

ABSTRACT
Postglacial environmental changes have influenced biodiversity and species evolution, yet the genomic and demographic responses of parasites remain underexplored. This study investigates the population genetics and demographic history of the flatworm Phyllodistomum umblae, a generalist trematode at the definitive host level infecting Coregonus spp. across perialpine and subarctic postglacial lakes. Additionally, we compare its demographic patterns to Proteocephalus fallax, a whitefish specialist tapeworm, to elucidate how ecological strategies shape evolutionary responses to environmental fluctuations. Genomic data from ddRAD sequencing revealed clear genetic differentiation in P. umblae between subarctic and perialpine regions, likely driven by geographic isolation during glacial cycles. Low genetic differentiation suggests hydrological connectivity and the parasite's ability to utilise several host species as definitive hosts. Demographic inference uncovered distinct evolutionary trajectories between P. umblae and Pr. fallax. During the Last Glacial Period (~115–11 kya), P. umblae populations underwent declines, followed by rapid postglacial expansions after the Last Glacial Maximum (~15–10 kya). In contrast, Pr. fallax exhibited older historical fluctuations, including pronounced bottlenecks during the Middle Pleistocene (~300 kya). Its populations remained stable during the LGP, likely due to host persistence in glacial refugia unavailable in earlier glaciation periods. These findings align with the taxon pulse concept within the Stockholm Paradigm, highlighting how glacial cycles triggered episodic population contractions and expansions. By integrating genomic and historical data, this study (1) underscores parasites as models for understanding ecological and evolutionary processes and (2) provides insights into biodiversity resilience and adaptation to past and future environmental changes.
Keywords: ddRAD‐seq, demographic inference, freshwater, population genetics, Trematoda
1. Introduction
Pleistocene glaciations, which occurred over the past 2.6 million years, were characterised by repeated cycles of glacial expansion and contraction that had a profound impact on the distribution and evolution of many organisms (Schluter 2000; Hewitt 2004; Schmitt 2007; Bernatchez et al. 2010). During glacial periods, species were often confined to refugia, that is, isolated areas where conditions remained favourable for survival (Willis and Whittaker 2000). These refugia served as reservoirs of genetic diversity, and during interglacial periods, the retreat of glaciers allowed species to expand into newly available habitats, triggering processes of colonisation and adaptation (Habel et al. 2010; Husemann et al. 2014). These glacial and postglacial dynamics align with the Stockholm Paradigm (Hoberg and Brooks 2015), a conceptual framework that links environmental change to patterns of diversification through four interacting processes: taxon pulse, ecological fitting, oscillation and geographic colonisation. Within this framework, the taxon pulse concept describes episodic expansions and contractions of species ranges in response to climatic fluctuations, leading to cycles of isolation, reconnection and adaptation (Erwin 1985; Agosta and Brooks 2020). This theoretical lens provides a valuable basis for interpreting how repeated glacial cycles may have shaped the evolutionary outcomes across species. Throughout the late Quaternary, significant environmental changes and dramatic climatic fluctuations profoundly impacted species distributions (Hewitt 2000; Smith et al. 2022). Thus, understanding how species evolved and adapted throughout these glacial and postglacial cycles is crucial for revealing broader patterns of biodiversity and evolution. Additionally, studying these past climatic shifts can provide critical information for anticipating future changes in the face of ongoing climatic change.
With the advent of novel DNA technologies, researchers can now uncover the genetic imprints of these historical shifts, offering powerful insights into the mechanisms underlying biodiversity and speciation (Cahill et al. 2013; Seehausen et al. 2014; Johannesson et al. 2020). Studies of the impact of Pleistocene glaciations have predominantly focused on free‐living organisms (e.g., Salvi et al. 2013; Cabanne et al. 2016; Nevado et al. 2018; Arcones et al. 2021), whereas the effect on parasites remains vastly underexplored. This lack of attention is problematic because parasite studies can potentially provide unprecedented insights not only into historical host geographical expansions and host–parasite interactions, but also into how ecosystems respond to dramatic climatic events. Parasites, due to their potentially higher mutation rates compared to their hosts, may accumulate genetic differences at a faster rate, which allows detecting demographic and evolutionary shifts with higher precision (Huyse et al. 2005). Thus, they can function as a ‘magnifying glass’, revealing subtle changes in host populations and environmental conditions that might not be easily detected focusing on the hosts themselves (Geraerts et al. 2022). Moreover, because parasite life cycles and dispersal are intricately tied to those of their hosts, parasites can provide valuable insights into the genetic structure of host populations, offering a broader understanding of ecological and evolutionary processes (Whiteman and Parker 2005; Gagne et al. 2022). Although previous studies have used parasites to infer host dynamics (Kmentová et al. 2019; Santoro et al. 2020), there has been limited focus on how parasites themselves respond to environmental changes. We propose that investigating the genomic diversity and population dynamics of parasites may provide key insights into their evolutionary trajectories, evaluate their resilience and identify their vulnerability to future environmental challenges, relevant in the current context of rapid global change.
Following the retreat of glaciers after the Last Glacial Maximum (LGM), ca.10,000–15,000 years ago, numerous post‐glacial lakes formed across Europe (Hewitt 2000; Brooks et al. 2011). The postglacial formation of these new environments created unique opportunities for studying evolutionary processes, serving as natural laboratories in which diversification occurred over shared timeframes (Schluter 1996; Hudson et al. 2011). The European whitefish ( Coregonus lavaretus species complex) has been extensively studied in this light (Østbye et al. 2005; Vonlanthen et al. 2012; Adams et al. 2016; Häkli et al. 2018; Selz et al. 2020; Crotti, Bean, et al. 2021; Crotti, Yohannes, et al. 2021). This species complex has undergone an intricate history of parallel diversification in lakes across the Northern Hemisphere, following the retreat of the ice shield after the LGM. Hence, whitefish exhibit an interesting evolutionary history across European freshwater systems (Østbye et al. 2005; Hudson et al. 2011) and are often used as a model system for studying rapid speciation and adaptive radiation (Hudson et al. 2007; Crotti, Bean, et al. 2021; Crotti, Yohannes, et al. 2021). However, the evolutionary trajectories of their parasitic fauna have been barely studied. Brabec et al. (2024) showed that the whitefish post‐glacial expansion and subsequent rapid diversification facilitated parasite colonisation and replicated spatial differentiation in these newly formed post‐glacial lakes. This whitefish diversification may have led to the creation of a variety of niches and ecological opportunities, providing parasites with diverse new resources to explore and exploit.
Here, we will focus on the population genomics of two parasitic platyhelminth species infecting European whitefish, Phyllodistomum umblae (Digenea) and Proteocephalus fallax (Eucestoda). The former is a generalist parasite at the definitive host level, infecting various salmoniform species of Coregonus, Salmo, Salvelinus and Thymallus (Moravec 2004; Soldánová et al. 2017; Faltýnková et al. 2020; Rochat et al. 2021). Although the life cycles of most Phyllodistomum species have not been fully elucidated, it is known that P. umblae successively infects freshwater bivalves (Sphaeriidae) as first intermediate hosts, and possibly Plecoptera nymphs as second intermediate hosts (Butorina et al. 2008; Petkevičiūtė et al. 2014, 2015; Siwertsson et al. 2016; Güven and Öztürk 2022) before reaching maturity in the definitive fish host. In contrast, Pr. fallax follows a more specific life cycle involving copepods (Cyclopidae and Diaptomidae) as intermediate hosts, with Coregonus spp. as its exclusive definitive host (Scholz 1999; Brabec et al. 2023; Scholz et al. 2024). The broader host range of P. umblae likely enhances its dispersal potential, as it can complete its life cycle in multiple fish species across different habitats. In contrast, the dependency of Pr. fallax on a single definitive host restricts its movement. Parasites infecting multiple definitive host species often show higher dispersal ability and reduced genetic structure (Huyse et al. 2005).
Brabec et al. (2024) explored the differentiation patterns of Pr. fallax across the same lakes covered in our study, showing that the external environment (i.e., abiotic environmental factors outside the host), host availability and microevolutionary forces, such as founder effects and genetic drift, shaped its evolution. These findings provide a valuable basis for comparing the differentiation patterns of Pr. fallax and P. umblae within the same postglacial ecosystems, which will be studied herein. The aim of this study is to elucidate the evolutionary dynamics underlying the demographic fluctuations of parasites in postglacial lakes and their different genetic signatures. To accomplish this, we used a double‐digestion restriction site‐associated DNA (ddRAD) sequencing protocol applied to P. umblae to obtain a high number of sufficiently variable molecular markers for a non‐model flatworm. In addition, we compare the demographic patterns between P. umblae and Pr. fallax based on previously published ddRAD data for the latter (Brabec et al. 2024). This enables a detailed investigation of population dynamics and demographic history for P. umblae and Pr. fallax across perialpine and subarctic postglacial lakes as independent replicates of colonisation events. We hypothesise that there will be low or no genetic differentiation within geographically proximate populations (i.e., within the same region) of the generalist flatworm P. umblae. The ability of the parasite to utilise diverse salmonid hosts would facilitate dispersal and gene flow across interconnected freshwater systems, hampering the emergence of significant local genetic structure. Additionally, and in line with the Pleistocene species range contractions and expansions (i.e., taxon pulses), we also hypothesise that lake populations of P. umblae and Pr. fallax have experienced founder events and population reductions followed up by demographic expansion driven by the availability and opportunity to exploit new hosts. Phyllodistomum umblae is expected to undergo rapid expansion post‐colonisation, whereas Pr. fallax would show a slower expansion due to its inability to utilise a broad host range for dispersion and establishment (Hoberg and Brooks 2008).
2. Methods
2.1. Data Acquisition, ddRAD Sequencing and Processing
Specimens of P. umblae were collected from Coregonus spp. across four perialpine lakes in Switzerland (Lakes Bienne, Brienz, Thun and Walen) and two subarctic lakes in northern Norway (Suohpatjávri and Langfjordvatn), as described in Brabec et al. (2024) (Figure 1). Genomic DNA from 136 specimens of P. umblae was extracted and processed for ddRAD library construction, following the protocol outlined by Brabec et al. (2024). Around 60 ng gDNA per sample was doubly digested with frequent cutters NlaII and MseI, before ligating the adaptors, tagged with 24 different barcodes. We applied 20 cycles of PCR amplification with reverse primers tagged with 16 different indexes. Fragments between 320 and 500 bp were selected for sequencing in six lanes of Illumina HiSeq 2500 with 150 bp paired‐end reads. Demultiplexing and trimming of ddRAD reads were performed using IpyRAD's first step (Eaton and Overcast 2020), followed by quality assessment using FASTQC (Andrews 2010). High‐quality reads were then mapped to the reference genome (see next section) using bwa‐mem2 (Vasimuddin et al. 2019). Then, SAMtools (Li et al. 2009) was used to filter out unmapped reads and to retain reads with a mapping quality score above 20, followed by BAM compression and indexing. After filtering, we kept 131 samples of P. umblae to carry out the population genetic and demographic analysis. The initial dataset for Pr. fallax retrieved from Brabec et al. (2024) comprised 510 specimens. After filtering for specimens with conditions matching those of our study, we retained 447 samples for the demographic analysis.
FIGURE 1.
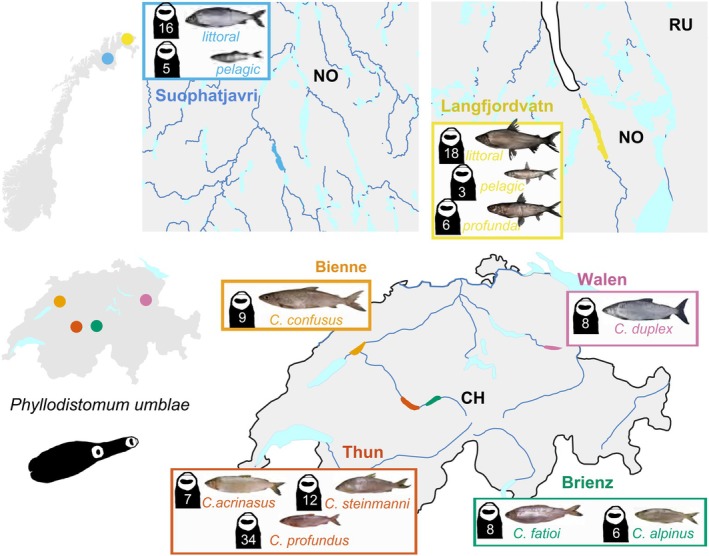
Sampling locations of Phyllodistomum umblae in the perialpine (four lakes in Switzerland ‐CH‐) and subarctic (two lakes in northern Norway ‐NO‐) regions (insets on the left). Lakes in each region are colour‐coded (enlarged maps: Suohpatjávri, blue; Langfjordvatn, yellow; Bienne, orange; Walen, pink; Thun, dark orange; Brienz, dark green). Illustrations of European whitefish species/ecotypes surveyed are accompanied by black silhouettes of P. umblae parasites, with the number of specimens genotyped from each host species indicated.
2.2. Parasite Whole Genome Sequencing and Assembly
Conducting genomic analyses on small organisms, such as parasitic helminths, presents notable challenges because of the limited amount of tissue for the acquisition of sufficient high‐molecular‐weight genomic DNA (gDNA) and the risk of contamination with host tissues (Thorn et al. 2023). To address these constraints, we selected five extractions with the highest DNA quality and fragment size from fresh adult specimens collected from Arctic charr ( Salvelinus alpinus (Linnaeus, 1758)) from Skogsfjordvatn (69°56′30″ N, 19°09′22″ E, Norway) for sequencing with short‐read and long‐read methods. This combination was necessary to balance read length and accuracy: short‐read sequencing ensures high base accuracy, whereas long‐read sequencing enables better assembly of repetitive regions and provides longer contiguity (Nowak et al. 2019).
One specimen with 800 ng gDNA extract was used to construct an Illumina TruSeq DNA PCR‐free library with insert sizes of ~550 bp following the Illumina standard protocol and subjected to paired‐end (2 × 150 bp) sequencing on a NovaSeq6000 platform (Illumina). We used two additional specimen extractions with 400 ng gDNA each separately for library preparation and MinION long‐read sequencing (Oxford NanoporeTM Technologies), with GUPPY base calling using the High‐Accuracy models. Finally, two specimen extractions were combined to a total of 600 ng gDNA for SMRTbell library preparation and PacBio HiFi (Pacific Biosciences) sequencing on a Sequel II. Read quality was assessed using FASTQC (Andrews 2010) and genome size estimation was carried out on Illumina reads using a k‐mer depth frequency distribution analysis performed with GenomeScope (Vurture et al. 2017). We performed a hybrid genome assembly (Nowak et al. 2019) combining short and long‐reads methods, to generate a reference genome that is as complete as possible. PacBio raw reads were assembled, and a first polishing step followed, integrating MinION raw reads using FLYE v2.9.1 (Kolmogorov et al. 2019). We used Illumina reads to perform a second polishing step using ntHits and ntEdit (Warren et al. 2019). Finally, the completeness of the genome was assessed using BUSCO v5 (Manni et al. 2021) with the Metazoa database (metazoa_odb9).
2.3. Extraction of Genetic Markers and Haplotype Network
To investigate the genetic diversity and evolutionary relationships of P. umblae, we analysed both nuclear and mitochondrial genetic markers, which are widely recognised for their utility in evolutionary studies and specimen identification (Blasco‐Costa et al. 2016). Partial sequences of the nuclear large ribosomal subunit RNA gene (lsrDNA) and mitochondrial cytochrome c oxidase subunit 1 (cox1) were extracted from both the reference genome and ddRAD samples. We used phylogenetic tree reconstructions based on both genes to (i) verify the integrity of P. umblae as a single species, which was corroborated through molecular identification of specimens (Supporting Information S1), (ii) assess evolutionary relationships among P. umblae populations, and (iii) evaluate evolutionary relationships of the specimens from whitefish with those infecting other salmonids.
Demultiplexed and trimmed reads of the ddRAD samples were mapped either to the cox1 or the lsrDNA reference sequences using bwa‐mem2 (Vasimuddin et al. 2019) and filtered using SAMtools (Li et al. 2009) (see details in Supporting Information S1). Consensus sequences corresponding to cox1 or lsrDNA loci from all samples were aligned with MAFFT (Katoh et al. 2002), and sites with 75% or more missing data were removed using Gblocks (Castresana 2000). The cox1 sequences, despite containing higher proportions of missing data (average of 23.5%, compared to 7.6% for lsrDNA), provided sufficient variation to allow further analyses. To examine haplotype relationships and assess genetic diversity across regions (Figure 1), we selected the cox1 sequences to build the haplotype network using PEGAS (Paradis 2010) and geneHapR (Zhang et al. 2023) R packages (R Core Team 2024). Finally, the most dominant cox1 haplotype (and the most complete sequence) from each lake, along with sequences of representative congeneric species of Phyllodistomum from GenBank, were used to generate a dataset for phylogenetic inference, focusing on evolutionary relationships within the genus (see Supporting Information S2).
2.4. Population Genetic Analyses
Since our ddRAD sequence coverage was low (3X–5X mean), we opted to work with ANGSD (Korneliussen et al. 2014), which is suitable to deal with low to medium sequencing depth data. This program operates within a probabilistic context using genotype likelihoods (i.e., probability of observing a particular sequence data given a particular genotype, at a particular location in the genome of a particular individual) (Korneliussen et al. 2014; Zhao et al. 2022), incorporating the uncertainty that may have been introduced in base calling, alignment, and assembly. We applied ANGSD to our ddRAD mapped reads to extract the genotype likelihoods, identify minor and major alleles, and call the SNPs. Only a subset of SNPs shared by at least 50% of the samples was retained. We used ngLSD (Fox et al. 2019) to estimate linkage disequilibrium among variable sites, and filtered them to keep r2 < 0.85. Genetic structure was investigated using PCAngsd (Meisner and Albrechtsen 2018), admixture proportions with NGSadmix (Skotte et al. 2013) for K values from 2 to 6 with up to 8000 iterations, and the most adequate number of clusters was evaluated with evalAdmix (Garcia‐Erill and Albrechtsen 2020). We estimated the identity by state (IBS) among samples and plotted it as a heatmap in R. We also estimated the site frequency spectrum (SFS) for each lake population and the fixation index (F ST) for every pair thereof with realSFS (Korneliussen et al. 2014).
2.5. Demographic History
Demographic changes in the last 700,000 years (beginning of the Middle Pleistocene) were investigated for P. umblae and Pr. fallax. Compressed fastq files for Pr. fallax sequences were retrieved from the data repository (NCBI's BioProject no. PRJNA910576, ddRAD accessions SAMN32132446–959, reference genome accession SAMN32134074) and were processed following the same pipeline as for P. umblae. Both species were sampled at the same time, locations, and came from the same host samples (Brabec et al. 2024). We used StairWay Plot 2 (Liu and Fu 2020) from the SFSs to infer demographic changes. We estimated the mutation rate following Lynch (2010), using a linear regression of log‐genome size and log‐mutation rate. This follows the assumption that mutation rates per nucleotide site per generation (u) scale with genome size (G). In addition to data provided by Lynch (2010), we included data from Taenia species (Wang et al. 2016) and from Ligula intestinalis (Nazarizadeh et al. 2023). The models yielded estimates of 3.66 × 10−9 and 5.06 × 10−9 substitutions/site/year for P. umblae and Pr. fallax, respectively. The contemporary effective population sizes and historical fluctuations were estimated for each lake population (six populations for both P. umblae and Pr. fallax). However, since the Stairway Plot relies on the informative power of the SFS (Terhorst and Song 2015; Reid and Pinsky 2022) the resolution and precision of the P. umblae dataset was insufficient to run the analysis at the lake level (see Supporting Information S5). Therefore, given the low F ST values within one region (see results below and Brabec et al. 2024), we treated each region as a ‘metapopulation’ to achieve higher resolution in the demographic analysis.
3. Results
3.1. De Novo Genome Assembly
The draft genome of P. umblae had a total length of 450 Mb, comparable to the k‐mer estimate from Illumina raw reads (350 Mb). Sequencing data included 226,262 PacBio reads with a mean read length of 11,989 bp, 249,845 MinIon reads averaging 7248 bp, and 383,555,354 Illumina TrueSeq reads. BUSCO metrics indicated moderate genome completeness, with 44.6% of expected genes identified as complete (see Table S3), which is consistent with expectations for non‐model flatworms.
3.2. Haplotype Diversity and Differentiation of Phyllodistomum umblae Populations
The partial cox1 and lsrDNA sequences extracted from the reference genomes were 1461 and 3873 bp long, respectively. The cox1 and lsrDNA sequences extracted from all ddRAD samples contained varying proportions of missing data, with average completeness of cox1 (23.5%) higher than that of lsrDNA (7.6%). The lsrDNA sequences from ddRAD samples were nearly identical across all samples (after removing sites with more than 5% missing data). The cox1‐based haplotype network revealed two distinct clusters of P. umblae separated by seven mutational steps (Figure 2). The first cluster corresponds to P. umblae populations from the subarctic region, whereas the second cluster includes populations from the perialpine region. Notably, the dominant haplotype in the perialpine cluster was also observed in three individuals from the subarctic Lake Suohpatjávri, highlighting a shared genetic variant between regions. Haplotypes within each cluster showed lower levels of differentiation, with few mutational steps separating them. Phylogenetic analyses depicted a strongly supported monophyletic clade, including representative sequences of P. umblae from whitefish from different lakes and conspecific sequences available from Genbank (Supporting Information S2).
FIGURE 2.

Haplotype network of Phyllodistomum umblae cox1 gene, sampled from European whitefish (Coregonus spp.) hosts in perialpine (Bienne, Brienz, Thun and Walen) and subarctic (Langfjordvatn and Suohpatjávri) lakes. The network was computed from uncorrected distances among 131 sequences, 504 nucleotides long, with geneHapR (Zhang et al. 2023). Hash marks represent mutational steps and the size of each circle is proportional to the haplotype frequency.
3.3. Population Genetic Structure of Phyllodistomum umblae
The quality metrics showed that over 90% of the reads had average Phred scores of 30 or higher, indicating high‐quality sequences suitable for downstream analyses. The ddRAD sequencing of P. umblae produced a total of 238,613,554 high‐quality trimmed reads (average ± standard deviation of 1,754,511 ± 570,393 reads per individual). Mapping to the newly generated reference genome yielded an average alignment rate of 41.9% after data cleaning. Using ANGSD, we identified 36,056 bi‐allelic SNPs shared by at least 50% of the samples. The draft genome used as reference showed moderate completeness (BUSCO 44.6%), which may have limited the recovery of some genomic regions and contributed to the proportion of unmapped reads. The genetic structure of P. umblae populations revealed a clear differentiation between subarctic and perialpine regions, as shown by distinct clusters in the PCA (Figure 3A, PC1: 48.29%) and admixture plots (Figure 3B, K = 4). Subarctic populations from Langfjordvatn and Suohpatjávri were differentiated in the admixture analysis, with no evidence of mixed ancestry between the two lakes. In contrast, the perialpine populations (Bienne, Brienz, Thun and Walen) showed minimal differentiation in the PCA (Figure 3A) and in the IBS heatmap (Figure 3C). However, the Lake Walen population appeared distinct from the other perialpine lakes (Figure 3B, but also see Supporting Information S4). Pairwise F ST values (Figure 3D) supported these findings, showing substantial differentiation between subarctic and perialpine regions (F ST ~ 0.2) and none to minimal genetic differentiation across lakes within each region (F ST ~ 0 to 0.1).
FIGURE 3.

Phyllodistomum umblae population genetic structure across all lakes and regions. (A) Principal component analysis showing PC1 and PC2, with each dot representing an individual and colour representing the different lakes. Only the first axis differentiated the populations between the subarctic and perialpine regions. (B) Ancestry proportions from NGSadmix analysis, where the most adequate number of clusters for P. umblae, was K = 4, according to evalAdmix. (C) Heatmap representing the pairwise distances based on the identity by state (IBS) matrix estimated with ANGSD. Colours correspond to similarity between two multilocus genotypes, from yellow (low) to dark red (high). The populations of P. umblae are closely related within each region, but differentiated between regions. (D) Matrix of pairwise FST values between lakes, estimating genetic differentiation among P. umbale populations. Higher FST values (darker shades) indicates stronger genetic differentiation, mainly between the subarctic and perialpine regions, while populations within each region show lower differentaition.
3.4. Demographic History
Demographic plots of P. umblae and Pr. fallax from the subarctic and perialpine metapopulations, spanning the Late Pleistocene (~126–11 kya) and Middle Pleistocene (~774–126 kya), revealed several demographic fluctuations (Figure 4). At the metapopulation level, the ranges of effective population size were 6–10 × 106 and 4–10 × 106 individuals for P. umblae and Pr. fallax, respectively. Historical fluctuations for P. umblae are inferred from the beginning of the Last Glacial Period (LGP; ~115–11 kya, see legend in Figure 4), whereas Pr. fallax exhibits evidence of earlier demographic events extending into the Middle Pleistocene. The demographic reconstructions are presented with confidence intervals: the dark grey line represents the 75% interval, and the light grey represents the 95%. These ranges illustrate the inherent uncertainty of the estimates, particularly regarding the timing of older demographic events, which should be interpreted with caution when using SFS‐based models such as Stairway Plot.
FIGURE 4.

Demographic history inference of ancestral metapopulations: (A) perialpine region of P. umblae, (B) subarctic region of P. umblae, (C) perialpine region of Pr. fallax, and (D) subarctic region of Pr. fallax. The X‐axis represents time in thousands of years before present (kya) and the Y‐axis displays the mean effective population size (N e) in millions of individuals, both axis are on a logarithmic scale. StairWay plots based on site frequency spectrum (SFS). The red line in the plots represents the mean, grey lines indicate 75% (dark) and 95% (light) confidence intervals. (E) Map showing the extent of ice sheets during the Saalian Glaciation (~300–130 kya) (in dark red) and the Last Glacial Maximum (~15–10 kya) (in dark blue) (Ehlers et al. 2023; Held et al. 2024), with pink dots indicating the locations of the study areas, and red boxes the estimated position of glacial refugia (Østbye et al. 2005).
A significant bottleneck occurred in Pr. fallax populations ~200 to ~300 kya in ancestors of the subarctic and perialpine metapopulations respectively (Figure 4B,D), coinciding with the Saalian Glaciation in Europe (~300–130 kya), followed by a rapid recovery in population size. During the LGP, ancestors of both metapopulations underwent another significant decline in population size between ~60 and 80 kya. In the ancestors of the perialpine metapopulation, this decline was followed by a relatively rapid recovery, whereas in those of the subarctic metapopulation, the population size remained low and relatively stable for several thousand years, until the LGM, when the population size grew. The demographic history of P. umblae is marked by continuous population declines during the LGP (Figure 4A,C). In the ancestors of the subarctic metapopulation, an early decline occurred at the start of the LGP, followed by a stabilisation period, interrupted by a small population expansion ~30 kya, lasting until the LGM. Following the LGM, a pronounced bottleneck occurred ~8 kya, followed by rapid population recovery and expansion between ~8 and 6 kya, after which the population stabilised. Among the ancestors of the perialpine metapopulation, P. umblae experienced a continuous decline throughout the LGP, with a pronounced bottleneck ~15 kya, followed by a modest recovery. After the LGM, a second bottleneck between ~8 and 7 kya was observed, followed by rapid population growth over a relatively short period. Thus, both species show a pattern of repeated declines, genetic diversity loss, and partial recoveries, with population sizes eventually stabilising.
4. Discussion
The population dynamics and genetic patterns of parasitic species in postglacial lake environments remain relatively underexplored, leaving significant gaps in our understanding of how these organisms respond to environmental changes. Our results of several P. umblae populations reveal marked genetic differentiation between the subarctic and perialpine regions, whereas genetic structure across lakes within each region was low or absent. The latter result supports our initial hypothesis that proximate spatial distribution of lakes within regions would lead to local factors, such as shared host species (generalist parasite) and interconnected water systems, to favour genetic exchange within regions, thereby maintaining gene flow among lake populations. Demographic inference indicated that both P. umblae (this study) and Pr. fallax (data from Brabec et al. 2024) experienced a series of demographic fluctuations over an extended timeframe, spanning the Middle and Late Pleistocene (~126–11 kya). In both species, cycles of ice sheet expansion and contraction caused reductions in effective population size and genetic diversity, resulting in bottlenecks that left small founding populations. The subsequent recovery and expansion, that is, taxon pulse, of these populations support our hypothesis of postglacial expansion driven by recolonisation from isolated refugia populations. Thus, our findings underscore how recurring glacial environmental cycles have profoundly shaped the demographic trajectories of P. umblae and Pr. fallax. By examining these dynamics in parasite species, our study demonstrates how such organisms may respond to large‐scale climatic events, providing a novel way to study the ecological and evolutionary history of parasitic taxa and their free‐living host species.
Phyllodistomum umblae parasitizing Coregonus spp. was confirmed to represent a single species across regions, aligning with findings of limited genetic divergence among parasite populations infecting various fish species. The genetic differentiation observed in P. umblae between subarctic and perialpine regions conforms with the pattern of European whitefish recolonisation of Western Europe from two distinct refugia (Østbye et al. 2005). These recolonisation events created geographical segregation among whitefish populations (Hudson et al. 2007; Crotti, Bean, et al. 2021; Crotti, Yohannes, et al. 2021), and the combination of geographical distance between regions and restricted gene flow further contributed to the differentiation of their parasitic symbionts. However, despite their different recolonisation origin, genetic exchange between subarctic and alpine populations might have occurred. This idea is supported by two lines of evidence: (i) the mitochondrial haplotype shared between Suohpátjavri and perialpine populations of P. umblae, and (ii) the lower genetic differentiation observed in the perialpine population of Lake Bienne, located furthest north among the studied perialpine lakes. One plausible scenario is that populations originating from the southern glacial refugium (i.e., ancestors of perialpine populations) migrated northwards during postglacial recolonisation. This movement could have occurred through meltwater drainages, such as the Rhine and Elbe river systems, which provided connections to the Baltic Ice Sea and to Northern Europe as glaciers retreated (Østbye et al. 2005). Occasional long‐distance gene flow could also have occurred through meltwater drainage systems that connected northern and central European basins during deglaciation, or via anthropogenic fish translocations between lakes.
Although P. umblae is a generalist parasite that infects several salmonid hosts (Moravec 2004), the pattern of population differentiation within each region is quite different. In the subarctic region, Langfjordvatn and Suohpatjávri, located c. 280 km apart in separate water basins with no current hydrological connection, may have been influenced by larger freshwater systems formed during the glacial melting period, such as those associated with the Baltic Ice Lake (Ehlers 2022). Such past connections, followed by periods of isolation as the ice retreated and freshwater systems reorganised, could have contributed to P. umblae population differentiation through restricted migration and dispersal (i.e., vicariance), as for whitefish (Præbel et al. 2013). In contrast, the absence of substantial genetic structure of P. umblae in the perialpine region may result from the interconnected nature of the lakes within the Rhine watershed (Yanites et al. 2013). This watershed connects various lakes through river capture and incision events, creating a network of waterways that may facilitate (or have facilitated) gene flow among parasite populations. In fact, patterns of minor genetic differentiation of P. umblae within the perialpine region correlated with the varying strengths of interlake connections, suggesting that the degree of hydrological linkage influences gene flow dynamics. Additionally, this pattern is consistent with the population structure of the hosts, where weak or absent genetic differentiation has been reported among some perialpine Coregonus populations, particularly those inhabiting hydrologically connected systems (e.g., Vonlanthen et al. 2012; Selz et al. 2020; Crotti, Bean, et al. 2021; Crotti, Yohannes, et al. 2021). For example, Lakes Thun and Brienz were part of a larger postglacial lake as the ice retreated (Selz et al. 2020), which would explain the homogeneity in parasite populations observed. By contrast, P. umblae populations in Lake Walen exhibited slight genetic differentiation. This lake is hydrologically connected to Lake Zurich, but remains relatively isolated from Lakes Thun and Brienz and traces its origins to a larger postglacial lake system shaped by recolonisation events from multiple glacial refugia (Østbye et al. 2005; Hudson et al. 2007). Thus, the lake isolation from other perialpine lakes, combined with its colonisation history and anthropogenic activities (such as stocking practices), may account for the weak differentiation pattern observed here.
The regional differentiation in P. umblae aligns with patterns observed in Pr. fallax (Brabec et al. 2024). Similar to P. umblae, Pr. fallax exhibits regional genetic differentiation likely influenced by the geographic separation of Coregonus spp. host populations resulting from recolonisation from distinct refugia. However, Pr. fallax shows higher local genetic structuring, probably driven by its strict specificity to Coregonus spp. and other ecological traits, such as variations in habitat use and trophic behaviour (Brabec et al. 2024). This difference underscores that while both parasites exhibit regional genetic divergence influenced by geography, Pr. fallax appears to display an additional layer of local structuring driven by host ecological factors. Despite these patterns, neither parasite seems to have diverged to the extent of their Coregonus hosts. Although we surveyed 12 Coregonus species (Figure 1) across both regions, only a single genetic lineage of each parasite persists.
The demographic dynamics of P. umblae and Pr. fallax across the late Quaternary were revealed through the integration of parasitic genetic data and glacial historical factors. The observed low genetic differentiation among lakes within regions in both parasite species permitted the regions to be treated as single ‘metapopulations’. Then, the inferred demographic fluctuations of P. fallax extend back to ~400 kya, encompassing two full glacial cycles—the Penultimate Glacial Period (PGP, ~194–135 kya) and the LGP (~115–11 kya) ‐ whereas those of P. umblae date to ~200 kya, covering the LGP. This discrepancy in the temporal depth of inferred histories likely reflects both biological and technical factors. The larger number of specimens analysed for Pr. fallax (447 vs. 131 for P. umblae) and its higher sequencing depth likely improved the recovery of rare variants and the resolution of coalescent signals, enhancing the depth and accuracy of demographic inference. These factors may have contributed to the inference of a larger ancestral population size and allowed the demographic history of Pr. fallax to be traced further back in time (Terhorst and Song 2015; Lapierre et al. 2017). Furthermore, the use of RADseq and reliance on the SFS may have limited our ability to detect low‐frequency alleles, potentially reducing resolution for recent demographic events (Terhorst and Song 2015).
The decline in effective population size observed in Pr. fallax metapopulations during the Middle Pleistocene (~770–130 kya) coincides with the Saalian Glaciation (~300–130 kya) in northern Europe. This glaciation period encompassed three glacial cycles, characterised by repeated contractions and expansions of the ice sheets over thousands of years, with the PGP (~194–135 kya) marking the last of these cycles (Lauer and Weiss 2018). The timing of advancing ice sheets and harsh climatic conditions is congruent with the pronounced demographic decline in both ancestral metapopulations of Pr. fallax observed. Following the retreat of glaciers after the PGP, a rapid population expansion of Pr. fallax is evident, suggesting successful colonisation as the environment stabilised and freshwater resources became available. During the LGP, the Scandinavian ice sheet extended over northern Europe, reaching as far south as Germany and Poland, whereas during the Saalian Glaciation, it expanded even further south, covering parts of western Europe, including the Netherlands and northern France (Makkaveyev et al. 2022) (Figure 4). This differential glacial advance implies that some refugia available during the LGP were likely inaccessible during the Saalian Glaciation (Figure 4E). For instance, several LGP refugia in the British Isles were covered by ice during this period (Ehlers et al. 2023), which could explain the first pronounced population decline observed in the ancestors of the subarctic metapopulation of Pr. fallax. It is plausible that northern Fennoscandia was recolonised after the glaciation by fish populations expanding from eastern or southeastern refugia (see below) (Østbye et al. 2005), although it remains unclear whether this followed local extinction or colonisation of deglaciation areas. To date, no study has examined the demographic history of Coregonus populations in the subarctic. Our results therefore highlight the potential for future comparative studies integrating host and parasite genomic data to better understand parallel responses to past climate events.
During the LGP, Pr. fallax, which exclusively parasitises Coregonus species as definitive hosts—a cold‐water fish adapted to low temperatures (Elliott and Bell 2011) –, could sustain its life cycle within glacial refugia, enabling stable populations to persist throughout the glacial period. Similarly, the ancestors of the subarctic metapopulation of P. umblae experienced a period of stability during the LGP, likely due to the confinement in glacial refugia. This underscores the role of these refugia in promoting parasite population persistence and acting as reservoirs of genetic diversity during periods of extensive ice coverage (Hoberg et al. 2017). However, the most recent population decline post‐LGM in the ancestral perialpine P. umblae metapopulation is unexpected and suggests demographic contraction despite environmental stabilisation. Such fluctuations may result from genetic drift and selective pressures during colonisation (Blakeslee et al. 2020; Sromek et al. 2023), such as founder events and extinctions in the refugia and its proximity. In addition, the timing of the most recent population expansions in both parasite species suggests that the subarctic region, despite being farther north, was colonised earlier (~9–10 kya) than its perialpine counterpart (Figure 4). This could be due to the proximity of glacial refugia to the newly deglaciated areas (Østbye et al. 2005). Subarctic populations appear to have originated from the northeastern refugium west to the Ural mountains, which provided early access to deglaciated regions via freshwater corridors, allowing colonisation and population expansions ~9–10 kya. In contrast, perialpine populations possibly originated in southern refugia, located south of 53° N, which matches the maximum south‐ward extension of Saalian glaciation (Østbye et al. 2005). Despite the earlier ice sheet retreat in the perialpine region (~15 kya), the greater distance and geographical barriers could have delayed recolonisation, resulting in a later population expansion around ~8–7 kya.
After the LGM (~15–10 kya), P. umblae's sharp population decline suggests founder events ~8 kya, followed by rapid population expansions likely driven by its ability to expand its host range and take advantage of available host opportunities (Hoberg and Brooks 2008; Araujo et al. 2015). In contrast, Pr. fallax populations, which depend on the survival and persistence of their exclusive definitive hosts, Coregonus spp., expanded more slowly (Brabec et al. 2024). These findings highlight how glacial cycles and environmental changes have differentially influenced the long‐term persistence, genetic structure, and evolutionary trajectories of parasite populations.
Parasites, despite being integral to ecosystem function (Brian 2023), remain understudied, particularly in how they respond to significant geological and climatic events. Our study addresses this gap, not only by illustrating how these organisms respond to past environmental changes, but by offering a framework for predicting future responses. Parasites may also serve as early indicators of ecosystem shifts, as their complex life cycles and sensitivity to host availability make them responsive to subtle environmental changes. In the Anthropocene, where rapid climate change and habitat loss are increasingly impacting ecosystems, such studies are essential, particularly for anticipating range shifts and the emergence of parasites (Kafle et al. 2020). The current loss of cold‐water habitats, essential for salmonid species and their parasites, underlines the urgency of understanding the cascading effects of climate change on these systems.
Overall, this work highlights the dual importance of parasites as both subjects of research and models for understanding broad‐scale ecological and evolutionary responses to environmental change. By integrating genetic data with historical climatic events, we revealed insights into the evolutionary history and vulnerability of parasite populations, providing a foundation for future conservation strategies and understanding biodiversity resilience in the face of global change.
Author Contributions
I.B.‐C., J.A.B., M.L.‐R. and J.I.L.‐L. conceived the study; I.B.‐C., R.K., J.B.r, O.S. and J.B. contributed to data collection. M.L.‐R. performed the analyses. M.L.‐R. led the writing, and I.B.‐C., J.A.B. and J.I.L.‐L. contributed to the writing of the manuscript. All authors contributed critically to the drafts and gave final approval for publication.
Disclosure
Data and Code Accessibility: All collapsed and paired‐end sequence data for samples sequenced in this study are available in compressed fastq format through NCBI's BioProject no. PRJNA1282119, together with rescaled and trimmed bam sequence alignments against the reference genome. All scripts used to perform the analyses presented in this paper are available through Zenodo and GitHub (10.5281/zenodo.15776244).
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Figure S1. Phylogenetic interrelationships of Phyllodistomum species parasitizing salmonids and the position of Phyllodistomum umblae within the group.
Figure S2. Phylogenetic interrelationships of Phyllodistomum species parasitizing salmonids and the position of Phyllodistomum umblae within the group.
Figure S3. Principal component analysis across perialpine lakes for Phyllodistomum umblae.
Figure S4. Principal component analysis across subarctic lakes for Phyllodistomum umblae.
Figure S5. Ancestry proportions from NGSadmix analysis, where the most adequate number of clusters for perialpine populations of P. umblae was K = 4 according to evalAdmix (residuals closest to 0).
Figure S6. Ancestry proportions from NGSadmix analysis, where the most adequate number of clusters for subarctic populations of P. umblae was K = 2 or K = 3 (both with the same probability) according to evalAdmix.
Figure S7. Demographic inference for each population (i.e., lake) in perialpine and subarctic region of Pr. fallax.
Figure S8. Estimation of base substitution rate per nucleotide site per generation with genome size.
Table S1. Phyllodistomum species accession information for lsrDNA gene, partial sequences retrived from Genbank.
Table S2. Phyllodistomum species accessions information for cox1 gene, partial sequences retrived from GenBank.
Table S3. Summary statistics of genetic diversity in Phyllodistomum umblae populations from each lake.
Table S4. Summary statistics and BUSCO results for the de‐novo genome assembly for Phyllodistomum umblae.
Table S5. Comparisons of Phyllodistomum umblae population genetic structure (F ST) among lakes.
Acknowledgements
We are thankful to the fisheries authorities responsible for each lake and to the fisherwomen and fishermen who have been essential in the collection of fish specimens. We also thank Oliver Selz, Kim Præbel, Laina Dalsbø, Karin Johannessen, Eloïse Rochat and Janik Pralong for their invaluable assistance in the field or the lab, and Nadir Alvarez and Jean Mariaux for their advice and informal discussions.
Handling Editor: Kayla King
Funding: This work received funding from the Swiss National Science Foundation (SNSF grant 169211 to IB‐C) and the Government of Spain MCIN/AEI/10.13039/501100011033 (PID2019‐104908GB‐I00 and Agencia Estatal de Investigación, PCI2023‐145950‐2 to JAB). MLR was supported by a pre‐doctoral contract from the Government of Spain (MCIN/AEI/10.13039/501100011033) and by the European Union Next Generation programme (EU/PRTR PRE2020‐095070).
Data Availability Statement
All clean data and the scripts used to perform the analyses presented in this manuscript are available through the following Zenodo DOI: 10.5281/zenodo.15776244. See Data Accessibility for raw data.
References
- Adams, C. E. , Bean C. W., Dodd J. A., et al. 2016. “Inter and Intra‐Population Phenotypic and Genotypic Structuring in the European Whitefish Coregonus lavaretus , a Rare Freshwater Fish in Scotland.” Journal of Fish Biology 88: 580–594. [DOI] [PubMed] [Google Scholar]
- Agosta, S. , and Brooks D. R.. 2020. “The Stockholm Paradigm.” In The Major Metaphors of Evolution: Darwinism Then and Now, 219–242. Springer International Publishing. [Google Scholar]
- Andrews, S. 2010. “FastQC A Quality Control Tool for High Throughput Sequence Data.”
- Araujo, S. B. L. , Braga M. P., Brooks D. R., et al. 2015. “Understanding Host‐Switching by Ecological Fitting.” PLoS One 10: e0139225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcones, A. , Ponti R., Ferrer X., and Vieites D. R.. 2021. “Pleistocene Glacial Cycles as Drivers of Allopatric Differentiation in Arctic Shorebirds.” Journal of Biogeography 48: 747–759. [Google Scholar]
- Bernatchez, L. , Renaut S., Whiteley A. R., et al. 2010. “On the Origin of Species: Insights From the Ecological Genomics of Lake Whitefish.” Philosophical Transactions of the Royal Society, B: Biological Sciences 365: 1783–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakeslee, A. M. H. , Haram L. E., Altman I., Kennedy K., Ruiz G. M., and Miller A. W.. 2020. “Founder Effects and Species Introductions: A Host Versus Parasite Perspective.” Evolutionary Applications 13: 559–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco‐Costa, I. , Cutmore S. C., Miller T. L., and Nolan M. J.. 2016. “Molecular Approaches to Trematode Systematics: ‘Best Practice’ and Implications for Future Study.” Systematic Parasitology 93: 295–306. [DOI] [PubMed] [Google Scholar]
- Brabec, J. , Gauthier J., Selz O. M., et al. 2024. “Testing the Radiation Cascade in Postglacial Radiations of Whitefish and Their Parasites: Founder Events and Host Ecology Drive Parasite Evolution.” Evolution Letters 8: 706–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabec, J. , Rochat E. C., Knudsen R., Scholz T., and Blasco‐Costa I.. 2023. “Mining Various Genomic Resources to Resolve Old Alpha‐Taxonomy Questions: A Test of the Species Hypothesis of the Proteocephalus longicollis Species Complex (Cestoda: Platyhelminthes) From Salmonid Fishes.” International Journal for Parasitology 53: 197–205. [DOI] [PubMed] [Google Scholar]
- Brian, J. I. 2023. “Parasites in Biodiversity Conservation: Friend or Foe?” Trends in Parasitology 39: 618–621. [DOI] [PubMed] [Google Scholar]
- Brooks, A. J. , Bradley S. L., Edwards R. J., and Goodwyn N.. 2011. “The Palaeogeography of Northwest Europe During the Last 20,000 Years.” Journal of Maps 7: 573–587. [Google Scholar]
- Butorina, T. E. , Shed'ko M. B., and Gorovaya O. Y.. 2008. “Specific Features of Ecology of Chars of the Genus Salvelinus (Salmonidae) From the Basin of Lake Kronotskoe (Kamchatka) According to Parasitological Data.” Journal of Ichthyology 48: 622–636. [Google Scholar]
- Cabanne, G. S. , Calderón L., Trujillo Arias N., et al. 2016. “Effects of Pleistocene Climate Changes on Species Ranges and Evolutionary Processes in the Neotropical Atlantic Forest.” Biological Journal of the Linnean Society 119: 856–872. [Google Scholar]
- Cahill, J. A. , Green R. E., Fulton T. L., et al. 2013. “Genomic Evidence for Island Population Conversion Resolves Conflicting Theories of Polar Bear Evolution.” PLoS Genetics 9: e1003345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana, J. 2000. “Selection of Conserved Blocks From Multiple Alignments for Their Use in Phylogenetic Analysis.” Molecular Biology and Evolution 17: 540–552. [DOI] [PubMed] [Google Scholar]
- Crotti, M. , Bean C. W., Gowans A. R., et al. 2021. “Complex and Divergent Histories Gave Rise to Genome‐Wide Divergence Patterns Amongst European Whitefish (Coregonus lavaretus).” Journal of Evolutionary Biology 34: 1954–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotti, M. , Yohannes E., Winfield I. J., Lyle A. A., Adams C. E., and Elmer K. R.. 2021. “Rapid Adaptation Through Genomic and Epigenomic Responses Following Translocations in an Endangered Salmonid.” Evolutionary Applications 14: 2470–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton, D. A. R. , and Overcast I.. 2020. “Ipyrad: Interactive Assembly and Analysis of RADseq Datasets.” Bioinformatics 36: 2592–2594. [DOI] [PubMed] [Google Scholar]
- Ehlers, J. 2022. “North Sea and Baltic Sea in the Ice Age.” In The Ice Age, edited by Ehlers J., 307–326. Springer. [Google Scholar]
- Ehlers, J. , Astakhov V., Gibbard P., Hughes P., Mangerud J., and Svendsen J.. 2023. Middle Pleistocene Glaciations in Eurasia. Elsevier. [Google Scholar]
- Elliott, J. A. , and Bell V. A.. 2011. “Predicting the Potential Long‐Term Influence of Climate Change on Vendace ( Coregonus albula ) Habitat in Bassenthwaite Lake, U.K.” Freshwater Biology 56: 395–405. [Google Scholar]
- Erwin, T. L. 1985. The Taxon Pulse: A General Pattern of Lineage Radiation and Extinction Among Carabid Beetles. Dr. W. Junk b.v. Publishers. [Google Scholar]
- Faltýnková, A. , Pantoja C., Skírnisson K., and Kudlai O.. 2020. “Unexpected Diversity in Northern Europe: Trematodes From Salmonid Fishes in Iceland With Two New Species of Crepidostomum Braun, 1900.” Parasitology Research 119: 2439–2462. [DOI] [PubMed] [Google Scholar]
- Fox, E. A. , Wright A. E., Fumagalli M., and Vieira F. G.. 2019. “ngsLD: Evaluating Linkage Disequilibrium Using Genotype Likelihoods.” Bioinformatics 35: 3855–3856. [DOI] [PubMed] [Google Scholar]
- Gagne, R. B. , Crooks K. R., Craft M. E., et al. 2022. “Parasites as Conservation Tools.” Conservation Biology 36: e13719. [DOI] [PubMed] [Google Scholar]
- Garcia‐Erill, G. , and Albrechtsen A.. 2020. “Evaluation of Model Fit of Inferred Admixture Proportions.” Molecular Ecology Resources 20: 936–949. [DOI] [PubMed] [Google Scholar]
- Geraerts, M. , Huyse T., Barson M., et al. 2022. “Mosaic or Melting Pot: The Use of Monogeneans as a Biological Tag and Magnifying Glass to Discriminate Introduced Populations of Nile Tilapia in Sub‐Saharan Africa.” Genomics 114: 110328. [DOI] [PubMed] [Google Scholar]
- Güven, A. , and Öztürk T.. 2022. “Morphological Features of Three Species of Phyllodistomum (Trematoda: Gorgoderidae) From Some Marine Fishes in the Southern Black Sea.” Marine Biological Journal 7: 46–54. [Google Scholar]
- Habel, J. C. , Drees C., Schmitt T., and Assmann T.. 2010. “Review Refugial Areas and Postglacial Colonizations in the Western Palearctic.” In Relict Species, 189–197. Springer. [Google Scholar]
- Häkli, K. , Østbye K., Kahilainen K. K., Amundsen P.‐A., and Præbel K.. 2018. “Diversifying Selection Drives Parallel Evolution of Gill Raker Number and Body Size Along the Speciation Continuum of European Whitefish.” Ecology and Evolution 8: 2617–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held, F. , Cheng H., Edwards R. L., Tüysüz O., Koç K., and Fleitmann D.. 2024. “Dansgaard‐Oeschger Cycles of the Penultimate and Last Glacial Period Recorded in Stalagmites From Türkiye.” Nature Communications 15: 1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt, G. 2000. “The Genetic Legacy of the Quaternary Ice Ages.” Nature 405: 907–913. [DOI] [PubMed] [Google Scholar]
- Hewitt, G. 2004. “Genetic Consequences of Climatic Oscillations in the Quaternary.” Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences 359: 183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoberg, E. P. , and Brooks D. R.. 2008. “A Macroevolutionary Mosaic: Episodic Host‐Switching, Geographical Colonization and Diversification in Complex Host–Parasite Systems.” Journal of Biogeography 35: 1533–1550. [Google Scholar]
- Hoberg, E. P. , and Brooks D. R.. 2015. “Evolution in Action: Climate Change, Biodiversity Dynamics and Emerging Infectious Disease.” Philosophical Transactions of the Royal Society, B: Biological Sciences 370: 20130553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoberg, E. P. , Cook J. A., Agosta S. J., et al. 2017. “Arctic Systems in the Quaternary: Ecological Collision, Faunal Mosaics and the Consequences of a Wobbling Climate.” Journal of Helminthology 91: 409–421. [DOI] [PubMed] [Google Scholar]
- Hudson, A. , Von lanthen P., and Seehausen O.. 2011. “Rapid Parallel Adaptive Radiations From a Single Hybridogenic Ancestral Population.” Proceedings of the Royal Society B: Biological Sciences 278: 58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, A. , Vonlanthen P., Müller R., and Seehausen O.. 2007. “Review: The Geography of Speciation and Adaptive Radiation in Coregonines.” Advances in Limnology 60: 111–146. [Google Scholar]
- Husemann, M. , Schmitt T., Zachos F. E., Ulrich W., and Habel J. C.. 2014. “Palaearctic Biogeography Revisited: Evidence for the Existence of a North African Refugium for Western Palaearctic Biota.” Journal of Biogeography 41: 81–94. [Google Scholar]
- Huyse, T. , Poulin R., and Théron A.. 2005. “Speciation in Parasites: A Population Genetics Approach.” Trends in Parasitology 21: 469–475. [DOI] [PubMed] [Google Scholar]
- Johannesson, K. , Butlin R. K., Panova M., and Westram A. M.. 2020. “Mechanisms of Adaptive Divergence and Speciation in Littorina saxatilis: Integrating Knowledge From Ecology and Genetics With New Data Emerging From Genomic Studies.” In Population Genomics: Marine Organisms, edited by Oleksiak M. F. and Rajora O. P., 277–301. Springer International Publishing. [Google Scholar]
- Kafle, P. , Peller P., Massolo A., et al. 2020. “Range Expansion of Muskox Lungworms Track Rapid Arctic Warming: Implications for Geographic Colonization Under Climate Forcing.” Scientific Reports 10: 17323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. , Misawa K., Kuma K., and Miyata T.. 2002. “MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform.” Nucleic Acids Research 30: 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kmentová, N. , Van Steenberge M., Thys van den Audenaerde D. F. E., et al. 2019. “Co‐Introduction Success of Monogeneans Infecting the Fisheries Target Limnothrissa miodon Differs Between Two Non‐Native Areas: The Potential of Parasites as a Tag for Introduction Pathway.” Biological Invasions 21: 757–773. [Google Scholar]
- Kolmogorov, M. , Yuan J., Lin Y., and Pevzner P. A.. 2019. “Assembly of Long, Error‐Prone Reads Using Repeat Graphs.” Nature Biotechnology 37: 540–546. [DOI] [PubMed] [Google Scholar]
- Korneliussen, T. S. , Albrechtsen A., and Nielsen R.. 2014. “ANGSD: Analysis of Next Generation Sequencing Data.” BMC Bioinformatics 15: 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapierre, M. , Lambert A., and Achaz G.. 2017. “Accuracy of Demographic Inferences From the Site Frequency Spectrum: The Case of the Yoruba Population.” Genetics 206: 439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer, T. , and Weiss M.. 2018. “Timing of the Saalian‐ and Elsterian Glacial Cycles and the Implications for Middle—Pleistocene Hominin Presence in Central Europe.” Scientific Reports 8: 5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker B., Wysoker A., et al. 2009. “The Sequence Alignment/Map Format and SAMtools.” Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , and Fu Y.‐X.. 2020. “Stairway Plot 2: Demographic History Inference With Folded SNP Frequency Spectra.” Genome Biology 21: 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M. 2010. “Evolution of the Mutation Rate.” Trends in Genetics 26: 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makkaveyev, A. N. , Faustova M. A., and Karpukhina N. V.. 2022. “Age and Position of the Maximum Boundary of the Scandinavian Ice Sheet in the Valdai (Weichselian, Vistulian) Glaciation and Relief in the Peripheral Area of the Ice Sheet.” Doklady Earth Sciences 507: S19–S22. [Google Scholar]
- Manni, M. , Berkeley M. R., Seppey M., and Zdobnov E. M.. 2021. “BUSCO: Assessing Genomic Data Quality and Beyond.” Current Protocols 1: e323. [DOI] [PubMed] [Google Scholar]
- Meisner, J. , and Albrechtsen A.. 2018. “Inferring Population Structure and Admixture Proportions in Low‐Depth NGS Data.” Genetics 210: 719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moravec, F. 2004. Metazoan Parasites of Salmonid Fishes of Europe. Academia. [Google Scholar]
- Nazarizadeh, M. , Nováková M., Loot G., et al. 2023. “Historical Dispersal and Host‐Switching Formed the Evolutionary History of a Globally Distributed Multi‐Host Parasite—The Ligula intestinalis Species Complex.” Molecular Phylogenetics and Evolution 180: 107677. [DOI] [PubMed] [Google Scholar]
- Nevado, B. , Contreras‐Ortiz N., Hughes C., and Filatov D. A.. 2018. “Pleistocene Glacial Cycles Drive Isolation, Gene Flow and Speciation in the High‐Elevation Andes.” New Phytologist 219: 779–793. [DOI] [PubMed] [Google Scholar]
- Nowak, R. M. , Jastrzębski J. P., Kuśmirek W., et al. 2019. “Hybrid De Novo Whole‐Genome Assembly and Annotation of the Model Tapeworm Hymenolepis Diminuta.” Scientific Data 6: 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Østbye, K. , Bernatchez L., Næsje T. F., Himberg K.‐J. M., and Hindar K.. 2005. “Evolutionary History of the European Whitefish Coregonus lavaretus (L.) Species Complex as Inferred From mtDNA Phylogeography and Gill‐Raker Numbers.” Molecular Ecology 14: 4371–4387. [DOI] [PubMed] [Google Scholar]
- Paradis, E. 2010. “Pegas: An R Package for Population Genetics With an Integrated–Modular Approach.” Bioinformatics 26: 419–420. [DOI] [PubMed] [Google Scholar]
- Petkevičiūtė, R. , Kudlai O., Stunžėnas V., and Stanevičiūtė G.. 2015. “Molecular and Karyological Identification and Morphological Description of Cystocercous Cercariae of Phyllodistomum Umblae and Phyllodistomum folium (Digenea, Gorgoderidae) Developing in European Sphaeriid Bivalves.” Parasitology International 64: 441–447. [DOI] [PubMed] [Google Scholar]
- Petkevičiūtė, R. , Stunžėnas V., and Stanevičiūtė G.. 2014. “Differentiation of European Freshwater Bucephalids (Digenea: Bucephalidae) Based on Karyotypes and DNA Sequences.” Systematic Parasitology 87: 199–212. [DOI] [PubMed] [Google Scholar]
- Præbel, K. , Knudsen R., Siwertsson A., et al. 2013. “Ecological Speciation in Postglacial European Whitefish: Rapid Adaptive Radiations Into the Littoral, Pelagic, and Profundal Lake Habitats.” Ecology and Evolution 3: 4970–4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . 2024. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. https://www.R‐project.org/. [Google Scholar]
- Reid, B. N. , and Pinsky M. L.. 2022. “Simulation‐Based Evaluation of Methods, Data Types, and Temporal Sampling Schemes for Detecting Recent Population Declines.” Integrative and Comparative Biology 62: 1849–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochat, E. C. , Brodersen J., and Blasco‐Costa I.. 2021. “Conspecific Migration and Environmental Setting Determine Parasite Infracommunities of Non‐Migratory Individual Fish.” Parasitology 148: 1057–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvi, D. , Harris D. J., Kaliontzopoulou A., Carretero M. A., and Pinho C.. 2013. “Persistence Across Pleistocene Ice Ages in Mediterranean and Extra‐Mediterranean Refugia: Phylogeographic Insights From the Common Wall Lizard.” BMC Evolutionary Biology 13: 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro, M. , Palomba M., Mattiucci S., Osca D., and Crocetta F.. 2020. “New Parasite Records for the Sunfish Mola mola in the Mediterranean Sea and Their Potential Use as Biological Tags for Long‐Distance Host Migration.” Frontiers in Veterinary Science 7: 579728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter, D. 1996. “Adaptive Radiation Along Genetic Lines of Least Resistance.” Evolution 50: 1766–1774. [DOI] [PubMed] [Google Scholar]
- Schluter, D. 2000. The Ecology of Adaptive Radiation. OUP Oxford. [Google Scholar]
- Schmitt, T. 2007. “Molecular Biogeography of Europe: Pleistocene Cycles and Postglacial Trends.” Frontiers in Zoology 4: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz, T. 1999. “Life Cycles of Species of Proteocephalus, Parasites of Fishes in the Palearctic Region: A Review.” Journal of Helminthology 73: 1–19. [PubMed] [Google Scholar]
- Scholz, T. , de Chambrier A., Brabec J., Knudsen R., and Blasco‐Costa I.. 2024. “Redescription of Proteocephalus Fallax La Rue, 1911 (Cestoda) and a List of Proteocephalid Tapeworms of Whitefish (Coregonus spp.).” Folia Parasitologica 71: 2024.019. [DOI] [PubMed] [Google Scholar]
- Seehausen, O. , Butlin R. K., Keller I., et al. 2014. “Genomics and the Origin of Species.” Nature Reviews. Genetics 15: 176–192. [DOI] [PubMed] [Google Scholar]
- Selz, O. M. , Dönz C. J., Vonlanthen P., and Seehausen O.. 2020. “A Taxonomic Revision of the Whitefish of Lakes Brienz and Thun, Switzerland, With Descriptions of Four New Species (Teleostei, Coregonidae).” Zookeys 989: 79–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siwertsson, A. , Refsnes B., Frainer A., Amundsen P.‐A., and Knudsen R.. 2016. “Divergence and Parallelism of Parasite Infections in Arctic Charr Morphs From Deep and Shallow Lake Habitats.” Hydrobiologia 783: 131–143. [Google Scholar]
- Skotte, L. , Korneliussen T. S., and Albrechtsen A.. 2013. “Estimating Individual Admixture Proportions From Next Generation Sequencing Data.” Genetics 195: 693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, M. L. , Wallace J., Tank D. C., Sullivan J., and Carstens B. C.. 2022. “The Role of Multiple Pleistocene Refugia in Promoting Diversification in the Pacific Northwest.” Molecular Ecology 31: 4402–4416. [DOI] [PubMed] [Google Scholar]
- Soldánová, M. , Georgieva S., Roháčová J., et al. 2017. “Molecular Analyses Reveal High Species Diversity of Trematodes in a Sub‐Arctic Lake.” International Journal for Parasitology 47: 327–345. [DOI] [PubMed] [Google Scholar]
- Sromek, L. , Ylinen E., Kunnasranta M., et al. 2023. “Loss of Species and Genetic Diversity During Colonization: Insights From Acanthocephalan Parasites in Northern European Seals.” Ecology and Evolution 13: e10608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terhorst, J. , and Song Y. S.. 2015. “Fundamental Limits on the Accuracy of Demographic Inference Based on the Sample Frequency Spectrum.” National Academy of Sciences of the United States of America 112: 7677–7682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorn, C. S. , Maness R. W., Hulke J. M., Delmore K. E., and Criscione C. D.. 2023. “Population Genomics of Helminth Parasites.” Journal of Helminthology 97: e29. [DOI] [PubMed] [Google Scholar]
- Vasimuddin, M. D. , Misra S., Li H., and Aluru S.. 2019. “Efficient Architecture‐Aware Acceleration of BWA‐MEM for Multicore Systems.” In 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS), 314–324. IEEE. [Google Scholar]
- Vonlanthen, P. , Bittner D., Hudson A. G., et al. 2012. “Eutrophication Causes Speciation Reversal in Whitefish Adaptive Radiations.” Nature 482: 357–362. [DOI] [PubMed] [Google Scholar]
- Vurture, G. W. , Sedlazeck F. J., Nattestad M., et al. 2017. “GenomeScope: Fast Reference‐Free Genome Profiling From Short Reads.” Bioinformatics 33: 2202–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Wang S., Luo Y., et al. 2016. “Comparative Genomics Reveals Adaptive Evolution of Asian Tapeworm in Switching to a New Intermediate Host.” Nature Communications 7: 12845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren, R. L. , Coombe L., Mohamadi H., et al. 2019. “ntEdit: Scalable Genome Sequence Polishing.” Bioinformatics 35: 4430–4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteman, N. K. , and Parker P. G.. 2005. “Using Parasites to Infer Host Population History: A New Rationale for Parasite Conservation.” Animal Conservation 8: 175–181. [Google Scholar]
- Willis, K. J. , and Whittaker R. J.. 2000. “The Refugial Debate.” Science 287: 1406–1407. [DOI] [PubMed] [Google Scholar]
- Yanites, B. J. , Ehlers T. A., Becker J. K., Schnellmann M., and Heuberger S.. 2013. “High Magnitude and Rapid Incision From River Capture: Rhine River, Switzerland.” Journal of Geophysical Research: Earth Surface 118: 1060–1084. [Google Scholar]
- Zhang, R. , Jia G., and Diao X.. 2023. “geneHapR: An R Package for Gene Haplotypic Statistics and Visualization.” BMC Bioinformatics 24: 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, L. , Nielsen R., and Korneliussen T. S.. 2022. “distAngsd: Fast and Accurate Inference of Genetic Distances for Next‐Generation Sequencing Data.” Molecular Biology and Evolution 39: msac119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Phylogenetic interrelationships of Phyllodistomum species parasitizing salmonids and the position of Phyllodistomum umblae within the group.
Figure S2. Phylogenetic interrelationships of Phyllodistomum species parasitizing salmonids and the position of Phyllodistomum umblae within the group.
Figure S3. Principal component analysis across perialpine lakes for Phyllodistomum umblae.
Figure S4. Principal component analysis across subarctic lakes for Phyllodistomum umblae.
Figure S5. Ancestry proportions from NGSadmix analysis, where the most adequate number of clusters for perialpine populations of P. umblae was K = 4 according to evalAdmix (residuals closest to 0).
Figure S6. Ancestry proportions from NGSadmix analysis, where the most adequate number of clusters for subarctic populations of P. umblae was K = 2 or K = 3 (both with the same probability) according to evalAdmix.
Figure S7. Demographic inference for each population (i.e., lake) in perialpine and subarctic region of Pr. fallax.
Figure S8. Estimation of base substitution rate per nucleotide site per generation with genome size.
Table S1. Phyllodistomum species accession information for lsrDNA gene, partial sequences retrived from Genbank.
Table S2. Phyllodistomum species accessions information for cox1 gene, partial sequences retrived from GenBank.
Table S3. Summary statistics of genetic diversity in Phyllodistomum umblae populations from each lake.
Table S4. Summary statistics and BUSCO results for the de‐novo genome assembly for Phyllodistomum umblae.
Table S5. Comparisons of Phyllodistomum umblae population genetic structure (F ST) among lakes.
Data Availability Statement
All clean data and the scripts used to perform the analyses presented in this manuscript are available through the following Zenodo DOI: 10.5281/zenodo.15776244. See Data Accessibility for raw data.