YJ and TL. YL, WQ, and JW analyzed the data. YF wrote the manuscript. LZ and ZY participated in the revision of the manuscript. All authors contributed to data interpretation and approved the final version.
Funding
This research was funded by the High Level Scientific and Technological Talents Project of Lvliang City (2023RC-2-5), Scientific Research Start Funds for Advanced Talent in Ningxia Province (2024BEH04076), Ningxia Agricultural Science and Technology Independent Innovation Special Project – Scientific and Technological Innovation Guidance Project (NKYG-25-22), and National Natural Science Foundation of China (NSFC, 72203117).
Data availability
The raw data supporting the conclusions of this article will be made available by the corresponding author, without undue reservation.
Declarations
Ethics approval
The animal trials were approved by the Animal Ethics Committee, Ningxia University, Yinchuan, P.R. China. Approval number: NXU-2023-083 (for mouse trials), NXU-2023-086 (for rat trials). All the experimental design, animal management, injection, and operations were strictly followed the Ethics Procedures and Guidelines of the People’s Republic of China and were supervised by the Animal Ethics Committee, Ningxia University, Yinchuan, P.R. China.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
ZhengQing Yu, Email: yuzhengqing@nxu.edu.cn.
TingLi Liu, Email: LTL1114@163.com.
References
- 1.Hill D, Dubey JP. Toxoplasma gondii: transmission, diagnosis and prevention. Clin Microbiol Infect. 2002;8(10):634–40. [DOI] [PubMed] [Google Scholar]
- 2.Montoya JG, Liesenfeld O, Toxoplasmosis. Lancet (London England). 2004;363(9425):1965–76. [DOI] [PubMed] [Google Scholar]
- 3.Robert-Gangneux F, Dardé ML. Epidemiology of and diagnostic strategies for toxoplasmosis. Clin Microbiol Rev. 2012;25(2):264–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tenter AM, Heckeroth AR, Weiss LM. Toxoplasma gondii: from animals to humans. Int J Parasitol. 2000;30(12–13):1217–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim K, Weiss LM. Toxoplasma gondii: the model apicomplexan. Int J Parasitol. 2004;34(3):423–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elbez-Rubinstein A, Ajzenberg D, Dardé M-L, et al. Congenital toxoplasmosis and reinfection during pregnancy: case report, strain characterization, experimental model of reinfection, and review. J Infect Dis. 2009;199(2):280–5. [DOI] [PubMed] [Google Scholar]
- 7.Montoya JG, Remington JS. Toxoplasmic chorioretinitis in the setting of acute acquired toxoplasmosis. Clin Infect Dis. 1996;23(2):277–82. [DOI] [PubMed] [Google Scholar]
- 8.Basit KA, Nasir S, Vohra E, et al. Toxoplasmosis in an immunocompetent patient. Pakistan J Med Sci. 2018;34(6):1579–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luft BJ, Remington JS. Toxoplasmic encephalitis in AIDS. Clin Infect Diseases: Official Publication Infect Dis Soc Am. 1992;15(2):211–22. [DOI] [PubMed] [Google Scholar]
- 10.Layton J, Theiopoulou DC, Rutenberg D, et al. Clinical spectrum, radiological findings, and outcomes of severe toxoplasmosis in immunocompetent hosts: a systematic review. Pathogens (Basel Switzerland). 2023;12(4):543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo M, Dubey JP, Hill D, et al. Prevalence and risk factors for Toxoplasma gondii infection in meat animals and meat products destined for human consumption. J Food Prot. 2015;78(2):457–76. [DOI] [PubMed] [Google Scholar]
- 12.Lyons RE, Mcleod R, Roberts CW. Toxoplasma gondii tachyzoite-bradyzoite interconversion. Trends Parasitol. 2002;18(5):198–201. [DOI] [PubMed] [Google Scholar]
- 13.Dittmar AJ, Drozda AA, Blader IJ. Drug repurposing screening identifies novel compounds that effectively inhibit. Toxoplasma Gondii Growth mSphere. 2016;1(2):e00042–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buxton D, A Innes E. A commercial vaccine for ovine toxoplasmosis. Parasitology. 1995;110(Suppl):S11–6. [DOI] [PubMed] [Google Scholar]
- 15.Hasan T, Nishikawa Y. Advances in vaccine development and the immune response against toxoplasmosis in sheep and goats. Front Veterinary Sci. 2022;9:951584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mévélec MN, Lakhrif Z, Dimier-Poisson I. Key limitations and new insights into the Toxoplasma gondii parasite stage switching for future vaccine development in human, livestock, and cats. Front Cell Infect Microbiol. 2020;10:607198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kur J, Holec-Gąsior L, Hiszczyńska-Sawicka E. Current status of toxoplasmosis vaccine development. Expert Rev Vaccines. 2009;8(6):791–808. [DOI] [PubMed] [Google Scholar]
- 18.Chu KB, Quan FS. Advances in Toxoplasma gondii vaccines: current strategies and challenges for vaccine development. Vaccines. 2021;9(5):413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng B, Lou D, Ding J, et al. GRA24-based DNA vaccine prolongs survival in mice challenged with a virulent Toxoplasma gondii strain. Front Immunol. 2019;10:418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X, Yuan H, Mahmmod YS, et al. Insight into the current Toxoplasma gondii DNA vaccine: a review Article. Expert Rev Vaccines. 2023;22(1):66–89. [DOI] [PubMed] [Google Scholar]
- 21.Harding CR, Meissner M. The inner membrane complex through development of Toxoplasma gondii and plasmodium. Cell Microbiol. 2014;16(5):632–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen AL, Kim EW, Toh JY et al. Novel components of the Toxoplasma inner membrane complex revealed by bioID. mBio. 2015, 6:e02357–14. [DOI] [PMC free article] [PubMed]
- 23.Mann T, Gaskins E, Beckers C. Proteolytic processing of TgIMC1 during maturation of the membrane skeleton of Toxoplasma gondii. J Biol Chem. 2002;277(43):41240–6. [DOI] [PubMed] [Google Scholar]
- 24.Gubbels MJ, Wieffer M, Striepen B. Fluorescent protein tagging in Toxoplasma gondii: identification of a novel inner membrane complex component conserved among apicomplexa. Mol Biochem Parasitol. 2004;137(1):99–110. [DOI] [PubMed] [Google Scholar]
- 25.Pati R, Shevtsov M, Sonawane A. Nanoparticle vaccines against infectious diseases. Front Immunol. 2018;9:2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rocha CV, Gonçalves V, Da Silva MC, et al. PLGA-based composites for various biomedical applications. Int J Mol Sci. 2022;23(4):2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Danhier F, Ansorena E, Silva JM, et al. PLGA-based nanoparticles: an overview of biomedical applications. J Controlled Release. 2012;161(2):505–22. [DOI] [PubMed] [Google Scholar]
- 28.Zhao D, Yu S, Sun B, et al. Biomedical applications of Chitosan and its derivative nanoparticles. Polymers. 2018;10(4):462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang S, Zhao G, Wang W, et al. Pathogenicity of two Toxoplasma gondii strains in chickens of different ages infected via intraperitoneal injection. Avian Pathol. 2014;43(1):91–5. [DOI] [PubMed] [Google Scholar]
- 30.Wang S, Fang Z, Huang X et al. The soluble tachyzoite antigen of Toxoplasma gondii has a protective effect on mouse allografts. Transpl. proc. 2013, 45(2):677–83. [DOI] [PubMed]
- 31.Liu Q, Singla LD, Zhou H. Vaccines against Toxoplasma gondii: status, challenges and future directions. Hum Vaccines Immunotherapeutics. 2012;8(9):1305–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kochanowsky JA, Koshy AA. Toxoplasma gondii. Curr Biol. 2018;28(14):R770–1. [DOI] [PubMed] [Google Scholar]
- 33.Rashidi S, Sánchez-Montejo J, Mansouri R, et al. Mining the proteome of Toxoplasma parasites seeking vaccine and diagnostic candidates. Animals. 2022;12(9):1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daher D, Shaghlil A, Sobh E, et al. Comprehensive overview of Toxoplasma gondii-induced and associated diseases. Pathogens (Basel Switzerland). 2021;10(11):1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elsheikha HM, Marra CM, Zhu XQ. Epidemiology, pathophysiology, diagnosis, and management of cerebral toxoplasmosis. Clin Microbiol Rev. 2021;34(1):e00115–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warner RC, Chapman RC, Davis BN, et al. Review of DNA vaccine approaches against the parasite Toxoplasma gondii. J Parasitol. 2021;107(6):882–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mamaghani AJ, Fathollahi A, Arab-Mazar Z, et al. Toxoplasma gondii vaccine candidates: a concise review. Ir J Med Sci. 2023;192(1):231–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li L, Petrovsky N. Molecular mechanisms for enhanced DNA vaccine immunogenicity. Expert Rev Vaccines. 2016;15(3):313–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Verma R, Khanna P. Development of Toxoplasma gondii vaccine: a global challenge. Hum Vaccines Immunotherapeutics. 2013;9(2):291–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang NZ, Chen J, Wang M, et al. Vaccines against Toxoplasma gondii: new developments and perspectives. Expert Rev Vaccines. 2013;12(11):1287–99. [DOI] [PubMed] [Google Scholar]
- 41.Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2003;55(3):329–47. [DOI] [PubMed] [Google Scholar]
- 42.Das A, Ali N. Nanovaccine: an emerging strategy. Expert Rev Vaccines. 2021;20(10):1273–90. [DOI] [PubMed] [Google Scholar]
- 43.Wang B, Dong Y, Cen Y, et al. PEI-PLGA nanoparticles significantly enhanced the immunogenicity of IsdB(137–361) proteins from Staphylococcus aureus. Immun Inflamm Dis. 2023;11(7):e928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keijzer C, Slütter B, Van Der Zee R, et al. PLGA, PLGA-TMC and TMC-TPP nanoparticles differentially modulate the outcome of nasal vaccination by inducing tolerance or enhancing humoral immunity. PLoS ONE. 2011;6(11):e26684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abd El Hady WE, Mohamed EA, Soliman OE, et al. In vitro-in vivo evaluation of chitosan-PLGA nanoparticles for potentiated gastric retention and anti-ulcer activity of diosmin. Int J Nanomed. 2019;14:7191–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Silva AL, Soema PC, Slütter B, et al. PLGA particulate delivery systems for subunit vaccines: linking particle properties to immunogenicity. Hum Vaccines Immunotherapeutics. 2016;12(4):1056–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roozbehani M, Falak R, Mohammadi M, et al. Characterization of a multi-epitope peptide with selective MHC-binding capabilities encapsulated in PLGA nanoparticles as a novel vaccine candidate against Toxoplasma gondii infection. Vaccine. 2018;36(41):6124–32. [DOI] [PubMed] [Google Scholar]
- 48.Layek B, Das S. Chapter 8 - Chitosan-based nanomaterials in drug delivery applications. In: Bera H, Hossain C, Saha M S, editors. Biopolymer-Based nanomaterials in drug delivery and biomedical applications. Cambridge: Academic; 2021. pp. 185–219. [Google Scholar]
- 49.Jafernik K, Ładniak A, Blicharska E, et al. Chitosan-based nanoparticles as effective drug delivery systems-a review. Molecules. 2023;28(4):1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li J, Cai C, Li J, et al. Chitosan-based nanomaterials for drug delivery. Molecules. 2018;23(10):2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiang SD, Wilson K, Day S, et al. Methods of effective conjugation of antigens to nanoparticles as non-inflammatory vaccine carriers. Methods (San Diego Calif). 2013;60(3):232–41. [DOI] [PubMed] [Google Scholar]
- 52.Allahyari M, Mohit E. Peptide/protein vaccine delivery system based on PLGA particles. Hum Vaccines Immunotherapeutics. 2016;12(3):806–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol. 2010;10(11):787–96. [DOI] [PubMed] [Google Scholar]
- 54.Schaffazick SR, Pohlmann AR, Dalla-Costa T, et al. Freeze-drying polymeric colloidal suspensions: nanocapsules, nanospheres and nanodispersion: A comparative study. Eur J Pharm Biopharm. 2003;56(3):501–5. [DOI] [PubMed] [Google Scholar]
- 55.Zorgi NE, Costa A, Galisteo AJ Jr., et al. Humoral responses and immune protection in mice immunized with irradiated T. gondii tachyzoites and challenged with three genetically distinct strains of T. gondii. Immunol Lett. 2011;138(2):187–96. [DOI] [PubMed] [Google Scholar]
- 56.Zhang Z, Li Y, Wang M, et al. Immune protection of Rhoptry protein 21 (ROP21) of Toxoplasma gondii as a DNA vaccine against toxoplasmosis. Front Microbiol. 2018;9:909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pagheh AS, Sarvi S, Gholami S, et al. Protective efficacy induced by DNA prime and Recombinant protein boost vaccination with Toxoplasma gondii GRA14 in mice. Microb Pathog. 2019;134:103601. [DOI] [PubMed] [Google Scholar]
- 58.Germann T, Bongartz M, Dlugonska H, et al. Interleukin-12 profoundly up-regulates the synthesis of antigen-specific complement-fixing IgG2a, IgG2b and IgG3 antibody subclasses in vivo. Eur J Immunol. 1995;25(3):823–9. [DOI] [PubMed] [Google Scholar]
- 59.Nabi H, Rashid I, Ahmad N, et al. Induction of specific humoral immune response in mice immunized with ROP18 nanospheres from Toxoplasma gondii. Parasitol Res. 2017;116(1):359–70. [DOI] [PubMed] [Google Scholar]
- 60.Suzuki Y, Sa Q, Gehman M, et al. Interferon-gamma- and perforin-mediated immune responses for resistance against Toxoplasma gondii in the brain. Expert Rev Mol Med. 2011;13:e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ahmed N, French T, Rausch S, et al. Toxoplasma Co-infection prevents Th2 differentiation and leads to a helminth-specific Th1 response. Front Cell Infect Microbiol. 2017;7:341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zheng B, Ding J, Lou D, et al. The virulence-related MYR1 protein of Toxoplasma gondii as a novel DNA vaccine against toxoplasmosis in mice. Front Microbiol. 2019;10:734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Murillo-Léon M, Bastidas-Quintero AM, Steinfeldt T. Decoding Toxoplasma gondii virulence: the mechanisms of IRG protein inactivation. Trends Parasitol. 2024;40(9):805–19. [DOI] [PubMed] [Google Scholar]
- 64.Ihara F, Yamamoto M. The role of IFN-γ-mediated host immune responses in monitoring and the elimination of Toxoplasma gondii infection. Int Immunol. 2024;36(5):199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dupont CD, Christian DA, Hunter CA. Immune response and immunopathology during toxoplasmosis. Semin Immunopathol. 2012;34(6):793–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tawfeek GM, Abou-El-Naga IF, Hassan EME, et al. Protective efficacy of Toxoplasma gondii infected cells-derived exosomes against chronic murine toxoplasmosis. Acta Trop. 2023;248:107041. [DOI] [PubMed] [Google Scholar]
- 67.Zhao M, Zhang R, Xu X, et al. IL-10 reduces levels of apoptosis in Toxoplasma gondii-infected trophoblasts. PLoS ONE. 2013;8(2):e56455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Doherty CM, Patterson PR, Emeanuwa JA et al. T lymphocyte-dependent IL-10 down-regulates a cytokine storm driven by Toxoplasma gondii GRA24. mBio. 2024;15(11):e0145524. [DOI] [PMC free article] [PubMed]
- 69.Silver JS, Stumhofer JS, Passos S, et al. IL-6 mediates the susceptibility of glycoprotein 130 hypermorphs to Toxoplasma gondii. J Immunol. 2011;187(1):350–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yan J, Wu B, Huang B, et al. Dectin-1-CD37 association regulates IL-6 expression during Toxoplasma gondii infection. Parasitol Res. 2014;113(8):2851–60. [DOI] [PubMed] [Google Scholar]
- 71.Muñoz M, Heimesaat MM, Danker K, et al. Interleukin (IL)-23 mediates Toxoplasma gondii-induced immunopathology in the gut via matrixmetalloproteinase-2 and IL-22 but independent of IL-17. J Exp Med. 2009;206(13):3047–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Silva JL, Rezende-Oliveira K, Da Silva MV et al. IL-17-expressing CD4⁺ and CD8⁺ T lymphocytes in human toxoplasmosis. Mediat inflamm. 2014;2014:573825. [DOI] [PMC free article] [PubMed]
- 73.Banchereau J, Briere F, Caux C, et al. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. [DOI] [PubMed] [Google Scholar]
- 74.Aerts-Toegaert C, Heirman C, Tuyaerts S, et al. CD83 expression on dendritic cells and T cells: correlation with effective immune responses. Eur J Immunol. 2007;37(3):686–95. [DOI] [PubMed] [Google Scholar]
- 75.Pinho MP, Migliori IK, Flatow EA, et al. Dendritic cell membrane CD83 enhances immune responses by boosting intracellular calcium release in T lymphocytes. J Leukoc Biol. 2014;95(5):755–62. [DOI] [PubMed] [Google Scholar]
- 76.Fooksman DR, Organizing. MHC class II presentation. Front Immunol. 2014;5:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the corresponding author, without undue reservation.