

ABSTRACT
The intrinsic characteristics of oligonucleotides pose a challenge for their assessment in conventional primary in vitro cardiac models, which were designed for the acute application of small molecule agents and are not suitable for transfection and extended culture periods. Conversely, human‐induced pluripotent stem cell‐derived cardiomyocytes (hiPSC‐CM) offer a viable platform for the evaluation of agents over prolonged application and recording times. Our previous experiments demonstrated that a chronic protocol of 48 h is necessary to discern the functional effects of a siRNA targeting hERG in a stable cell line heterologously expressing hERG. To investigate whether a targeted hERG siRNA induces delayed repolarization in hiPSC‐CM, we recorded field potentials (FPs) using a multielectrode array. FP duration (FPD) prolongation was noted as early as 10 min after exposure to moxifloxacin, whereas pentamidine required 24 h to induce FPD prolongation. Transfection with hERG‐targeting siRNA reduced mRNA expression at 6 h post‐transfection. However, FPD prolongation was only observed after 24 h post‐transfection, with significantly larger effects at 48 h, which is indicative of the time needed for turnover of the hERG protein on the plasma membrane. Our findings provide compelling evidence that MEA recordings in hiPSC‐CM can accurately detect disruptions in cardiac repolarization due to various mechanisms that impair hERG channel function, including direct channel blockade, inhibition of protein trafficking, and gene silencing via siRNA. The findings also indicate that indirect mechanisms of hERG knockdown, including gene silencing, require assessment at least 48 h following treatment to detect delayed repolarization in the hiPSC‐CM model.
Keywords: hERG, hiPSC‐CM, multielectrode array, oligonucleotide, PCR, siRNA
Summary.
- What is the current knowledge on the topic?
-
○Evaluating oligonucleotides in conventional primary in vitro cardiac models is challenging due to their inherent characteristics, which are unsuitable for transfection and extended culture periods. These models were designed for the acute application of agents.
-
○
- What question did this study address?
-
○Are human‐induced pluripotent stem cell‐derived cardiomyocytes (hiPSC‐CM) a feasible alternative model for assessing agents over the extended application and recording durations that are required for the mechanism of gene silencing by siRNA.
-
○
- What does this study add to our knowledge?
-
○Our research demonstrates that MEA recordings in hiPSC‐CM can precisely identify disruptions in cardiac repolarization caused by various mechanisms impairing hERG channel function, such as direct blockage, inhibition of protein trafficking, and gene silencing via siRNA.
-
○
- How might this change clinical pharmacology or translational science?
-
○This study demonstrates strong evidence that hiPSC‐CM provides comprehensive risk assessment in understanding and mitigating the proarrhythmic risk of oligonucleotide therapeutics. As the field progresses, this model will continue to evolve, incorporating new data and technologies, ultimately ensuring the safe and effective use of these innovative therapies.
-
○
1. Introduction
Oligonucleotides are synthetic nucleic acid molecules that can modulate the expression of target genes through various mechanisms, such as antisense, RNA interference, or gene editing [1, 2]. Oligonucleotides (ONTs) have emerged as a promising class of drugs for the treatment of various diseases, especially those with genetic origins. As of June 2024, 21 ONTs have been approved in the EU and/or US [3, 4, 5, 6, 7] and many more are in development which sparks hope for patients with rare and fatal diseases [8, 9]. However, the inherent properties of oligonucleotides present obstacles to their evaluation in traditional primary in vitro cardiac models, which are typically designed for the short‐term application of agents and are unsuitable for transfection and prolonged culture durations.
Human stem cell‐derived cardiomyocytes (SC‐CM) [10], especially those derived from the induced pluripotent stem (iPS) cells (iPSC‐CM [11]), have provided an opportunity to assess the cardiac safety pharmacology profile of new agents as one of the new approach methodologies (NAM). Human iPSC‐CMs are widely available and can serve as in vitro models of human cardiac cells for drug safety and efficacy testing and may be useful for addressing species differences and support improved translation from preclinical models to clinical outcomes, alleviating reliance on primary heart tissues which are limited in availability [12]. A variety of platforms are being used to study the electrophysiological profile of hiPSC‐CMs, with multiple technologies available for screening applications including patch clamp, [13, 14] fluorescent imaging techniques, [15, 16, 17, 18, 19] and multielectrode array (MEA, e.g., [18, 20, 21]).
MEA electrophysiology measures electrical changes in cardiomyocytes using arrays of microscopic electrodes distributed over a small surface area at the bottom of wells in a multiwell plate. Cells are cultured on top of the electrodes and form cohesive networks over time. MEA is used to record induced or spontaneous field potentials (FPs) rather than directly measuring action potentials, capturing the spatiotemporal electrical activity of cell clusters, which represents a summation of all ionic processes. These FPs, akin to clinical electrocardiograms, allow for the reconstruction of waveforms to extract vital physiological parameters and infer cardiac ion channel effects. MEA is particularly useful for real‐time and medium‐throughput measurement of hiPSC‐CM electrical activity, providing valuable data on potential proarrhythmic risks by measuring FP duration (FPD) [18, 20, 21]. The combination of hiPSC‐CM with MEA technology for the assessment of cardiovascular risk at the preclinical stage of drug development is a valuable tool for lead optimization and candidate selection [22]. MEA systems can capture fast‐acting events, such as cardiac depolarization (initial FP spike), and provide a measure of cardiac repolarization which is represented by the time from the initial FP spike to the peak of T‐wave, termed the FPD and used as a surrogate for QT interval. MEA systems offer the distinct advantage of continuous data collection over an extended time period and the ability to perform repeated measures from the same culture on multiple occasions, thus offering a comprehensive evaluation of cardiac function over time [23].
In our previous study [24], a siRNA specifically targeting hKCNH2 (positive control) was utilized to investigate the time course of siRNA‐mediated inhibition of hERG function and gene expression, i.e., knockdown. To determine whether MEA recordings in hiPSC‐CMs can detect repolarization delay by the positive tool siRNA targeting hKCNH2 and the time course of such an effect, we evaluated the hKCNH2 gene expression and repolarization duration using FPD as a biomarker in hiPSC‐CM with and without hKCNH2 siRNA transfection. In addition, a direct hERG channel blocker, moxifloxacin, and a hERG trafficking inhibitor, pentamidine, were also included to compare time‐dependent effects on FPDc and hKCNH2 gene expression.
2. Methods
2.1. Cell Culture and Transfection
hiPSC‐CM, also known as iCell Cardiomyocytes,2 were purchased from FUJIFILM Cellular Dynamics (Madison, WI). At day 0, MEA plates were coated with fibronectin diluted in 0.1% gelatin, and cells were seeded over the recording electrodes at a density of 50k cells/well in 5 μl of plating medium (iCell Cardiomyocytes Plating Medium, FUJIFILM Cellular Dynamics) in the Cytoview 48‐well plates (Axion Biosystems Inc., Atlanta, GA). The MEA plate with the seeded cardiomyocytes was incubated in a cell culture incubator at 37°C, 5% CO2 for 1 h, and 300 μl fresh maintenance medium was added to each well of the MEA plates. At day 1 and day 4, the medium was changed, and incubation continued under the same conditions.
At day 5, DharmaFECT 4 (Horizon Discovery, Catalog ID: T‐2005‐01) transfection reagent was employed to transfect siRNAs into iPSC‐CMs. The process began with the preparation of the siRNA transfection mixture. Specifically, the hKCNH2 siRNA and DharmaFECT 4 were diluted in OptiMEM (Invitrogen) separately to achieve the desired final concentration.
For optimal results, the dilution steps were critical. First, the hKCNH2 siRNA was carefully measured and diluted in OptiMEM. To reach 10 nM siRNA in the transfection mixture, siRNA was diluted with 10 μl siRNA to 500 μl OptiMEM; to reach 30 nM siRNA in the transfection mixture, siRNA was diluted with 30 μl siRNA to 500 μl OptiMEM. Concurrently, DharmaFECT 4 was separately diluted. To reach 10 nM siRNA in the transfection mixture, DharmaFECT 4 was diluted with 2 μl DharmaFECT 4–500 μl OptiMEM; to reach 30 nM siRNA in the transfection mixture, DharmaFECT 4 was diluted with 6 μl DharmaFECT 4–500 μl OptiMEM. The diluted siRNA and DharmaFECT 4 were incubated separately for 5 min. After the incubation period, the diluted siRNA and DharmaFECT 4 reagent were combined into a single mixture with 1:1 ratio. Then, this mixture was incubated for 25 min.
Once the transfection mixture was ready after 25 min incubation, 30 μL of this mixture was pipetted into each well containing the iPSC‐CMs. The plates were then incubated at 37°C under an atmosphere of 5% CO2 for 24 h. Post‐transfection 24 h, the medium was carefully removed and replaced with fresh maintenance media.
The plates were frozen at −70°C after each recording for RNA isolation.
2.2. FP Recordings and Analysis in hiPSC‐CM
FP recordings were performed using a Maestro Pro multiwell multielectrode array (MEA) (Axion Biosystems Inc.) at days 5, 6, and 7. When the cells were confluent and formed one beating syncytium, the FP was recorded for 2 min as a baseline. For moxifloxacin and pentamidine treatment, recordings were performed at 10 min, 6, and 24 h after addition. For siRNA transfection, recordings were performed at 6, 24, and 48 h after siRNA transfection. The medium was changed to a fresh medium after the 24 h recording.
When recordings were performed, Cytoview plates were connected to the Maestro Pro multiwell MEA system and FPs were collected for 2 min per well. The FP was analyzed and quantified using the Cardiac Analysis Tool (Axion Biosystems Inc.) to determine the FPD (msec) and beat period (BP, msec). Since BP was affected by treatment, FPD was corrected using the Fridericia equation using the following formula, FPDc = (FPD in msec)/(BeatPeriod in msec)1/3.
Data are presented as the mean and standard deviation of the mean. Statistical analysis was performed with one‐way ANOVA with Dunnett's multiple comparisons (GraphPad Prism, GraphPad Software, MA, USA). Significant differences were identified with p < 0.05.
2.3. RNA Isolation and qPCR
Cells were harvested with RLT (RNeasy lysis of cells and tissues) buffer, and QIAGEN RNeasy Mini Kit was used for RNA purification following the manufacturer's instructions (QIAGEN). After RNA purification, total RNA was treated with RQ1 DNase (Promega) to eliminate genomic DNA contamination. The evaluation of qPCR reaction was performed using TaqMan RNA‐to‐CT 1‐Step Kit (Applied Biosystems).
KCNH2 expression was evaluated by qPCR. HPRT1 was measured as a housekeeping gene in iPSC‐CMs and used for the normalization of KCNH2 expression levels. Samples from multiple Cytoview plate wells with the same treatment were pooled together and used for KCNH2 expression analysis. The assay IDs of the probes used for quantifying the human gene expressions are KCNH2 Hs00542479_g1 and housekeeping HPRT1 Hs02800695_m1.
2.4. Reagents
Moxifloxacin (151096‐09‐2) and pentamidine (100‐33‐4) were purchased from SigmaAldrich (MO, US). The stock solutions of both agents were prepared in DMSO and diluted in iPSC‐CM maintenance medium with a final concentration of 0.1% DMSO. The final testing concentrations of moxifloxacin were 3, 10, and 30 μM, and the final testing concentrations of pentamidine were 1, 3, and 10 μM.
Four tool siRNAs (KCNH2 siRNA) were investigated in this study to understand concentration‐ and time‐dependent effects on KCNH2 gene expression and repolarization in hiPSC‐CM. The KCNH2 siRNAs were used to specifically knockdown hERG expression and served as the positive control. These molecules have sense strand sequences shown below that target the nucleotides of c‐linker regions as well as the S5 side of the P‐loop of hERG.
Homo sapiens KCNH2 transcript variant 1 mRNA (NM_000238.4).
siRNA 1 targeting location: 2015–2033: CGCGGAAGCUGGAUCGCUA; c‐linker region
siRNA 2 targeting location: 1844–1862: ACGAGGAGGUGGUCAGCCA; between S5P and P
siRNA 3 targeting location: 2175–2193: GGGCGACCAGAUAGGCAAA: c‐linker region
siRNA 4 targeting location: 2140–2158: CACAUGGACUCACGCAUCG: c‐linker region
Both KCNH2 siRNA and nontargeting control negative siRNA were ordered from Horizon Discovery (Dharmacon). (Catalog ID: L‐006233‐00‐0002 for KCNH2 siRNA; Catalog ID: D‐001810‐01‐05 for nontargeting control negative siRNA). The nontargeting control negative siRNA has the sequence of UGGUUUACAUGUCGACUAA.
3. Results
3.1. Effects of an Acute hERG Blocker, Moxifloxacin
To investigate the concentration and time‐dependent effects of a direct hERG blocker on FPDc in hiPSC‐CM, moxifloxacin treatment was performed at 3, 10, and 30 μM and FPDc was recorded after 10 min, 6, and 24 h application. FPDc is shown in Figure 1 as the percent change from baseline at each condition, including no treatment and treatment with vehicle (0.1% DMSO). Statistical analysis was performed by comparing each drug treatment with DMSO application. Moxifloxacin significantly prolonged the FPDc in a concentration‐dependent manner. Notably, significant effects were observed at concentrations of 10 and 30 μM, which were consistent with reported hERG inhibition potency (IC50 = 62 μM, [25]). The FPDc prolongation at 10 μM was statistically significant (p < 0.01) at 10 min, 6 and 24 h with a slight increase in magnitude with longer incubation (13.4%, 19.6%, and 29.2%, respectively). While the effect at 24 h was significantly different from 10 min (p < 0.05), it was not significantly different from 6 h. At 30 μM, there was no significant difference among the 3 time points, which is consistent with a mechanism of an acute effect through direct channel blockade.
FIGURE 1.

Moxifloxacin increased beat rate corrected field potential duration (FPDc) in hiPSC‐CM. Percent changes from baseline and the application time of 10 min, 6, and 24 h are presented under different conditions as labeled. Compared to vehicle control (DMSO), statistically significant prolongation of FPDc was observed at 10 and 30 μM moxifloxacin at all time points. The number of wells, from the same plate, in each condition are indicated, and statistical significance is indicated as **p < 0.01; 5.**p < 0.001; ****p < 0.0001.
Moxifloxacin treatment also increased the BP in comparison with the 0.1% DMSO control as shown in Figure S1, i.e., the beat rate decreased at the same concentrations that increased FPDc.
After 24 h of treatment, the expression level of KCNH2 (determined from the MEA test plate) was not affected by moxifloxacin at any of the testing concentrations (Figure 3).
FIGURE 3.

Lack of effects on hKCNH2 expression by treatments with moxifloxacin and pentamidine for 24 h. These were the same cells after the MEA recordings. Each measurement was performed with four to six wells pooled together from the same plate. The hKCNH2 expression was normalized to HPRT, a housekeeping gene in hiPSC‐CM.
3.2. Effects of a hERG Trafficking Inhibitor, Pentamidine
The concentration‐ and time‐dependent effects of pentamidine on hiPSC‐CM FPDc were evaluated at multiple concentrations (1, 3, and 10 μM) and timepoints (10 min, 6, and 24 h) after treatment. As shown in Figure 2, no effects were detected at 10 min and 6 h. At 24 h, a mean 21.9% prolongation of FPDc by 1 μM pentamidine was observed, which was statistically significant (p < 0.01) when compared to 0.1% DMSO application. A greater prolongation of FPDc was achieved at 3 μM (73.4% mean increase), while at 10 μM, irregular beating of hiPSC‐CM made it difficult to derive the FPDc, as an example waveform shown in Figure S2.
FIGURE 2.

Pentamidine increased beat rate corrected field potential duration (FPDc) in hiPSC‐CM. Effects are observed at 24 h after application while having no effect on FPDc at 10 min and 6 h after treatment. Percent changes from baseline are presented under different conditions as labeled. At 24 h after application, compared to vehicle control (DMSO), statistically significant prolongation of FPDc was achieved at 1 and 3 μM of pentamidine. The number of wells, from the same plate, in each condition are indicated, and statistical significance is indicated as *p < 0.05; ****p < 0.0001.
Treatment with pentamidine also increased the BP as shown in Figure S3, i.e., the beat rate was decreased at the same concentration time points as FPDc increases. While 10 min treatment with 10 μM pentamidine decreased the BP significantly compared to the DMSO application, the effect was less than 4%, which was not considered biologically meaningful.
After 24 h of treatment, the expression level of KCNH2 (determined from the MEA test plate) was not affected by pentamidine at any of the testing concentrations (Figure 3).
3.3. Effects of KCNH2 siRNAs in hiPSC‐CM
Transfection of four individual KCNH2 siRNAs (positive controls) was initially investigated to look for optimal inhibition of KCNH2 expression and compared with two negative siRNAs (negative controls) in hiPSC‐CM. In addition, a mixture of all four KCNH2 siRNAs was transfected to evaluate if a combination of siRNAs would produce greater effects than an individual siRNA. The hKCNH2 mRNA level relative to HPRT1 mRNA (a housekeeping gene in hiPSC‐CM) was used to determine the knockdown efficiency of the tested siRNAs. Figure 4 left panel showed the relative hKCNH2 expression under different transfection conditions at 48 and 72 h. The hKCNH2 expression levels after transfection with the two negative control siRNAs were similar to mock transfection, confirming they had no effect on hKCNH2 expression and could be used as negative controls. Negative siRNA #1 was selected as the negative control for later experiments. In contrast, hKCNH2 expression was greatly reduced by transfection with 10 nM of each individual KCNH2 siRNA or a mixture of the four siRNAs (2.5 nM each). The magnitude of hKCNH2 reduction with hKCNH2 siRNA #1 transfection (90% at 48 h) was similar to that of the mixture of four siRNAs (89% at 48 h) and was larger than the effect achieved individually with KCNH2 #2, #3, or #4 siRNAs (75%–76% at 48 h). No further decrease in hKCNH2 mRNA levels was observed at 72 h. Based on these findings, hKCNH2 siRNA #1 was selected as the positive control siRNA for the next set of studies investigating the effect of knockdown on FPD and is simply referred to as hKCNH2 siRNA.
FIGURE 4.
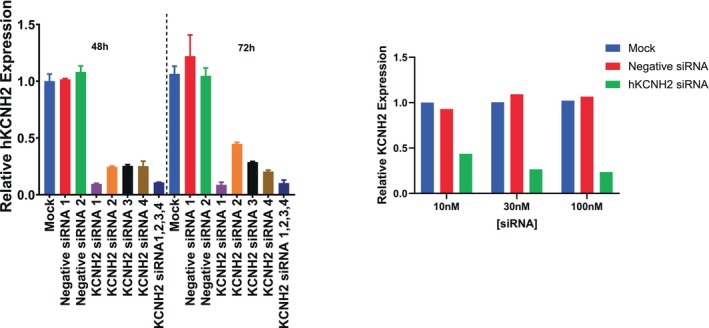
Selection of positive hKCNH2 siRNA, negative siRNA, and the testing concentration. Left panel: Expression level of hKCNH2 relative to HPRT1 following transfection of hiPSC‐CM with negative and positive KCNH2 siRNAs. Relative hKCNH2 expression after 48 and 72 h transfection with negative sRNAs (1 and 2), and KCNH2 siRNA (1, 2, 3, 4, and a mixture of all 4 were shown). Each individual siRNA was tested at 10 nM and the concentration of each siRNA in the mixture was 2.5 nM. Each condition was tested twice, and three wells of cells from the same plate were pooled together. Right panel: Relative expression level of hKCNH2 was decreased by KCNH2 siRNA transfection in a concentration‐dependent manner while there was no effect of Negative KCNH2 siRNA 48 h after transfection in hiPSC‐CM. Each sample was from eight wells pooled together from one plate.
To select the testing concentration of hKCNH2 siRNA, we evaluated the impact of selected hKCNH2 siRNA on hKCNH2 mRNA levels by exploring concentration dependence using transfection concentrations of 10, 30, and 100 nM. Post 48 h transfection, hKCNH2 mRNA levels were measured and compared against either mock transfection or negative siRNA transfection. Figure 4 right panel shows a pronounced reduction in hKCNH2 expression across all tested concentrations with greater decreases observed at 30 and 100 nM (76% and 78%, respectively) compared to 10 nM (53%). Based on this result, 30 nM was selected for the next experiment.
To investigate the time‐dependent effect of hKCNH2 siRNA on repolarization in hiPSC‐CM, 30 nM hKCNH2 was used to transfect hiPSC‐CM. Recordings of FPD were performed at 6, 24, and 48 h after siRNA transfection. As shown in Figure 5, no effects were observed 6 h after hKCNH2 siRNA transfection. At 24 h, mean FPDc was prolonged by 38.6%, and further prolongation of FPDc was observed at 48 h (mean prolongation of 185.9%). Both effects were statically significant (p < 0.0001). In addition, at 48 h, irregular beats were observed in multiple wells making the measurement of FPD difficult and only measurable in five wells. An example waveform is shown in Figure S4.
FIGURE 5.

Time‐dependent effects of KCNH2 siRNA on beat rate corrected field potential duration (FPDc) in hiPSC‐CM. Percent change from baseline is presented under different conditions as labeled. The tested concentration of siRNA was 30 nM. No effects were observed at 6 h, statistically significant prolongation of FPDc was achieved at 24 and 48 h after transfection with greater effects observed at 48 h. The number of wells in each condition are indicated, one plate was used for each time point. Statistical significance is indicated as ****p < 0.0001.
The transfection of hKCNH2 siRNA also increased BP at 24 and 48 h as shown in Figure S5, i.e., beat rate was decreased at the same concentrations and time points as FPDc increases. At 6 h, the BP was not impacted by KCNH2 siRNA, and differences from mock transfection were similar to that of transfection with negative siRNA.
To correlate hKCNH2 mRNA levels with functional changes in FPD, qPCR was performed on the same hiPSC‐CM at 6, 24, and 48 h following transfection with siRNA after completion of MEA recordings. Figure 6 displays the KCNH2 mRNA levels normalized to HPRT1. The KCNH2 mRNA levels were similar between mock and negative siRNA transfection at all three timepoints. Compared to mock and negative siRNA transfection, the mRNA levels were markedly reduced at all three timepoints in the KCNH2 siRNA transfected cells.
FIGURE 6.

Time‐dependent effects of KCNH2 siRNA on relative hKCNH2 expression levels. The measurement of qPCR was performed with the same cells after MEA recordings. The cells were harvested from five to six wells and pooled from the same plate (three samples at each condition). Each symbol represented one sample and the bar represented the mean value.
To illustrate the time‐dependent effects, the relative KCNH2 mRNA expression with KCNH2 siRNA transfection was normalized to the relative expression with negative siRNA transfection and expressed as % reduction (Figure 7). The time‐dependent effects of KCNH2 siRNA on FPDc in hiPSC‐CM are overlayed and show that reductions in mRNA expression occurred earlier than the effect on FPDc, an endpoint for protein function. For example, the percent reduction in KCNH2 mRNA was 86% at 6 h following transfection, a time when there were minimal effects on FPDc. The full functional impact of the mRNA knockdown is not observed until 48 h following transfection, likely reflecting the time required for turnover of hERG protein expressed on the plasma membrane.
FIGURE 7.

Gene silencing occurs before functional changes. Overlay plots of percent changes in beat rate corrected field potential duration (FPDc) and KCNH2 mRNA level caused by KCNH2 siRNA transfection in hiPSC‐CM.
4. Discussion
To our best knowledge, the current study is the first investigating time‐dependent prolongation of FPD by a specific hKNH2 siRNA in hiPSC‐CM, as compared to a direct hERG channel blocker and a hERG trafficking inhibitor. Our results strongly suggest that MEA recordings in hiPSC‐CM can truthfully identify perturbations of cardiac repolarization caused by various mechanisms that reduce hERG channel function, including direct hERG channel blockage, inhibition of hERG protein trafficking, and gene silencing through siRNA. In addition, the time‐dependent reduction of hERG mRNA and prolongation of FPDc by the tool hERG siRNA in hiPSC‐CM correlate well with the time‐dependent inhibition of hERG mRNA expression and function in the more traditional hERG patch clamp assay performed in a heterologous cell expression system [24]. The capability to maintain stable hiPSC‐CM function over extended periods (days to months) and to introduce test agents via transfection offers significant advantages over traditional in vitro cardiac models for detecting different modes of hERG modulation including direct pore block and changes in cell surface expression.
4.1. Time‐ and Concentration‐Dependent Effects of Moxifloxacin and Pentamidine Are Consistent
Our results with moxifloxacin and pentamidine treatment are consistent with previous publications and their known mechanisms of action. Moxifloxacin is a fluoroquinolone antibiotic, and its clinical usage has been associated with acquired long QT syndrome [26]. Moxifloxacin inhibits the hERG channel by interacting directly with the amino acids in the channel's inner cavity [27] and is commonly used as a positive control in the evaluation of hERG function and QT assessment in the clinic [28]. The ICH E14/S7B training materials data [29] show that moxifloxacin inhibits hERG with an IC50 of 62 μΜ (95% confidence interval: 38 μM, 104 μM) and actual QTc prolongation (10 ms) occurs at a free human plasma exposure level of 2.8 μM [25]. Our study showed that moxifloxacin prolonged FPDc of hiPSC‐CM in a concentration‐dependent manner and significant effects were observed at 10 and 30 μM, consistent with hERG inhibition potency, but three‐fold less sensitive than clinical findings. In addition, the magnitude of FPDc increase remained relatively unchanged with longer incubation periods (up to 24 h), consistent with a mechanism of an acute inhibition caused by a direct channel blocker.
Pentamidine is an antiprotozoal drug that causes acquired long QT syndrome in the clinic, which is associated with QT interval prolongation, tachycardias, and sudden cardiac arrest [30]. Pentamidine reduces the surface expression of hERG by binding to a folding intermediate, which arrests channel maturation in a conformational state that cannot be exported from the endoplasmic reticulum [31]. Therefore, pentamidine has been investigated extensively as a small molecule anti‐chaperone or trafficking inhibitor of hERG. The therapeutic exposure (total) of pentamidine is in the range of 0.3–1.4 μg/mL (0.9–4.1 μM; [30]). With 69% protein bound [32], the free therapeutic plasma exposure range is 0.3–1.3 μM. In the current study, prolongation of FPDc was observed at 24 h with 1 μM pentamidine, a concentration similar to the free therapeutic plasma concentration, while no changes were observed at 10 min and 6 h, consistent with its mechanism of action and previous publications [18].
4.2. Human iPSC‐CMs as an In Vitro Proarrhythmia NAM
Induced pluripotent stem cell‐derived cardiomyocytes (iPSC‐CM) have gained prominence as a transformative tool in cardiac research and various applications, particularly serving as in vitro models to study proarrhythmic risks [33]. However, a significant limitation of iPSC‐CM lies in their immature phenotype when compared to adult cardiomyocytes. These cells display characteristics akin to fetal cardiomyocytes, such as underdeveloped sarcomeres, reduced expression of ion channels, and distinct electrophysiological properties. For example, hiPSC‐CM have a more positive maximum diastolic potential, a slower upstroke velocity, and an almost absent notch [33, 34]. One of the most remarkable differences between hiPSC‐CM and native adult human primary cardiomyocytes is that hiPSC‐CM (including ventricular‐like and atrial‐like cells) display innate automaticity resulting in spontaneous beating similar to fetal human cardiomyocytes [35]. The rate of spontaneous beating is significantly decreased in the presence of hERG inhibition as shown in this study and previous publications [18, 36, 37], due to the dominant role that hERG plays in hiPSC‐CM repolarization. This immaturity can potentially impact the precision of proarrhythmic risk evaluations [33, 34].
Despite their electrical and mechanical immaturity, the gene expression of KCNH2 and the protein expression of hERG in hiPSC‐CM have been observed at levels comparable to those in the human heart [18, 38]. Moreover, hiPSC‐CM respond robustly to hERG channel‐blocking drugs leading to APD prolongation and arrhythmias. Consistently reported in the literature, specific small molecule blockers of the hERG current not only induce AP prolongation, but also arrhythmogenic events such as early afterdepolarizations (EADs) in hiPSC‐CM. FPD in multielectrode array recordings is sensitive to hERG channel blockade by small molecules [18, 21, 39, 40, 41] with a repolarization delay in hiPSC‐CM at a pharmacological concentration range that also inhibits hERG channel function. These findings provide strong evidence in supporting the use of hiPSC‐CM as a human tissue model for detecting QTc interval prolongation risk induced by small molecules that inhibit hERG channel function.
One of the important advantages of using hiPSC‐CM over scarcely available primary heart cells is their capacity for extended culture and study duration, ranging from days to months. This extended timeframe presents the opportunity to evaluate agents with chronic mechanisms of hERG inhibition, such as protein trafficking blockers [18] and the experimental oligonucleotides demonstrated in this study. For example, two negative nontargeting siRNAs were tested, and no effects were observed, which indicated that siRNA transfection did not impact hERG expression and subsequent function through nonspecific interaction. The experimental observations in hiPSC‐CM showed that a 48‐h interval following transfection was optimal to detect the inhibitory effect of a KCNH2 siRNA on cardiac repolarization. This is consistent with the time course for siRNA inhibition of hERG function through a gene silencing mechanism in a heterologous cell expression system [24] and agrees with previous research that a longer time window (48–96 h) is necessary for siRNA‐mediated gene silencing [42, 43].
A recent FDA report described real‐world case examples of cardiotoxicity assessments that leveraged hiPSC‐CM‐based assays and reinforces the potential of this human‐relevant model to transform regulatory decision‐making [44]. While there are challenges to overcome, these context‐of‐use case examples highlight the promise of hiPSC‐CM‐based assays to provide accurate, efficient, and clinically relevant cardiac safety data. As scientific experience and confidence grow, the acceptance and integration of hiPSC‐CM‐based assays into regulatory frameworks will likely become a reality, paving the way for employing this NAM for more effective drug development.
4.3. Evaluating New Chemical Entities for Proarrhythmic Risk Using hiPSC‐CM
Oligonucleotides (ONTs) are designed to specifically target and modulate genetic sequences, offering a high degree of single‐target specificity [2, 45] which minimizes their off‐target liability, including cardiac dysfunction and proarrhythmic risk. Therefore, the nonspecific off‐target potential of inhibiting hERG function and causing QTc prolongation is expected to be low for ONTs compared to conventional small molecules [46]. Some ONTs are engineered to deliver their payload to specific organs and tend to have minimal exposure in the heart, factors that further lower the risk of disrupting the intracellular machinery of cardiomyocytes, including hERG channel synthesis pathways [4, 47]. A recent review of 29 ONTs (17 approved; 12 experimental) found that as a drug class, ONTs have no risk for QTc interval prolongation or pro‐arrhythma (e.g., Torsades de Pointes) based on nonclinical (ICH S7B) and clinical (ICH E14) studies and postapproval adverse event reporting [5]. The mounting evidence suggests that ONTs would benefit from alternative cardiac safety approaches during preclinical and clinical development, given that the proarrhythmia risk is low and resembles that of large molecule drugs [44]. The ICH S7B guideline recommends a tiered approach for the assessment of small molecule drug‐induced delayed repolarization risk that incorporates two core assays: in vitro hERG and in vivo QTc assays. Evaluating an ONT in this paradigm is challenging and not recommended because both core assays were designed to detect acute or immediate effects associated with maximal drug exposure (Cmax). A more practical and effective approach would be to integrate high‐quality telemetry methods into a repeat dose toxicology study to detect potential QTc prolongation following chronic drug exposure [46]. If evidence of a cardiac safety signal emerged following chronic exposure to a novel ONT (e.g., QTc prolongation is seen in a repeat dose toxicology study), the mechanism of proarrhythmic risk could be addressed in a follow‐up hiPSC‐CM assay with various incubation times to detect either direct (e.g., channel block) or indirect (e.g., hERG knockdown) drug effects, based on the findings in the present report. This approach would be rationale, evidence‐based and perhaps more informative to the need and/or design of subsequent clinical QTc studies, especially with respect to the timing of ECG data collection relative to dosing.
4.4. Limitation of the Study: hKCNH2 siRNA Is Not a Clinically Relevant Positive Control
In the safety pharmacology assessment space, it is the best practice to include positive controls in the same modality to validate assays or methods. Ideally, a therapeutic agent is the best option because translation to clinical findings could be achieved. For example, when testing a small molecule in the hERG assay, dofetilide or moxifloxacin, is a common and clinically relevant positive control. However, E‐4031, as a tool compound used solely for research purposes, is also a common positive control, although it is not developed for clinical use. For new chemical modalities like siRNA, the limited number of siRNA that have attained clinical approval does not inhibit hERG function. To understand what kind of protocol is appropriate for testing an siRNA in the hERG assay, the only scientifically relevant chemical tool to use for validation is a specific siRNA that targets hERG. The mechanism and kinetics of siRNA in silencing hERG gene expression should be the same for many types of siRNA molecules; thus, the protocol we identified using the hERG‐targeting siRNA tool compound should be representative of the modality of oligonucleotides.
5. Conclusion
Human iPSC‐CMs provide a promising in vitro platform for studying the proarrhythmic potential of conventional small molecules and novel modalities and elucidating the various mechanisms involved in regulating hERG function. A significant benefit of current iPSC‐CMs is their capacity to be cultured for extended durations. This facilitates transfection procedures, prolonged drug exposure, and comprehensive cardiac safety pharmacology evaluations, allowing for the characterization of delayed responses over longer time periods.
Author Contributions
Y.Q. wrote the manuscript, designed the research, and analyzed the data; W.G. wrote the manuscript, designed the research, performed the research, and analyzed the data; B.W. and B.G. contributed new reagents; J.V.N., J.C., and H.M.V. designed the research and analyzed the data.
Conflicts of Interest
All authors are employees of Amgen Inc.
Supporting information
Figure S1.–S5.
Acknowledgments
We thank our Amgen colleague, Dr. Renee Hukkanen (Pathology Director, Nonclinical Safety Science) for reviewing and providing valuable suggestions for this manuscript.
Qu Y., Guo W., Wu B., et al., “Delayed Repolarization Caused by hERG Block With Different Drug Modalities Can Be Detected in Stem Cell‐Derived Cardiomyocytes: Incubation Time Matters,” Clinical and Translational Science 18, no. 9 (2025): e70283, 10.1111/cts.70283.
Funding: This study was supported by Amgen Inc.
Yusheng Qu and Wei Guo contributed equally to this work.
References
- 1. Fire A., Xu S., Montgomery M., Kostas S. A., Driver S. E., and Mello C. C., “Potent and Specific Genetic Interference by Double‐Stranded RNA in Caenorhabditis elegans ,” Nature 391 (1998): 806–811. [DOI] [PubMed] [Google Scholar]
- 2. Elbashir S. M., Martinez J., Patkaniowska A., Lendeckel W., and Tuschl T., “Functional Anatomy of siRNAs for Mediating Efficient RNAi in Drosophila melanogaster Embryo Lysate,” EMBO Journal 20 (2001): 6877–6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roberts T. C., Langer R., and Wood M. J. A., “Advances in Oligonucleotide Drug Delivery,” Nature Reviews. Drug Discovery 19 (2020): 673–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang M. M., Bahal T., Rasmussen T. P., Manautou J. E., and Zhong X. B., “The Growth of siRNA‐Based Therapeutics: Updated Clinical Studies,” Biochemical Pharmacology 189 (2021): 114432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Qu Y., Henderson K. A., T. A. Harper, Jr. , and Vargas H. M., “Scientific Review of the Proarrhythmic Risks of Oligonucleotide Therapeutics: Are Dedicated ICH S7B/E14 Studies Needed for Low‐Risk Modalities?,” Clinical Pharmacology and Therapeutics 116 (2024): 96–105. [DOI] [PubMed] [Google Scholar]
- 6. Al Shaer D., Al Musaimi O., Albericio F., and de la Torre B. G., “2023 FDA TIDES (Peptides and Oligonucleotides) Harvest,” Pharmaceuticals 17 (2024): 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Verdin P., “FDA New Drug Approvals in Q2 2024,” Nature Reviews. Drug Discovery 23 (2024): 573. [DOI] [PubMed] [Google Scholar]
- 8. Lauffer M. C., van Roon‐Mom W., Aartsma‐Rus A., and N = 1 Collaborative , “Possibilities and Limitations of Antisense Oligonucleotide Therapies for the Treatment of Monogenic Disorders,” Communication & Medicine 4 (2024): 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Egli M. and Manoharan M., “Chemistry, Structure and Function of Approved Oligonucleotide Therapeutics,” Nucleic Acids Research 51 (2023): 2529–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kehat I., Kenyagin‐Karsenti D., Snir M., et al., “Human Embryonic Stem Cells Can Differentiate Into Myocytes With Structural and Functional Properties of Cardiomyocytes,” Journal of Clinical Investigation 108 (2001): 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Takahashi K. and Yamanaka S., “Induction of Pluripotent Stem Cells From Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors,” Cell 126 (2006): 663–676. [DOI] [PubMed] [Google Scholar]
- 12. Burnett S. D., Blanchette A. D., Chiu W. A., and Rusyn I., “Human Induced Pluripotent Stem Cell (iPSC)‐Derived Cardiomyocytes as an In Vitro Model in Toxicology: Strengths and Weaknesses for Hazard Identification and Risk Characterization,” Expert Opinion on Drug Metabolism & Toxicology 17, no. 8 (2021): 887–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Qu Y., Gao B., Fang M., and Vargas H. V., “Human Embryonic Stem Cell Derived Cardiac Myocytes Detect hERG‐Mediated Repolarization Effects, but Not Nav1.5 Induced Depolarization Delay,” Journal of Pharmacological and Toxicological Methods 68 (2013): 74–81. [DOI] [PubMed] [Google Scholar]
- 14. Haechl N., Ebner J., Hilber K., Todt H., and Koenig X., “Pharmacological Profile of the Bradycardic Agent Ivabradine on Human Cardiac Ion Channels,” Cellular Physiology and Biochemistry 53 (2019): 36–48. [DOI] [PubMed] [Google Scholar]
- 15. Cerignoli F., Charlot D., Whittaker R., et al., “High Throughput Measurement of Ca2+ Dynamics for Drug Risk Assessment in Human Stem Cell‐Derived Cardiomyocytes by Kinetic Image Cytometry,” Journal of Pharmacological and Toxicological Methods 66, no. 3 (2012): 246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Patel D., Stohlman J., Dang Q., Strauss D. G., and Blinova K., “Assessment of Proarrhythmic Potential of Drugs in Optogenetically Paced Induced Pluripotent Stem Cell‐Derived Cardiomyocytes,” Toxicological Sciences 170 (2019): 167–179. [DOI] [PubMed] [Google Scholar]
- 17. Hortigon‐Vinagre M. P., Zamora V., Burton F. L., and Smith G. L., “The Use of Voltage Sensitive Dye di‐4‐ANEPPS and Video‐Based Contractility Measurements to Assess Drug Effects on Excitation–Contraction Coupling in Human‐Induced Pluripotent Stem Cell–Derived Cardiomyocytes,” Journal of Cardiovascular Pharmacology 77, no. 3 (2021): 280–290. [DOI] [PubMed] [Google Scholar]
- 18. Blinova E., Stohlman J., Vicente J., et al., “Comprehensive Translational Assessment of Human‐Induced Pluripotent Stem Cell Derived Cardiomyocytes for Evaluating Drug‐Induced Arrhythmias,” Toxicological Sciences 155, no. 1 (2017): 234–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kilfoil P., Feng S. L., Bassyouni A., et al., “Characterization of a High Throughput Human Stem Cell Cardiomyocyte Assay to Predict Drug‐Induced Changes in Clinical Electrocardiogram Parameters,” European Journal of Pharmacology 912 (2021): 174584. [DOI] [PubMed] [Google Scholar]
- 20. Pradhapan P., Kuusela J., Viik J., Aalto‐Setälä K., and Hyttinen J., “Cardiomyocyte MEA Data Analysis (CardioMDA)—A Novel Field Potential Data Analysis Software for Pluripotent Stem Cell Derived Cardiomyocytes,” PLoS One 8 (2013): e73637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qu Y. and Vargas H. M., “Proarrhythmia Risk Assessment in Human Induced Pluripotent Stem Cell‐Derived Cardiomyocytes Using the Maestro MEA Platform,” Toxicological Sciences 147, no. 1 (2015): 286–295. [DOI] [PubMed] [Google Scholar]
- 22. Gintant G., Kaushik E. P., Feaster T., et al., “Repolarization Studies Using Human Stem Cell‐Derived Cardiomyocytes: Validation Studies and Best Practice Recommendations,” Regulatory Toxicology and Pharmacology 117 (2020): 104756. [DOI] [PubMed] [Google Scholar]
- 23. Narkar A., Willard J. M., and Blinova K., “Chronic Cardiotoxicity Assays Using Human Induced Pluripotent Stem Cell‐Derived Cardiomyocytes (hiPSC‐CMs),” International Journal of Molecular Sciences 23, no. 6 (2022): 3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qu Y., Kirby R., Davies R., et al., “Time Is a Critical Factor When Evaluating Oligonucleotide Therapeutics in hERG Assays,” Nucleic Acid Therapeutics 33, no. 2 (2022): 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Leishman D. J., Brimecombe J., Crumb W., et al., “Supporting an Integrated QTc Risk Assessment Using the hERG Margin Distributions for Three Positive Control Agents Derived From Multiple Laboratories and on Multiple Occasions,” Journal of Pharmacological and Toxicological Methods 128 (2024): 1056–8719. [DOI] [PubMed] [Google Scholar]
- 26. Shah R., “Drugs, QT Interval Prolongation and ICH E14,” Drug Safety 28 (2005): 115–125. [DOI] [PubMed] [Google Scholar]
- 27. Alexandrou A. J., Duncan R. S., Sullivan A., et al., “Mechanism of hERG K+ Channel Blockade by the Fluoroquinolone Antibiotic Moxifloxacin,” British Journal of Pharmacology 147, no. 8 (2006): 905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Florian J. A., Tornøe C. W., Brundage R., Parekh A., and Garnett C. E., “Population Pharmacokinetic and Concentration—QTc Models for Moxifloxacin: Pooled Analysis of 20 Thorough QT Studies,” Journal of Clinical Pharmacology 51 (2011): 1152–1162. [DOI] [PubMed] [Google Scholar]
- 29. US Food and Drug Administration , “ICH E14/S7B Q&as Training Material Examples Supplemental File,” (2022).
- 30. Sands M., Kron M. A., and Brown R. B., “Pentamidine: a review,” Reviews of Infectious Diseases 7 (1985): 625–634. [DOI] [PubMed] [Google Scholar]
- 31. Dennis A. T., Wang L., Wan H., Nassal D., Deschenes I., and Ficker E., “Molecular Determinants of Pentamidine‐Induced hERG Trafficking Inhibition,” Molecular Pharmacology 81, no. 2 (2012): 198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Navin T. R., Gretz J. E., Melby P. C., et al., “Effect of Azotemia in Dogs on the Pharmacokinetics of Pentamidine,” Journal of Infectious Diseases 155 (1987): 1020–1026. [DOI] [PubMed] [Google Scholar]
- 33. Garg P., Garg V., Negroni S., et al., “Human Induced Pluripotent Stem Cell–Derived Cardiomyocytes as Models for Cardiac Channelopathies: A Primer for Non‐Electrophysiologists,” Circulation Research 123 (2018): 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pourrier M. and Fedida D., “The Emergence of Human Induced Pluripotent Stem Cell‐Derived Cardiomyocytes (hiPSC‐CMs) as a Platform to Model Arrhythmogenic Diseases,” International Journal of Molecular Sciences 21 (2020): 657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bishop S. P., Zhang J., and Ye L., “Cardiomyocyte Proliferation From Fetal‐ To Adult‐ and From Normal‐ To Hypertrophy and Failing Hearts,” O Biologico 11, no. 6 (2022): 880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kitaguchi T., Moriyama S., Taniguchi T., et al., “CSAHi Study: Evaluation of Multi‐Electrode Array in Combination With Human iPS Cell‐Derived Cardiomyocytes to Predict Drug‐Induced QT Prolongation and Arrhythmia–Effects of 7 Reference Compounds at 10 Facilities,” Journal of Pharmacological and Toxicological Methods 78 (2016): 93–102. [DOI] [PubMed] [Google Scholar]
- 37. Yamamoto W., Asakura K., Ando H., et al., “Electrophysiological Characteristics of Human iPSC‐Derived Cardiomyocytes for the Assessment of Drug‐Induced Proarrhythmic Potential,” PLoS One 11, no. 12 (2016): e0167348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kodama M., Furutani K., Kimura R., et al., “Systematic Expression Analysis of Genes Related to Generation of Action Potentials in Human iPS Cell‐Derived Cardiomyocytes,” Journal of Pharmacological Sciences 140, no. 4 (2019): 325–330. [DOI] [PubMed] [Google Scholar]
- 39. He J. Q., Ma Y., Lee Y., Thomson J. A., and Kamp T. J., “Human Embryonic Stem Cells Develop Into Multiple Types of Cardiac Myocytes: Action Potential Characterization,” Circulation Research 93 (2003): 32–39. [DOI] [PubMed] [Google Scholar]
- 40. Peng S., Lacerda A. E., Kirsch G. E., Brown M., and Bruening‐Wright A., “The Action Potential and Comparative Pharmacology of Stem Cell‐Derived Human Cardiomyocytes,” Journal of Pharmacological and Toxicological Methods 61 (2010): 277–286. [DOI] [PubMed] [Google Scholar]
- 41. Honda M., Kiyokawa J., Tabo M., and Inoue T., “Electrophysiological Characterization of Cardiomyocytes Derived From Human Induced Pluripotent Stem Cells,” Journal of Pharmacological Sciences 117 (2011): 149–159. [DOI] [PubMed] [Google Scholar]
- 42. Bartlett D. W. and Davis M. E., “Insights Into the Kinetics of siRNA‐Mediated Gene Silencing From Live‐Cell and Live‐Animal Bioluminescent Imaging,” Nucleic Acids Research 34 (2006): 322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Raab R. M. and Stephanopoulos G., “Dynamics of Gene Silencing by RNA Interference,” Biotechnology and Bioengineering 88 (2004): 121–132. [DOI] [PubMed] [Google Scholar]
- 44. Yang X., Ribeiro A. J. S., Pang L., and Strauss D. G., “Use of Human iPSC‐CMs in Nonclinical Regulatory Studies for Cardiac Safety Assessment,” Toxicological Sciences 190, no. 2 (2022): 117–126. [DOI] [PubMed] [Google Scholar]
- 45. Tuschl T., Zamore P. D., Lehmann R., Bartel D. P., and Sharp P. A., “Targeted mRNA Degradation by Double Stranded RNA In Vitro,” Genes & Development 13 (1999): 3191–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Abernathy M. M. and Leishman D. J., “The Efficacy, Effectiveness, and Efficiency of Integrated QTc Assessment: Rationalizing Approaches to New Drug Modalities,” Clinical Pharmacology and Therapeutics 116 (2024): 22–25. [DOI] [PubMed] [Google Scholar]
- 47. Setten R. L., Rossi J. J., and Han S. P., “The Current State and Future Directions of RNAi‐Based Therapeutics,” Nature Reviews. Drug Discovery 18 (2019): 421–446. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.–S5.